
Abstract
Severe headache and meningism provide clear evidence for the activation of trigeminal neurotransmission in meningitis. The authors assessed the antiinflammatory potential of 5HT1B/D/F receptor agonists (triptans), which inhibit the release of proinflammatory neuropeptides from perivascular nerve fibers. In a 6-hour rat model of pneumococcal meningitis, zolmitriptan and naratriptan reduced the influx of leukocytes into the cerebrospinal fluid, and attenuated the increase of regional cerebral blood flow. Elevated intracranial pressure as well as the brain water content at 6 hours was reduced by triptans. These effects were partially reversed by a specific 5HT1D as well as by a specific 5HT1B receptor antagonist. Meningitis caused a depletion of calcitonin gene-related peptide (CGRP) and substance P from meningeal nerve fibers, which was prevented by zolmitriptan and naratriptan. In line with these findings, patients with bacterial meningitis had significantly elevated CGRP levels in the cerebrospinal fluid. In a mouse model of pneumococcal meningitis, survival and clinical score at 24 hours were significantly improved by triptan treatment. The findings suggest that, besides mediating meningeal nociception, meningeal nerve fibers contribute to the inflammatory cascade in the early phase of bacterial meningitis. Adjunctive treatment with triptans may open a new therapeutic approach in the acute phase of bacterial meningitis.
The early pathophysiologic events of bacterial meningitis are the influx of leukocytes into the cerebrospinal fluid (CSF) the breakdown of the blood—brain and blood—CSF barrier, brain edema, increased CSF outflow resistance, and cerebrovascular complications, all contributing to the disastrous outcome (Quagliarello and Scheld, 1992). The combination of the acute inflammatory response, secondary effects of the altered physiology, and bacterial factors that may cause neuronal injury (Braun et al., 1999) results in 28% mortality (Durand et al., 1993) and permanent sequelae in up to 50% of the survivors (Bohr et al., 1984). Consequently, adjunctive treatment strategies to improve the outcome of bacterial meningitis are aimed at reducing the inflammatory response.
Headache and meningism are the leading clinical features of bacterial meningitis and provide clear evidence for the activation of sensory nerve fibers. These symptoms are mediated through sensory nerve fibers that originate from the trigeminal nerve and innervate the meninges. In addition to their ability to mediate pain, sensory nerve fibers can release vasoactive factors including the proinflammatory neuropeptides substance P (SP) and calcitonin gene-related peptide (CGRP). Apart from their role in afferent nociception, trigeminal nerve fibers participate in the vasomotor innervation of meningeal blood vessels (Suzuki et al., 1989), forming the so-called trigeminovascular system (TVS) (May and Goadsby, 1999). Unmyelinated sensory C-fibers release vasoactive neuropeptides from perivascular terminals in response to nociceptive stimuli (Edvinsson et al., 1990), including the presence of an inflammatory environment (Ebersberger et al., 1999). Of these neuropeptides, CGRP and SP are potent dilators of cerebral arteries (Edvinsson et al., 1987;McCulloch et al., 1986), and SP leads to increased vascular permeability and plasma protein extravasation (Pernow, 1985;O'Shaughnessy and Connor, 1993). In the dura mater, these phenomena have been summarized as neurogenic inflammation. Recent anatomic studies have revealed the presence of nerve fibers immunoreactive for several neuropeptides including CGRP and SP also within the leptomeninx (Fricke et al., 1997), arguing that the proinflammatory role of neuropeptide release is not confined to the dura mater, but may affect the subarachnoid space and possibly the adjacent cortical tissue. Thus, modulation of neuropeptide release may offer a novel path to decrease meningeal inflammation.
Calcitonin gene-related peptide is a potent vasodilator in the cerebral circulation and has been linked to hyperemia during central nervous system inflammation (Brian et al., 1995). Both SP and CGRP promote neutrophil adherence to endothelial cells in vitro (Sung et al., 1992;Zimmerman et al., 1992). Increased blood flow, activation of endothelial cells and neutrophil adhesion to the endothelium are initial features of bacterial meningitis.
