
Abstract
Very little is known regarding the mechanism of action for the endothelium-derived hyperpolarizing factor (EDHF) response in cerebral vessels. The authors tested two hypotheses: (1) activation of the cytoplasmic form of phospholipase A2 (cPLA2) is involved with EDHF-mediated dilations in rat middle cerebral arteries; and (2) activation of the cPLA2 involves an increase in endothelial Ca2+ through activation of phospholipase C. Middle cerebral arteries were isolated from the rat, pressurized to 85 mm Hg, and luminally perfused. The EDHF response was elicited by luminal application of uridine triphosphate (UTP) after NO synthase and cyclooxygenase inhibition (10−5 mol/L N-nitro-
Keywords
In arteries and arterioles there is at least one factor or mechanism, not involving nitric oxide/endothelium-derived relaxing factor (NO/EDRF) or cyclooxygenase metabolites (most often prostacyclin), that is responsible for dilations when endothelial receptors are stimulated. The factor or mechanism is termed endothelium-derived hyperpolarizing factor (EDHF) and is characterized by the following: (1) it requires endothelium, (2) it is distinct from both NO/EDRF and prostacyclin, (3) it relaxes by hyperpolarizing the vascular smooth muscle, and (4) it involves calcium-activated potassium channels (KCa) (Golding et al., 2002). The EDHF response can be elicited in cerebral vessels by ATP, ADP, UTP, substance P, A23187 (Ca2+ ionophore), and acetylcholine (see Golding et al., 2002 for a brief review).
Very little is known regarding the mechanism of action of the EDHF response in cerebral vessels. There are, however, three observations that support the idea that the EDHF mechanism is different in cerebral and peripheral vessels. First, the EDHF-mediated relaxations in guinea pig middle cerebral arteries (MCAs) and mesenteric resistance vessels were differently affected by a number of pharmacological inhibitors, including those that inhibit cytochrome P450 enzymes and potassium channels (Dong et al., 2000). Second, the presence of basal concentrations of NO attenuated the EDHF response by 50% to 100%, depending on the agonist and concentration in peripheral arteries (Bauersachs et al., 1996). In rat MCAs, however, basal or above basal concentrations of NO had no effect on the EDHF response (Schildmeyer and Bryan Jr., 2002). Third, estrogen downregulated the EDHF response in cerebral vessels (Golding and Kepler, 2001, Xu et al., 2001) while it upregulated the response in peripheral vessels (Tagawa et al., 1997; McCulloch and Randall, 1998). Therefore, the mechanism for the EDHF response in cerebral vessels is unique and cannot be extrapolated from peripheral mechanisms. It must be determined independently from that in peripheral vessels.
The purpose of the present study was to determine whether activation of phospholipase A2 (PLA2) is involved with EDHF-mediated dilations. One important pathway for the activation of cytoplasmic PLA2 (cPLA2) involves an increase in Ca2+ through the activation of phospholipase C (PLC) and subsequent liberation of inositol 1,4,5-triphosphate (IP3). After determining that activation of PLA2 is involved, subsequent goals were to determine whether the PLC and an increase in cytoplasmic Ca2+ were also steps in the EDHF response.
MATERIALS AND METHODS
The Animal Protocol Review Committee at Baylor College of Medicine approved the experimental protocol. MCAs were harvested from male Long Evans rats (250 to 350 g) after isoflurane (3%) anesthesia (Bryan Jr. et al., 1996). Micropipettes were inserted into the ends of each MCA segment, and the vessel was secured in place using nylon ties. Each MCA was bathed luminally and abluminally in a 37°C physiologic salt solution (PSS), which was equilibrated with a gas mixture of 20% O2/5% CO2/balance N2. The pH of the PSS was 7.4, P
MCAs were pressurized to 85 mm Hg by raising PSS-containing reservoirs, connected to the micropipettes by tubing, above the vessel. Pressure transducers on either side of the MCA allowed measurement of the perfusion pressure across the vessel. Luminal flow was adjusted to 150 μL/min by setting inflow and outflow reservoirs at different heights (Bryan Jr. et al., 1996). Each MCA was visualized after magnification using a video monitor. Diameters were continuously measured using Optimus image-analysis software (Bothell, WA, U.S.A.).
After warming and pressurizing to 85 mm Hg, MCAs developed spontaneous tone over the course of an hour by constricting to ∼75% of their initial diameters.
The EDHF response was elicited by the luminal application of UTP after inhibition of nitric oxide synthase and cyclooxygenase with 10−5 mol/L N-nitro-
Measurement of Ca2+ in endothelium
Ca+ concentrations in the cytoplasm of the endothelium and vessel diameter were simultaneously measured as previously described (Marrelli, 2000, 2001). Briefly, fura-2 AM (0.67 μmol/L final concentration) was added to the luminal perfusate (5-minute exposure). Addition of fura-2 AM through the lumen at the above concentration and duration selectively loads the endothelium. For Ca2+ measurements, the vessels were illuminated with excitation light alternating between wavelengths of 340 and 380 nm using a xenon arc lamp, appropriate filters, and a filter changer (Intracellular Imaging, Cincinnati, OH, U.S.A.). Additionally, red light from a separate lamp was used to trans-illuminate the vessels for diameter measurements. The light was collected with a quartz objective (Nikon 10X, NA = 0.5) and subsequently split with a dichroic mirror to separate the fluorescence and red light signals. The red light was diverted to a CCD for diameter measurements, and the remainder of the light continued to a photomultiplier after passing through a 510-nm narrow bandpass filter. Intensities of the 510-nm fluorescence light were used to quantitate intracellular Ca + according to the following equation:
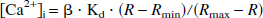
where [Ca2+]i was the intracellular Ca2+ concentration in the endothelium, β was the ratio of the 380-nm fluorescence intensity for Ca2+-unbound fura 2 over Ca2+-bound fura 2, R was the ratio of light intensity at 510 nm when excited at 340 nm to the intensity when excited at 380 nm (340/380 ratio) at a given condition (i.e., luminal addition of UTP), Rmin was the 340/380 ratio with zero [Ca2+]i, Rmax was the 340/380 ratio when [Ca2+]i was sufficiently high to saturate fura 2, and Kd was 282 nmol/L. β, Rmin and Rmax were determined in a separate group of vessels as previously described (Marrelli, 2000).
Drugs and reagents
UTP,
AACOCF3, AACOCH3, PACOCF3, PACOCH3, and ETYA were dissolved in ethanol; U73122, U73343, HELSS, and RHC-80267 were dissolved in dimethyl sulfoxide (DMSO). Fura 2-AM (50 μg) was dissolved in 75 μL of DMSO (containing 14% pluronic). Indomethacin was prepared by dissolving it in a 15-mmol/L Na2CO3 solution. Other reagents were dissolved in distilled water.
All blockers were administered 30 minutes prior to the addition of UTP. The composition of the PSS was as previously described (Bryan Jr. et al., 1996).
Statistical analysis
All data are reported as mean ± standard deviation. For concentration-response curves, the results are presented as the percentage of the maximum diameter and calculated by the following equation:
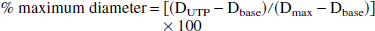
where DUTP was the diameter after luminal administration of UTP, Dbase was the baseline diameter before addition of UTP, and Dmax was the maximum diameter at 85 mm Hg (measured using Ca-free/EGTA buffer).
For statistical analysis, one- or two-way repeated measures ANOVA was used followed by Tukey test for individual comparisons when appropriate. P < 0.05 defined the acceptable level of significance.
RESULTS
A total of 141 MCAs were studied. At a pressure of 85 mm Hg, the maximum outside diameter (Ca2+-free/EGTA buffer) was 290 ± 20 μm and, after developing spontaneous tone, was 221 ± 23 μm. The diameter after spontaneous tone represented 76 ± 5% of the maximum diameter.
In the presence of NO synthase and cyclooxygenase inhibitors, 10−5 mol/L
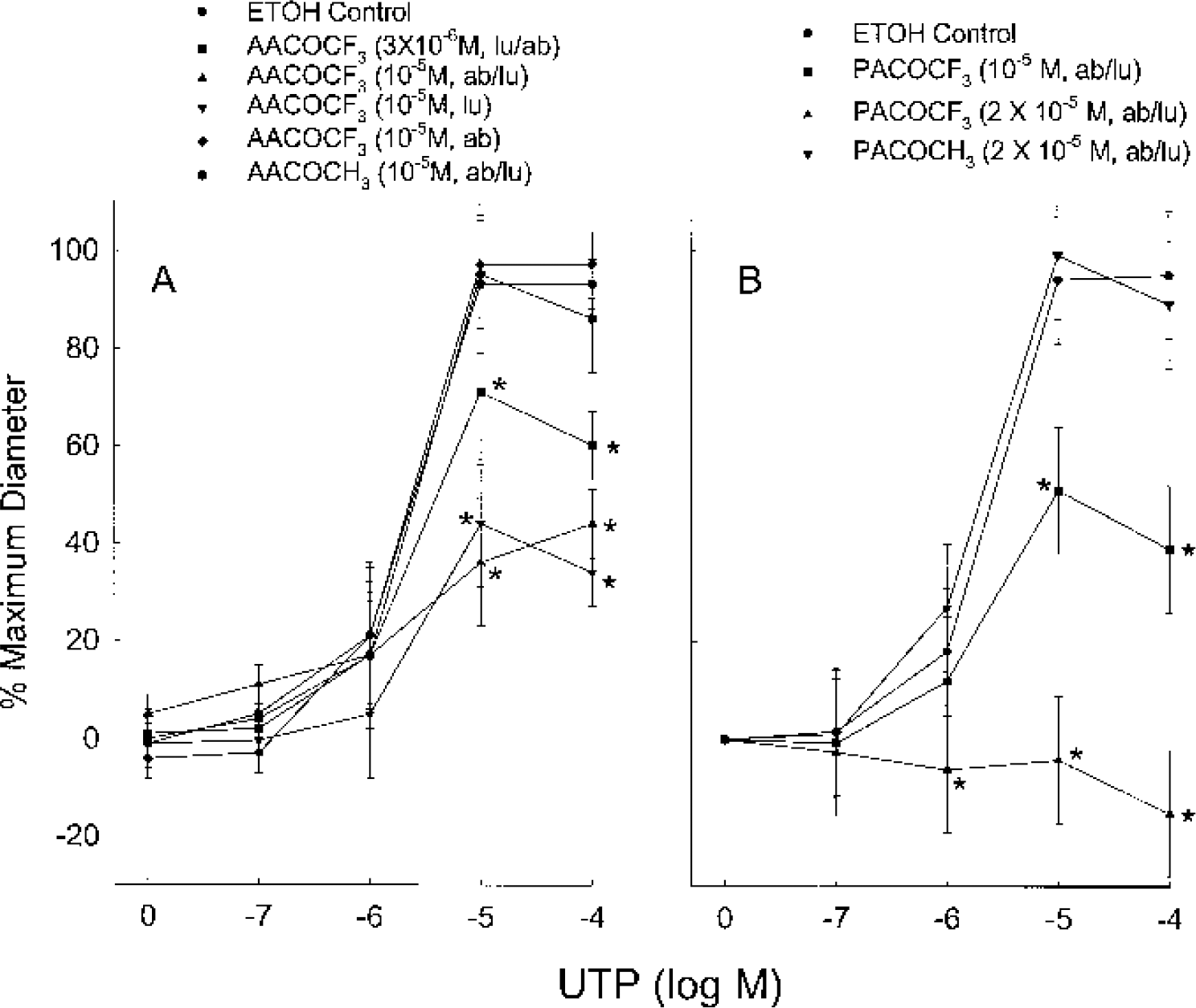
The effects of AACOCF3
When 10−5 mol/L AACOCF3 was added to the luminal compartment, the EDHF response was attenuated to the same degree as when the same concentration was added to both the luminal and abluminal compartments. However, the addition of 10−5 mol/L AACOCF3 to the abluminal compartment alone had no effect on the EDHF response (Fig. 1A). The addition of pharmacological agents to the luminal compartment has preference to endothelium, while addition to the abluminal compartment has preference to vascular smooth-muscle cells.