Sectioning the nasociliary nerve 2 weeks before meningitis induction significantly reduced the rise in regional cerebral blood flow (rCBF) on the denervated side (Weber et al., 1996). These experiments suggest a critical role for the TVS, most likely due to the depletion of neuropeptides otherwise released locally from the trigeminal nerve into the perivascular space (Suzuki et al., 1989;Weber et al., 1996). Similarly, early pial vasodilation in experimental meningitis of the rat was reduced by local treatment with an SP antagonist (Pfister et al., 1995), as was the inflammatory response in murine meningoencephalitis caused by Trypanosoma brucei (Kennedy et al., 1997).
Neuropeptide release is inhibited by agonist action on presynaptic 5HT1B and 5HT1D receptors coexpressed on sensory neurons (Arvieu et al., 1996;Durham and Russo, 1999). Stimulation of these receptors by specific agonists (triptans) effectively blocks vasodilation and protein extravasation from meningeal blood vessels in response to nociceptive stimuli (Buzzi et al., 1991;Buzzi and Moskowitz, 1990, 1991;Connor et al., 1997;Huang et al., 1993;Martin et al., 1997;Messlinger et al., 1997). In the present study, we hypothesized a potential proinflammatory role of the trigeminovascular system in bacterial meningitis. Here we demonstrate that the 5HT1B/D/F receptor agonists zolmitriptan and naratriptan reduce leukocyte influx, the increase of cerebral blood flow and intracranial pressure, brain edema, and prolong survival in experimental pneumococcal meningitis. We conclude that besides mediating pain, sensory nerve fibers of the meninges add to the early inflammatory host response.
MATERIALS AND METHODS
All animal experimental designs were reviewed and approved by the Senate of Berlin.
Rat experiments
Table 1 outlines the experimental group design used in the study. The general experimental procedure was as described earlier (Weber et al., 1996). Experiments were performed on 51 male Wistar rats (280 to 330 g) anesthetized with intraperitoneal sodium thiopental (100 and 20 mg/kg every 2 hours; Trapanal, Byk Gulden, Konstanz, Germany). Animals were tracheotomized and mechanically ventilated (AP-10; K. Effenberger, Paffing, Germany). End-tidal CO2 was monitored continuously (Artema MM204; Heyer, Bad Ems, Germany). Body temperature was measured by a rectal probe and maintained at 37.8°C ± 0.4°C using a heating pad. A transducer (Statham P109 EZ, Spectramed, Oxnard, CA, U.S.A.) connected to a catheter placed in the left femoral artery measured mean arterial blood pressure (MABP) continuously. From this catheter, arterial blood samples were analyzed for Pao2, Paco2, and pH at 0, 2, 4, and 6 hours. The left femoral vein was cannulated for infusion of triptans or saline. A 3 × 3-mm area of the parietal bone lateral to the sagittal suture was thinned to allow laser Doppler flow (LDF) measurements (Periflux 4001 Master, Järfälla, Sweden) in cortical blood vessels (Lindauer et al., 1993;Weber et al., 1996). A catheter was placed into the cisterna magna through an occipital burr hole and connected to a pressure transducer (Statham P109 EZ, Spectramed) for continuous intracranial pressure (ICP) measurement. One hundred microliters CSF was removed from this catheter and replaced by 100 μL saline in controls or 100 μL pneumococcal cell wall (PCW) suspension. Instillation of PCW induces an inflammatory response similar to bacterial meningitis (Pfister et al., 1992). At the end of the experiment, CSF samples were obtained to determine the CSF leukocyte count. Animals were then killed by exsanguination. Brains were removed and heat dehydrated for 18 hours at 180°C. Wet and dry brain weights were compared to calculate the water content as an indicator of the presence of brain edema.
Experimental groups and parameters of inflammation at 2, 4, and 6 hours after the beginning of the experiment

Data are given as mean ± SD. In the posttreated zolmitriptan group, IV treatment was started 2 hours after PCW instillation.
IC, intracisternal; IV, intravenous, ΔICP, increase of intracranial pressure compared with baseline; CSF, cerebrospinal fluid; PCW, pneumococcal cell wall suspension.