The selective inhibitor of the Ca2+ insensitive form of PLA2 (iPLA2), HELSS had no effect on the EDHF response (i.e,
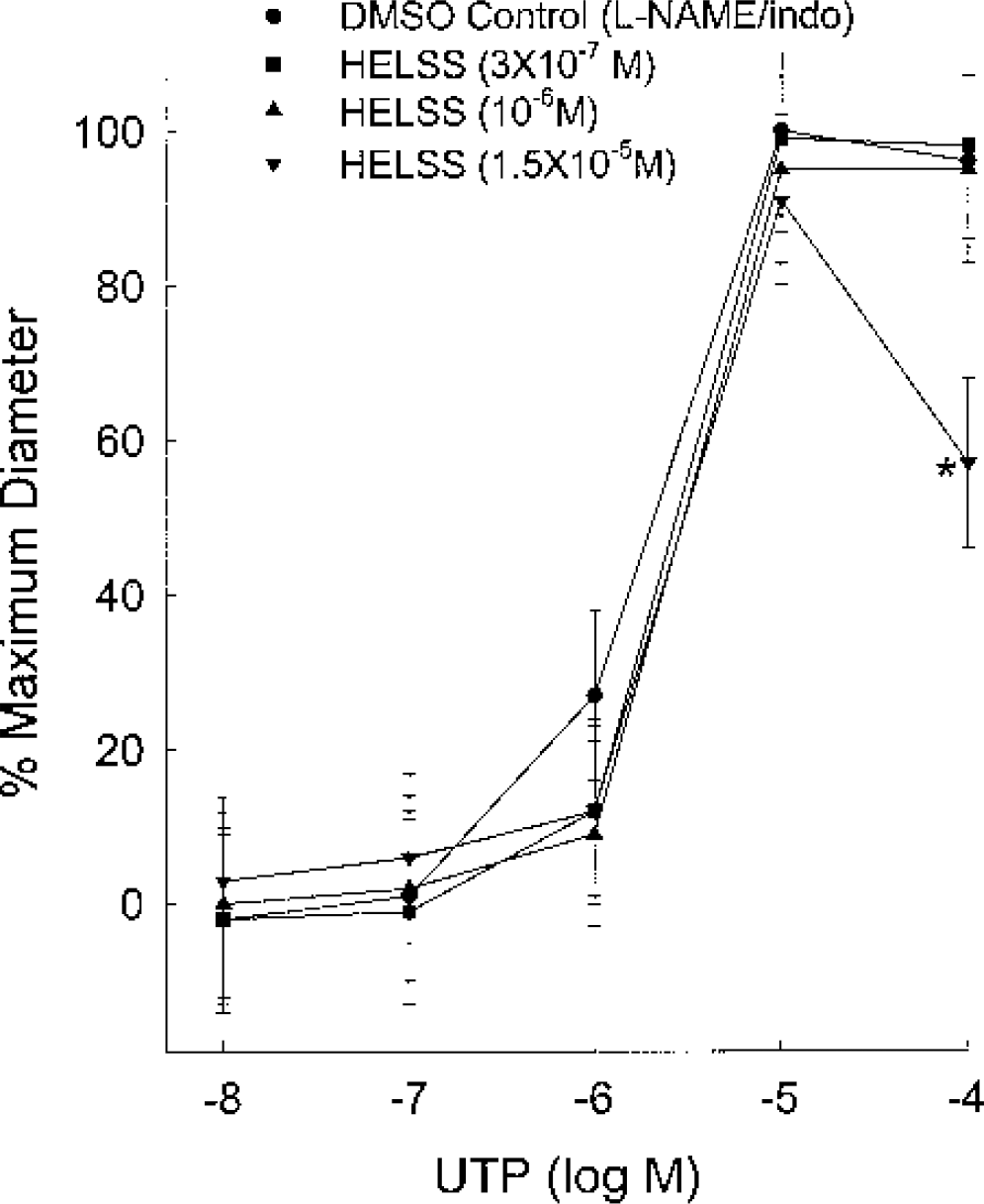
The effects of haloenol lactone suicide substrate (HELSS), a selective inhibitor of the Ca2+-insensitive form of phospholipase A2, on the endothelium-derived hyperpolarizing factor-mediated dilations to UTP in the rat middle cerebral artery. All experiments were conducted in the presence of 10−5 mol/L
In the presence of
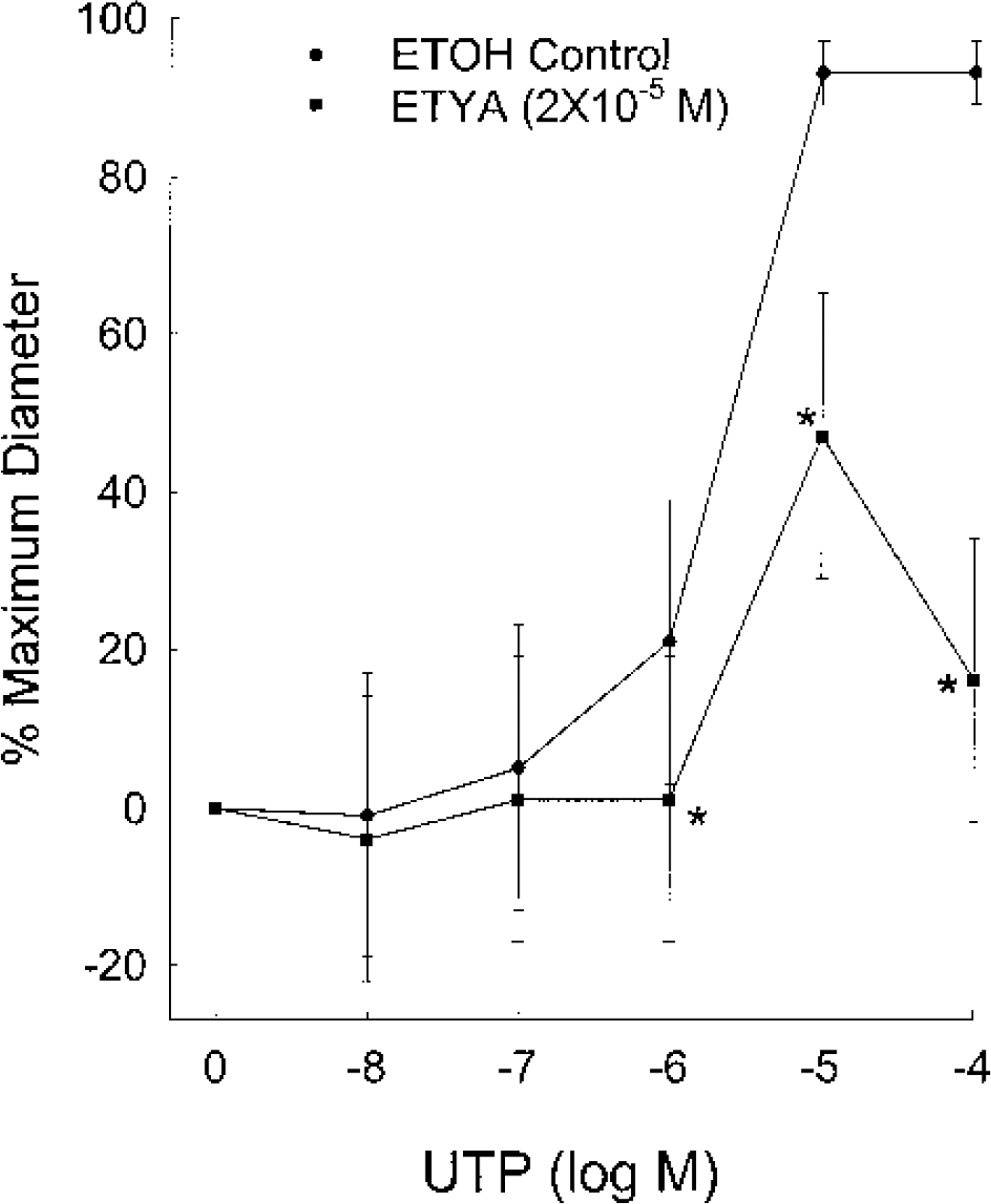
The effects of 2 × 10−5 mol/L ETYA, an inhibitor of phospholipase A2, cyclooxygenase, and lipoxygenases, on the endothelium-derived hyperpolarizing factor-mediated dilations to UTP in the rat middle cerebral artery. All experiments were conducted in the presence of 10−5 mol/L
The diacylglycerol lipase inhibitor, RHC-80267 (10−5 mol/L) (Sutherland and Amin, 1982), had minimal effects on the EDHF response (n = 8 for DMSO controls and 6 for the RHC-80267 group, data not shown). In the presence of
Addition of the PLC inhibitor, U73122 (10−6 mol/L), to both the luminal and abluminal compartments of the MCA preparation significantly attenuated the EDHF response compared to the DMSO control or after the addition of 10−6 mol/L U73343, an analog of U73122 that only slightly inhibits PLC (Fig. 4). Maximum dilations for the U73343 control and the U73122 group were 100 ± 0.6% and 42 ± 40% respectively. The concentration of UTP required to elicit one half of the maximum response was 2.6 ± 1.2 μmol/L for the U73343 control and 19 ± 18 μmol/L for the U73122 group. Two-way repeated measures ANOVA revealed a group difference (P = 0.001), a concentration difference (P < 0.001), and a significant interaction between group and concentration (P = 0.011).