Indicates a significant difference (P < 0.05) compared with untreated meningitis;
indicates a significant difference compared with animals treated with saline intracisternally (ANOVA, Duncan post hoc analysis).
Triptan treatment of rats.
Zolmitriptan (a generous gift from Zeneca Pharmaceuticals, Macclesfield, U.K.) and naratriptan (a generous gift from Glaxo Wellcome UK Ltd., Uxbridge, U.K.) were obtained in pure form without auxiliary substances. For intravenous application, zolmitriptan was dissolved in DMSO and diluted with 0.9% saline to a final concentration of 0.5 mg/mL. All substances were given intravenously as an initial bolus at the time of meningitis induction followed by a continuous infusion for the remainder of the experiment. Naratriptan was applied as a bolus of 7.5 mg/kg followed by 2.5 mg · kg−1·h−1; for zolmitriptan, dosages were 3 mg/kg and 1 mg · kg−1 · h−1, respectively. These doses were selected as effective doses from a limited dose-finding series. In a delayed-treatment group, zolmitriptan was started 2 hours after the induction of meningitis using the same dosages. In animals not receiving triptans, saline infusions were given instead. A further series of zolmitriptan-treated animals were given the selective 5HT1B antagonist SB 216641 (Biotrend GmbH, Köln, Germany; n = 4), the selective 5HT1D antagonist BRL 15572 (Biotrend GmbH; n = 4), or both (n = 5) prior to induction of meningitis in an attempt to restore inflammation and confirm the specificity of the observed effects. For this purpose, SB 216641 (10 mg/kg) was dissolved in purified water; BRL 15572 (10 mg/kg) was dissolved in DMSO. Both substances were given intraperitoneally 10 minutes before the beginning of zolmitriptan infusion.
Immunochemistry of rat dura mater.
For demonstration of CGRP and SP within meningeal nerve fibers, immunochemistry was performed in rats injected intracisternally with saline (n = 2), animals with untreated meningitis (n = 2), and animals with naratriptan- or zolmitriptan-treated meningitis (n = 5). Skulls were immersed in 4% paraformaldehyde in 0.1 mol/L phosphate-buffered saline (PBS) for 2 hours immediately after removal of the brains. After overnight incubation in 20% sucrose, dura mater was removed from the bone and immunostained using a free-floating technique. Following repeated washes in PBS, specimens were blocked with 2% normal goat serum and 0.2% Triton X in PBS and then incubated overnight with a polyclonal rabbit antibody against rat CGRP (1:1,000 in blocking solution) or Substance P (1:500) at 4°C. For visualization, an antirabbit antibody conjugated to Texas red was used (1:200, 1 hour). Omission of the primary antibody was used to control for unspecific binding of the secondary antibody. All antibodies were purchased from Sigma (Sigma-Aldrich, Deisenhofen, Germany).
Mouse model of meningitis
Mice experiments were conducted using a modification of a previously published model (Tang et al., 1996). In brief, male 129S6 mice (20 g) were anesthetized with intraperitoneal ketamine (100 mg/kg; Ketanest, Parke-Davis GmbH, Freiburg, Germany) and xylazine (20 mg/kg; Rompun, Bayer AG, Leverkusen, Germany). A skin incision was made exposing the lumbar spine. Using a 30-gauge needle and a Hamilton syringe, 50 μL of a suspension containing 5 × 105 colony-forming units (cfu) live pneumococci in sterile PBS were slowly injected into the spinal canal at the level of L2 or L3. The skin incision was closed using dermal clips. Immediately after inoculation, animals received either zolmitriptan (3 mg/kg, n = 10) or vehicle (n = 10) intraperitoneally. Animals were then allowed to wake up and given free access to food and water. Absence of pareses and adequate waking were verified. Mice were kept under constant surveillance beginning at 12 hours after inoculation. At this time, intraperitoneal injections of zolmitriptan or vehicle were repeated. The duration of the experiment was limited to 24 hours. Primary end points were time of death and a clinical score at 24 hours in the surviving animals. The score was defined as follows: normal activity, 5 points; inactivity, but normal locomotion after stimulation, 4 points; ataxic gait, 3 points; delayed righting, 2 points; unable to right, 1 point; dead, 0 points. Immediately at the time of death or at the termination of the experiment at 24 hours, CSF was collected for determination of bacterial titers. Using a modification of previously described methods (Carp et al., 1971;Meyding-Lamade et al., 1996) a skin incision was made over the head and neck. After dissection of the suboccipital muscles under a preparation microscope, the cisterna magna was punctured and CSF withdrawn using a 27-gauge butterfly cannula connected to a Luer lock Hamilton syringe. For the determination of bacterial titers, serial dilutions of the CSF in sterile PBS were plated on sheep blood agar and grown overnight at 37°C with 5% CO2.