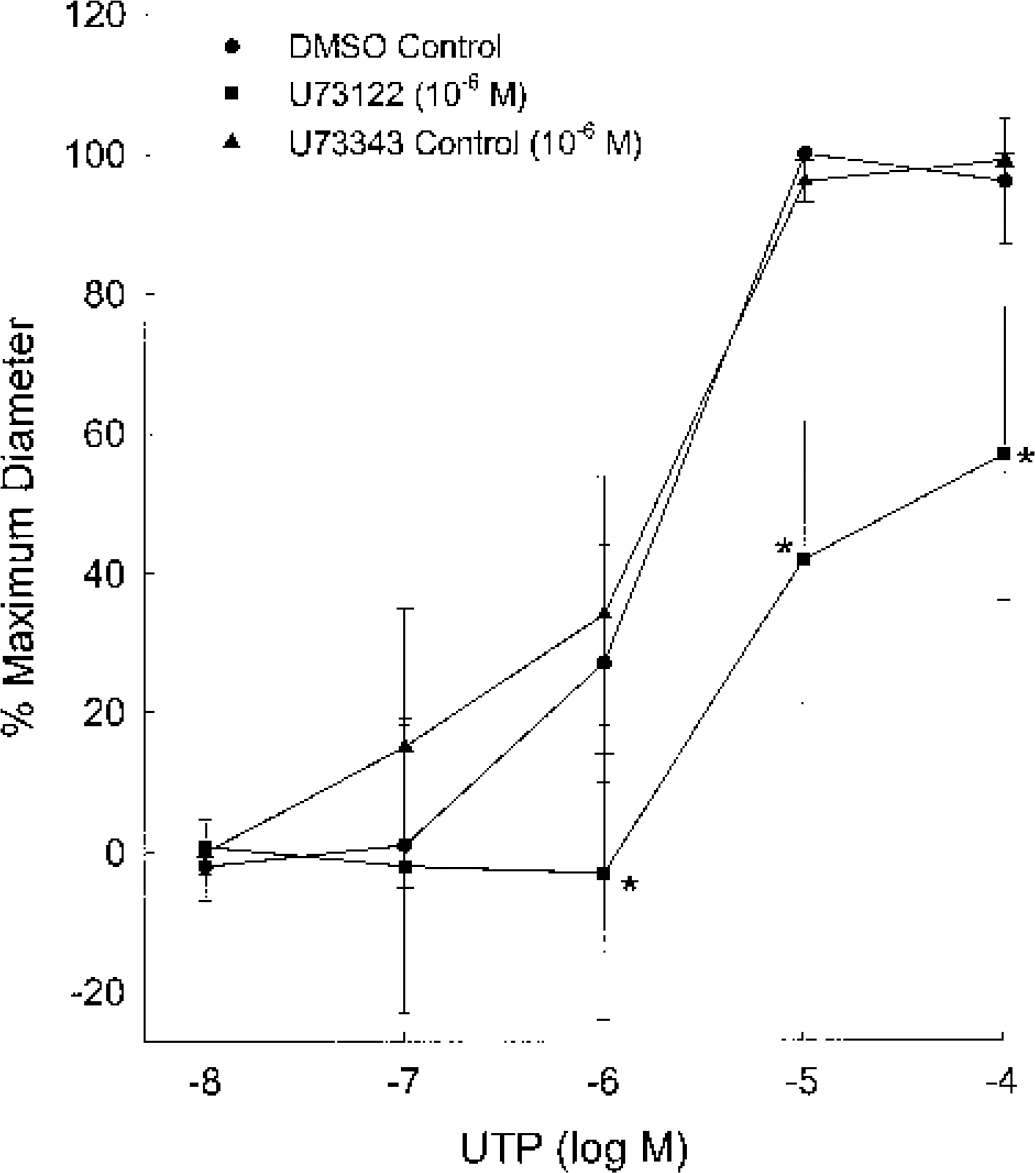
The effects of 10−6 mol/L U73122, a phospholipase C inhibitor, on the endothelium-derived hyperpolarizing factor-mediated dilations to UTP in the rat middle cerebral artery. All experiments were conducted in the presence of 10−5 mol/L
The effects of luminally administered UTP on the concentration of free Ca2+ in endothelium and diameter are shown in Fig. 5. Figure 5A shows endothelial Ca2+ and diameter measured simultaneously from an individual MCA after inhibition of NO synthase and cyclooxygenase. Note that the addition of 10−6 mol/L UTP increased endothelial Ca2+ by approximately 100 nmol/L over baseline with very little effect on diameter. With the addition of 10−5 mol/L UTP, Ca2+ further increased to greater than 500 nmol/L and the MCA dilated. Figure 5B shows the summary data from 5 MCAs. The group data further demonstrate that 10−6 mol/L UTP increased endothelial Ca2+ from 154 ± 33 to 246 ± 34 nmol/L (n = 5) with little effect on the MCA diameter. Application of 10−5 mol/L UTP increased Ca2+ to 468 ± 113 nmol/L and produced a near-maximal dilation of the MCA.
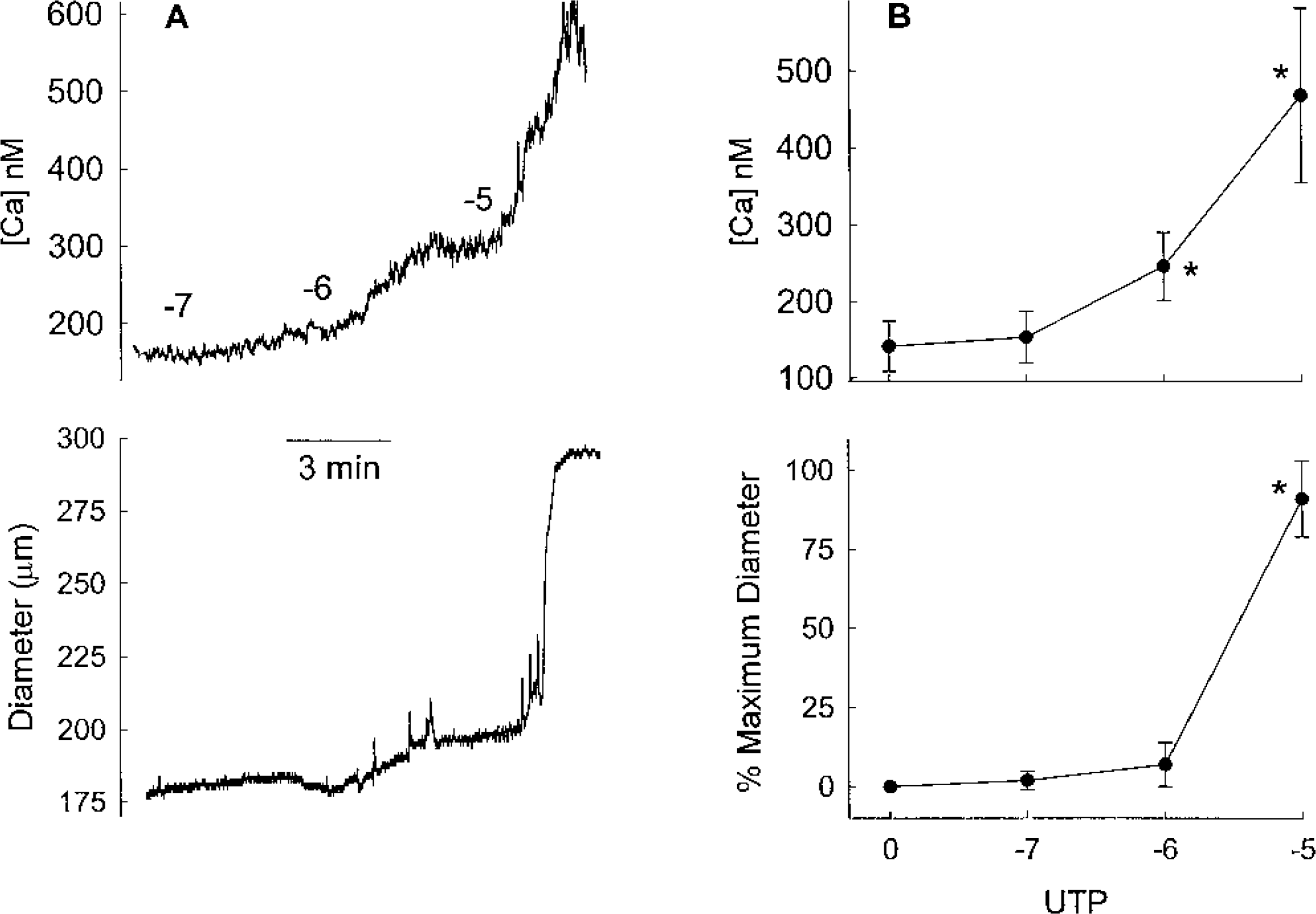
(A) Endothelial Ca2+ concentration and diameter, measured simultaneously from a single middle cerebral artery (MCA), during the luminal infusion of UTP at the concentrations noted (log M). The experiment was conducted in the presence of 10−5 mol/L
In order to determine whether the attenuation of the EDHF response by blockers as described above was a general inhibition of dilation, the dilator response to SNAP, an NO donor, was studied in the presence of AACOCF3, ETYA, and U73122. The inhibitors were added to both the luminal and abluminal compartments and were added at the highest concentration used in the studies described above. In the presence of
DISCUSSION
ATP and UTP, which are potent agonists for P2Y2 receptors, are found in plasma in physiologically relevant concentrations (Bryan Jr., 2002). There is strong evidence that these agonists are important regulators of blood flow during normal physiologic states and during pathologic conditions. Administration of UTP or ATP, P2Y2 agonists, to the lumen of rat cerebral arteries and arterioles dilates in part by the EDHF response (You et al., 1999a, b ; Bryan Jr. et al., 2001a,b). In the present study, we have demonstrated that the EDHF response in rat MCAs involves activation of cPLA2. We further provide evidence that cPLA2 activation is accompanied by activation of PLC and an increase in endothelial Ca2+.