Bacterial preparations
For the preparation of live bacteria, encapsulated pneumococci from a clinical isolate (strain D39, kindly provided by Dr. E. Tuomanen, St. Jude Children's Research Hospital, Memphis, TN, U.S.A.) were grown in standard C+Y culture medium (Lacks and Hotchkiss, 1960) overnight at 37°C with 5% CO2. After centrifugation at 10,000 g for 2 minutes, the pellet was resuspended in sterile PBS. Using a standard curve, the number of cfu per milliliter in this preparation was determined photometrically. Aliquots for the inoculation of animals were then prepared using adequate dilutions in sterile PBS. Correctness of cfu calculations was verified by plating serial dilutions of the inoculate.
Pneumococcal cell walls, which permit study of the inflammatory host response in the absence of bacterial metabolic effects, were prepared as follows. Unencapsulated pneumococci (Strain PnR 527, Jena, Germany) were cultivated overnight on Columbia agar plates, suspended in pyrogen-free saline, and heat inactivated. After disintegration by ultrasound, pneumococcal cell walls were produced and modified as described earlier (Tuomanen et al., 1985;Weber et al., 1995). The concentration of PCW corresponded to 107 cfu/mL. Absence of lipopolysaccharides was ensured using a chromogenic limulus amebocyte lysate test (BioWhittaker, Walkersville, MD, U.S.A.).
Measurement of calcitonin gene-related peptide concentration in human cerebrospinal fluid
Diagnostic CSF samples from 10 patients with bacterial meningitis and 14 samples from controls (i.e., patients in whom CSF studies revealed no inflammatory changes) were included in the analysis. For a diagnosis of bacterial meningitis, CSF pleocytosis of >1,000 cells/μL with >90% neutrophils was required. The CGRP concentrations were measured using an enzyme-linked immunoassay (human CGRP EIA kit, SPI-BIO, Massy, France) according to the manufacturer's instructions.
Tumor necrosis factor-α bioassay
The tumor necrosis factor-α (TNF-α) bioactivity was measured as described previously (Freyer et al., 1999). In brief, a modified L 929 cytotoxicity assay (Flick and Gifford, 1984) was performed by adding 100 μL CSF per well to the culture medium in the presence of 1 μg/mL actinomycin D. After incubation at 37°C for 20 hours, cell viability was quantified by the uptake of crystal violet in living cells, which was determined spectrophotometrically (595 nm) using an ELISA reader (Dynatech, Denkendorf, Germany). Equivalent concentrations of rat TNF-α (a gift from Dr. P. Scholz, Schering AG, Berlin, Germany) were used as a standard.
Statistical analysis
For descriptive statistics, data are expressed as mean values and standard deviations. For the rat experiments, comparisons between groups were performed using one-way ANOVA with post hoc testing by the Duncan multiple-range test. In the mice experiments, a log-rank test was performed to test for significance of the difference in survival time between meningitis treated with vehicle or zolmitriptan, while nonparametric Mann-Whitney U tests were used to test for a statistically significant difference in clinical score and CSF bacterial titers at the time of death between the two groups. Mann-Whitney U tests were also used to test for a statistically significant difference of CSF CGRP concentrations between patients and 12 controls and of TNF-α bioactivity in rat CSF between PCWinduced meningitis and naratriptan-treated meningitis. Statistical tests were performed using SPSS 10 statistical software (SPSS Inc., Chicago, Illinois, U.S.A.).
RESULTS
Rat experiments
Values of MABP, Pao2, Paco2, and pH were within normal ranges during the entire experiment, and no biologically relevant differences were found between the experimental groups (Table 2). In animals intracisternally injected with saline, there was no significant change in rCBF or ICP during the 6-hour experimental period (Table 1, Figs. 1A and 2). Brain water content at the end of the experiment was 80.99% ± 0.63% and leukocyte count in the CSF was 20 ± 21 cells/μL. In animals treated with intracisternal saline, no significant effect of triptan treatment could be demonstrated on ICP, brain water content, or CSF leukocyte count.
Physiologic parameters at 2, 4, and 6 hours after intracisternal challenge
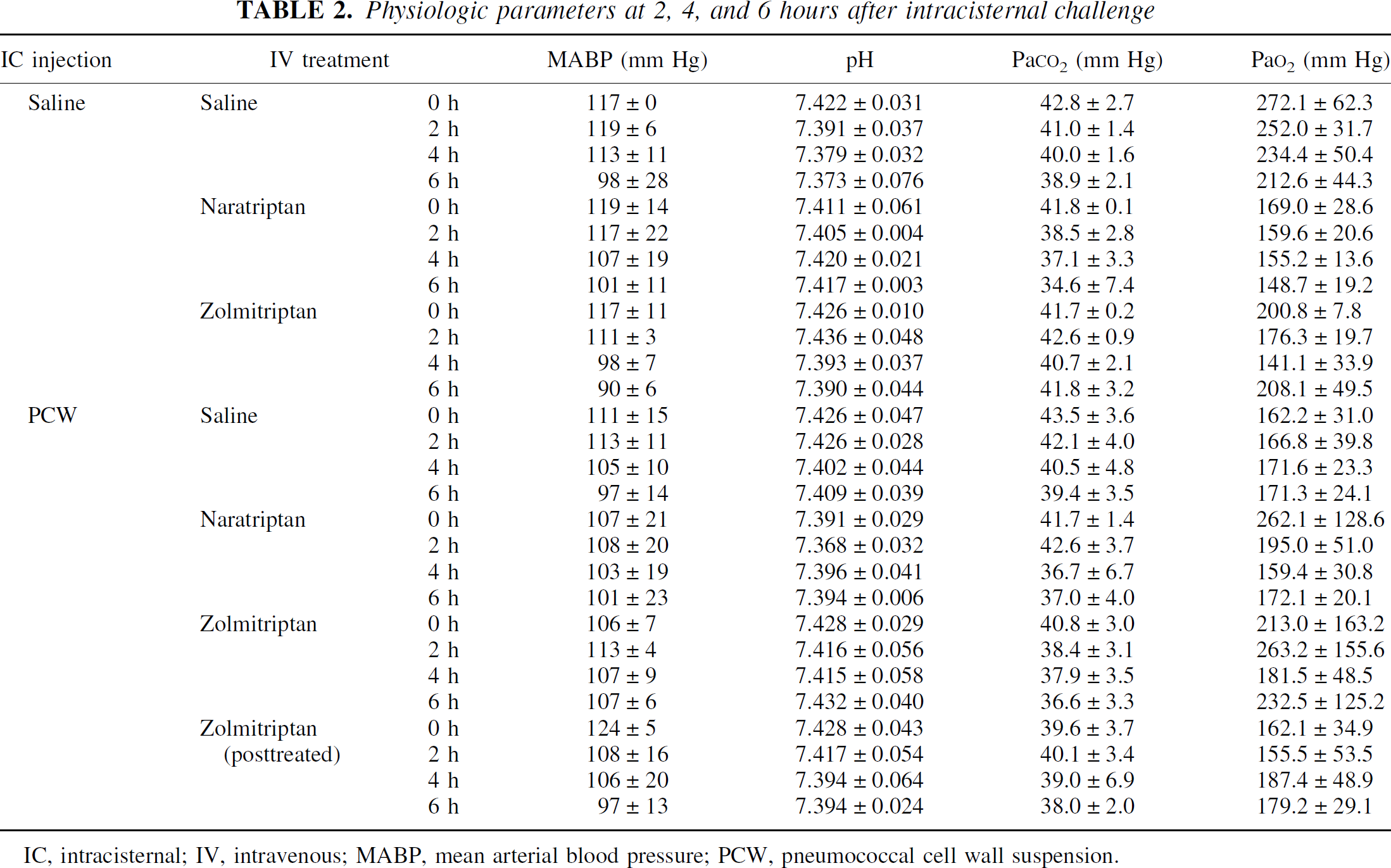
IC, intracisternal; IV, intravenous; MABP, mean arterial blood pressure; PCW, pneumococcal cell wall suspension.