Involvement of cytoplasmic form of phospholipase A2 in the endothelium-derived hyperpolarizing factor response
PLA2, a lipase that catalyzes the hydrolysis of the sn−2 linkage of diacyl glycerophosphates, can be subdivided into three major classes: (1) sPLA2 or the secreted form, which requires millimolar concentrations of Ca2+ for activation; (2) cPLA2, which is activated by submicromolar concentrations of Ca2+, and (3) iPLA2, which is calcium insensitive. cPLA2 and iPLA2 are active within the cell whereas, as the name suggests, the sPLA2 is secreted and its catalytic activity occurs extracellularly. Because of the fact that increases in endothelial Ca2+ appeared to be involved with the EDHF response (Marrelli, 2001), we suspected that cPLA2 might be involved. Our results from this series of studies demonstrated that our hypothesis was valid.
AACOCF3 and PACOCF3, inhibitors of iPLA2 and cPLA2 (Conde-Frieboes et al., 1996; Balsinde and Dennis, 1997), dose dependently blocked the EDHF response in rat MCAs (Fig. 1). For both inhibitors, a concentration of 10−5 mol/L (or 10 μmol/L) blocked approximately one half of the dilation produced by UTP. We obtained a complete inhibition of the dilation with 2 × 10−5 mol/L PACOCF3 (Fig. 1B). Inhibition of the EDHF dilation in our studies is consistent with reported values for the IC50, the inhibitor concentration necessary to inhibit 50% of the PLA2 activation. IC50 values reported for AACOCF3 range from 5 to 50 μmol/L for cPLA2 and 0.5 to 15 μmol/L for iPLA2. IC50 reported for PACOCF3 was 45 μmol/L for cPLA2 and 3 μmol/L for iPLA2 (Ackermann et al., 1995; Conde-Frieboes et al., 1996; Handlogten et al., 2001).
In order to determine which form of the PLA2 enzyme, iPLA2 or cPLA2, was involved, we further used HELSS, a selective inhibitor of iPLA2. HELSS, which is 1,000-fold more selective for iPLA2 over cPLA2, has IC50 values for iPLA2 of 1 μmol/L or less (Hazen et al., 1991; Ackermann et al., 1995). HELSS had no effect at 10−6 mol/L (1 μmol/L) and at a concentration of 1.5 × 10−5 mol/L (15 μmol/L) attenuated the EDHF response at a UTP concentration of 10−4 mol/L but not at 10−5 mol/L UTP (Fig. 2). We have consistently observed that the vasomotor response produced by 10−4 mol/L UTP is often difficult to interpret since UTP, which has a constrictor response when in direct contact with smooth muscle, could be gaining access to the smooth muscle at this higher concentration. Note that the HELSS did not inhibit the EDHF-mediated dilations at 10−5 mol/L UTP. We consider this lack of inhibition at 10−5 mol/L UTP to be more insightful than the apparent inhibition at 10−4 mol/L UTP. Regardless of whether our interpretation regarding the apparent attenuation of the response is correct or not, iPLA2 does not seem to be involved at UTP concentrations up to and including 10−5 mol/L UTP. We therefore conclude that the predominant, if not the exclusive, form of PLA2 involved in the EDHF response is cPLA2.
Although these studies cannot conclusively distinguish between endothelial and smooth-muscle cPLA2, our data would suggest the endothelium as the site of PLA2 activation. Intraluminal application of the inhibitor AACOCF3 alone inhibited the EDHF response to the same degree as the combination of the luminal/extra-luminal application of the inhibitor at the same concentration. When AACOCF3 was administered only abluminally, no inhibition occurred (Fig. 1A). As noted earlier, the luminal application of a drug has preferential access to the endothelium and the abluminal application has preferential access to the vascular smooth muscle. While these data support the idea that cPLA2 in endothelium is involved with the EDHF response, additional studies are required to support this conclusion.
AACOCF3 has been reported to abolish or inhibit the EDHF response in some peripheral arteries but failed to have an effect in others (Fulton et al., 1996; Adeagbo and Henzel, 1998; Hutcheson et al., 1999; Drummond et al., 2000; Hutcheson and Griffith, 2000). The above studies implicating PLA2 are supported by inhibition of the EDHF response with quinacrine, a less specific PLA2 inhibitor (Bauersachs et al., 1994; Fukao et al., 1997). Interestingly, Dong et al. (2000) reported that 1 μmol/L AACOCF3 had no effect on guinea pig mesenteric arteries but inhibited the EDHF response in guinea pig MCAs by 48%. These latter authors did not conduct further studies to distinguish between cPLA2 and iPLA2. Similar to the guinea pig MCA and other peripheral vessels, the EDHF response in rat MCAs appears to rely heavily, if not exclusively, on the activation of PLA2 (specifically cPLA2).
Involvement of phospholipase C
A possible involvement of PLC was tested using the inhibitor, U73122. U73122 (10−6 mol/L or 1 μmol/L) produced a 50% to 60% inhibition of the EDHF response (Fig. 4). This attenuation is consistent with reported IC50 concentrations in the μmol/L range (Bleasdale et al., 1990) and previously reported concentrations for inhibition of the EDHF response in the porcine coronary artery (20 μmol/L abolished the EDHF response) (Weintraub et al., 1995) and rabbit mesenteric artery (0.5 μmol/L produced greater than 50% inhibition) (Hutcheson et al., 1999). In guinea pig, 1 μmol/L U73122 had no effect on the EDHF response in the mesenteric artery but produced a 39% inhibition in the MCA (Dong et al., 2000). Thus, while PLC, as determined by inhibitor studies with U73122, appears to be involved with the EDHF response in a number of arteries including the rat MCA, it may not be involved with the EDHF response in all vascular systems.