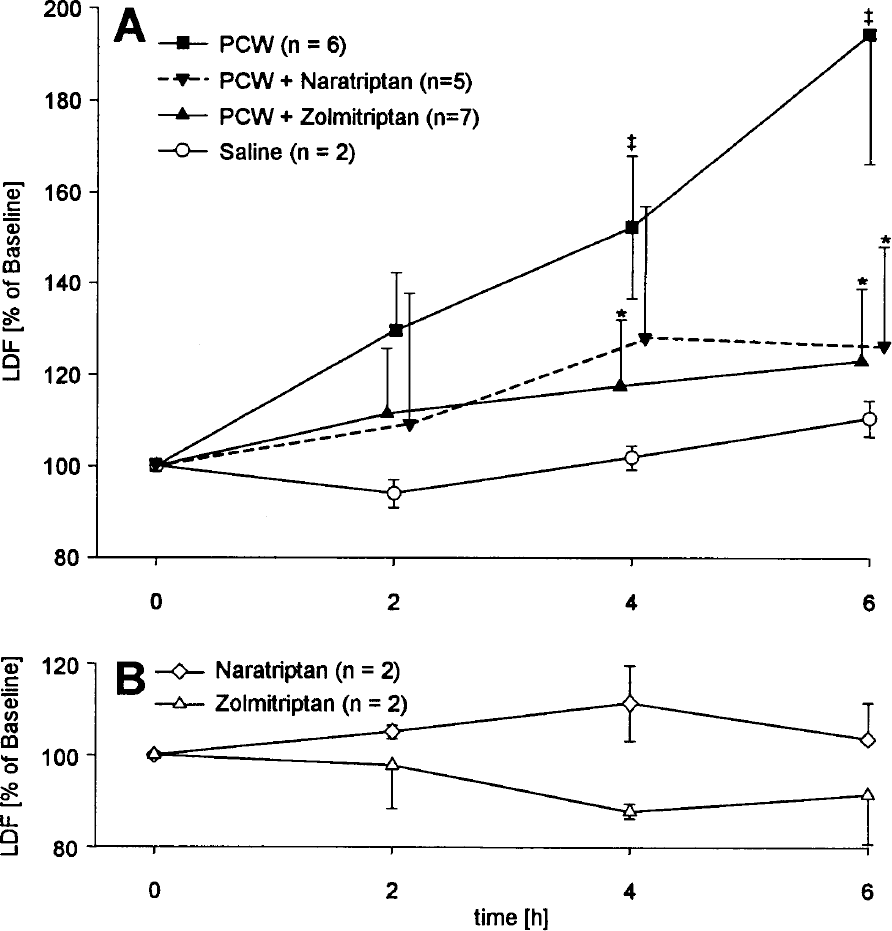
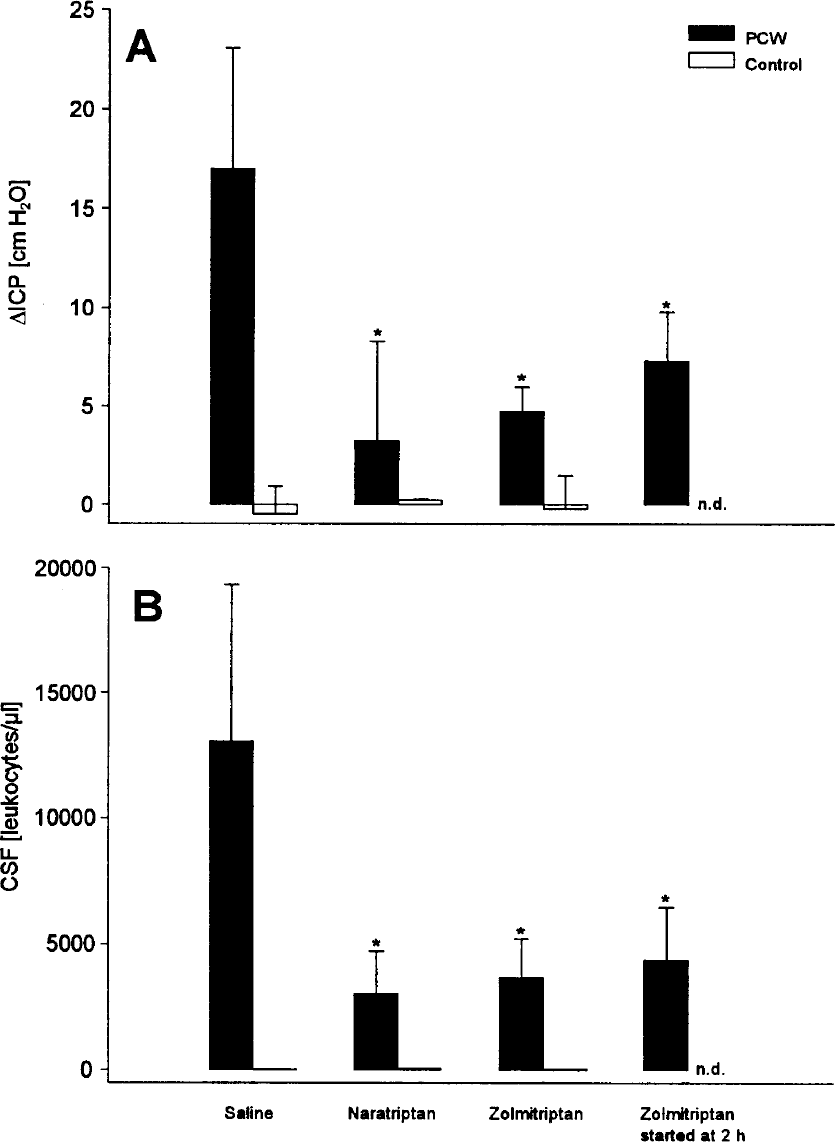
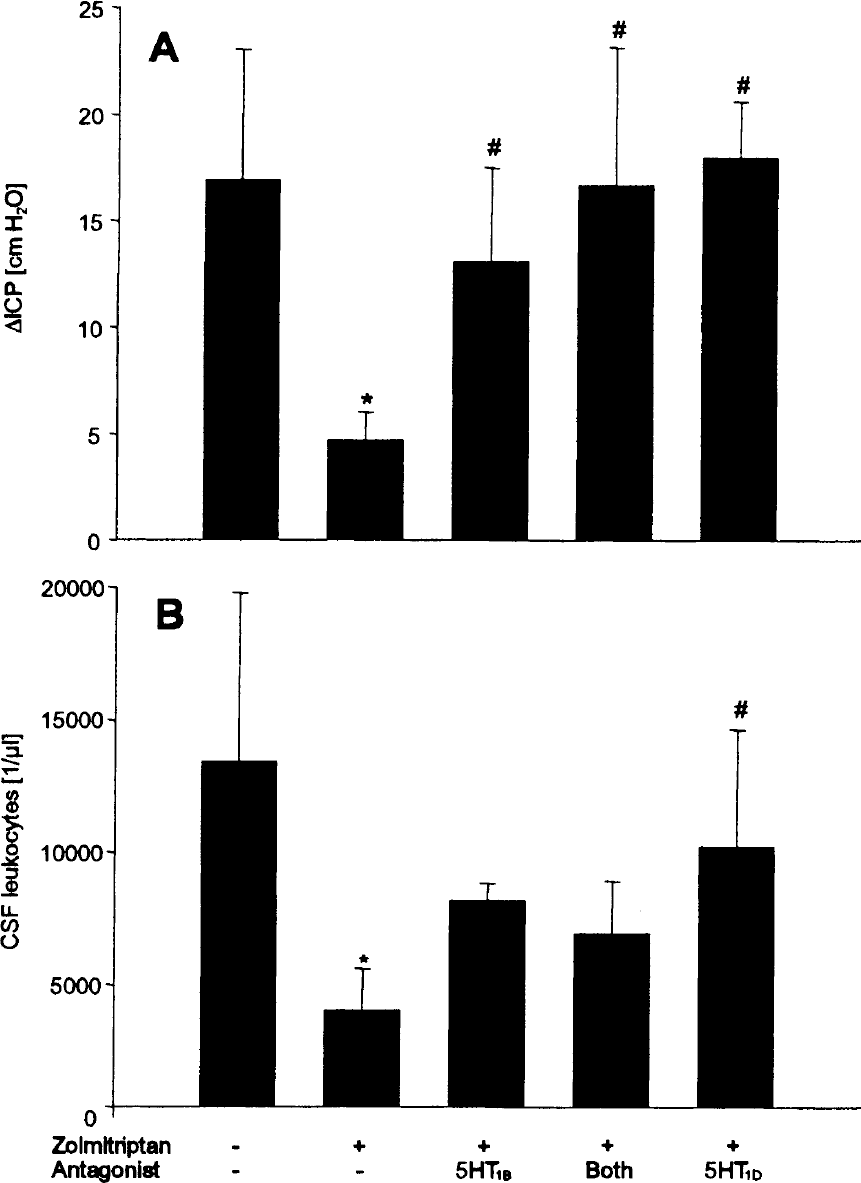
In the untreated meningitis group, a significant increase in rCBF as well as an increase in ICP compared to animals intracisternally injected with saline was observed (Table 1, Figs. 1 and 2B). These changes were accompanied by a significant influx of leukocytes into the CSF and raised brain water content (Table 1, Fig. 2B). Treatment of PCW-challenged animals with zolmitriptan or naratriptan significantly reduced the increase in blood flow, ICP, and the formation of brain edema, as well as the leukocytes in the CSF (Table 1, Figs. 1 and 2). Zolmitriptan treatment 2 hours after meningitis induction still significantly reduced LDF and ICP increase, brain edema, and CSF leukocytosis. The