Involvement of Ca2+ in the endothelium-derived hyperpolarizing factor response
The regulation of cPLA2 is both complex and interesting. While this enzyme is catalytically active under resting conditions, it can be further activated through serine phosphorylation (MAP kinase) and/or possibly tyrosine phosphorylation (Leslie, 1997; Balsinde et al., 1999; Hirabayashi and Shimizu, 2000). Although increases in Ca2+ do not catalytically activate cPLA2, Ca2+ increases the efficiency of the reaction by promoting cPLA2 binding to the membrane, the site of the phospholipid substrate (Balsinde et al., 1999; Hirabayashi and Shimizu, 2000; Leslie, 1997). Phosphatidylinositol 4,5-bisphosphate (PIP2) may also promote PLA2 binding to membrane, but it has not been as extensively studied as Ca2+ (Balsinde et al., 1999; Hirabayashi and Shimizu, 2000).
Although circumstantial evidence has indicated a second messenger role for Ca2+ in the EDHF response, direct proof of that has only been recently demonstrated (Marrelli, 2001). The endothelial Ca2+ concentration necessary to elicit the EDHF response was ∼330 nmol/L in the rat MCA (Marrelli, 2001). This concentration is 100 nmol/L greater than the Ca2+ threshold for activation of NO synthase (Marrelli, 2001). Consistent with these previous studies, we demonstrate that the EDHF response does not occur at an endothelial Ca2+ concentration of 246 nmol/L but appears to be fully active at 468 nmol/L (Fig. 5).
This magnitude for the Ca2+ increase is consistent with the concentration of Ca2+ necessary to promote cPLA2 binding to the membrane. For example, at a concentration of 450 nmol/L, 70% of the cPLA2 was lost from the soluble fraction in a macrophage cell line indicating binding to the membrane. This loss in the soluble fraction was accompanied by activation of cPLA2 to a similar magnitude (Channon and Leslie, 1990). Not only does our study demonstrate that endothelial Ca2+ increases in the MCA upon application of UTP, but also that it increases to levels that are required for activation of cPLA2.
Events downstream of cytoplasmic form of phospholipase A2 in the endothelium-derived relaxing factor response
A critical step that remains to be determined regarding the EDHF response in rat MCAs, is the event(s) downstream from PLA2. The product of the cPLA2 reaction could be the EDHF itself, could be metabolized into the EDHF, or could simply be a step in the signal process in the EDHF response.
There are a number of possibilities for the product of the cPLA2 reaction being involved in the EDHF response. Arachidonic acid immediately comes to mind since it has been the most studied product of the enzymatic reaction and PLA2 has a preference for arachidonic acid containing phospholipids. Metabolites of arachidonic acid, through the lipoxygenase or epoxygenase pathway, have been implicated in the EDHF response in peripheral arteries (Campbell et al., 1996; Pfister et al., 1998; Fisslthaler et al., 1999; Zink et al., 2001). In line with peripheral vessels, arachidonic acid dilated the rat basilar artery through activation of KCa and hyperpolarization of the vascular smooth muscle (Faraci et al., 2001). The dilation produced by addition of the arachidonic acid appeared to be due to conversion to 12(S)-hydroxyeicosatetraenoic acid, a metabolite of the lipoxygenase pathway.
The products of these pathways could be synthesized on demand following activation of cPLA2 and the subsequent liberation of arachidonic acid. Alternatively, these products could be stored in the phospholipid pool and liberated directly from the sn−2 position of the glycerol backbone by the action of cPLA2. Metabolites of arachidonic acid through the epoxygenase and lipoxygenase pathway (epoxyeicosatrienoic acids, hydroxyeicosatetraenoic acids, and others) can be incorporated into the sn−2 position of the phospholipid and released upon activation of PLA2 (Richards et al., 1986; VanRollins et al., 1993; Weintraub et al., 1997, 1999; Zink et al., 2001; Fleming, 2001).
We must also consider, however, that there are fatty acids, other than arachidonic acid, or metabolites in the sn−2 position of the glycerol phosphate backbone that, when cleaved by PLA2, could be involved with the EDHF process in the rat MCA. Often overlooked in the PLA2 enzymatic reaction are lysophospholipids, the molecule remaining after catalytic action at the sn−2 position of the glycerol backbone. Lysophospholipids can be converted to lysophosphatidic acid by action of phospholipase D. Lysophosphatidic acid is rapidly emerging as an important signaling molecule (Hla et al., 2001) and appears to be a dilator in the rat MCA (unpublished observation).
Although the product of the cPLA2 reaction, which is involved with EDHF dilations, is not known, we must point out that our studies do not preclude proposed mechanisms for EDHF such as K+ from endothelium or gap junctions (Edwards et al., 1998; Chaytor et al., 1998). The product of the cPLA2 reaction could be the EDHF itself, could be metabolized into the EDHF, or could be a step in the signaling process leading to release of K+ from endothelium or gap junction involvement.
In summary, we have demonstrated that activation of PLC, cPLA2, and an increase in endothelial Ca2+ are key steps in mediating the EDHF response in rat MCAs. The increase in endothelial Ca2+ was sufficient to promote binding of cPLA2 to the membrane, the location of the phospholipid substrate.