inhibition of the inflammatory response in zolmitriptan-treated meningitis was partially reversed by pretreatment with the 5HT1B antagonist SB 216641, the 5HT1D antagonist BRL 15572, or a combination of these antagonists. Data for cell count and intracranial pressure are summarized in Figs. 3A and 3B. Increase in rCBF at 6 hours in PCW-induced meningitis, which was reduced from 196% ± 16% at baseline to 121% ± 21% with zolmitriptan, was restored to 142% ± 45% with SB 216641, to 315% ± 98% with BRL 15572, and to 206% ± 29% with a combination of both antagonists. Zolmitriptan led to a decrease in brain water content from 83.06% ± 0.31% in untreated meningitis to 81.40% ± 0.45% (P < 0.05), which was reversed by BRL 15572 14 to 83.40% ± 0.44% (P < 0.05), by SB 216641 to 82.57% ± 0.37% (P < 0.05), and by the addition of both substances to 82.35% ± 0.44% (P < 0.05). In animals treated with saline intracisternally and saline intravenously, no significant effect of SB 216641 or BRL 15572 on LDF, ICP, leukocyte influx, or brain water content after was observed after 6 hours (data not shown).
The inhibition of intracranial pressure (ICP) increase
Immunochemistry of rat dura mater.
In rats treated with saline intracisternally, immunostaining for CGRP and SP was observed in nerve fibers of different caliber (Figs. 4A and 4D). In comparison, immunoreactivity for both neuropeptides was markedly reduced in the untreated-meningitis group (Fig. 4B, E). Depletion of CGRP and SP was inhibited in zolmitriptan-treated meningitis (Fig. 4C, F). Neuropeptide release was also attenuated by naratriptan (data not shown).
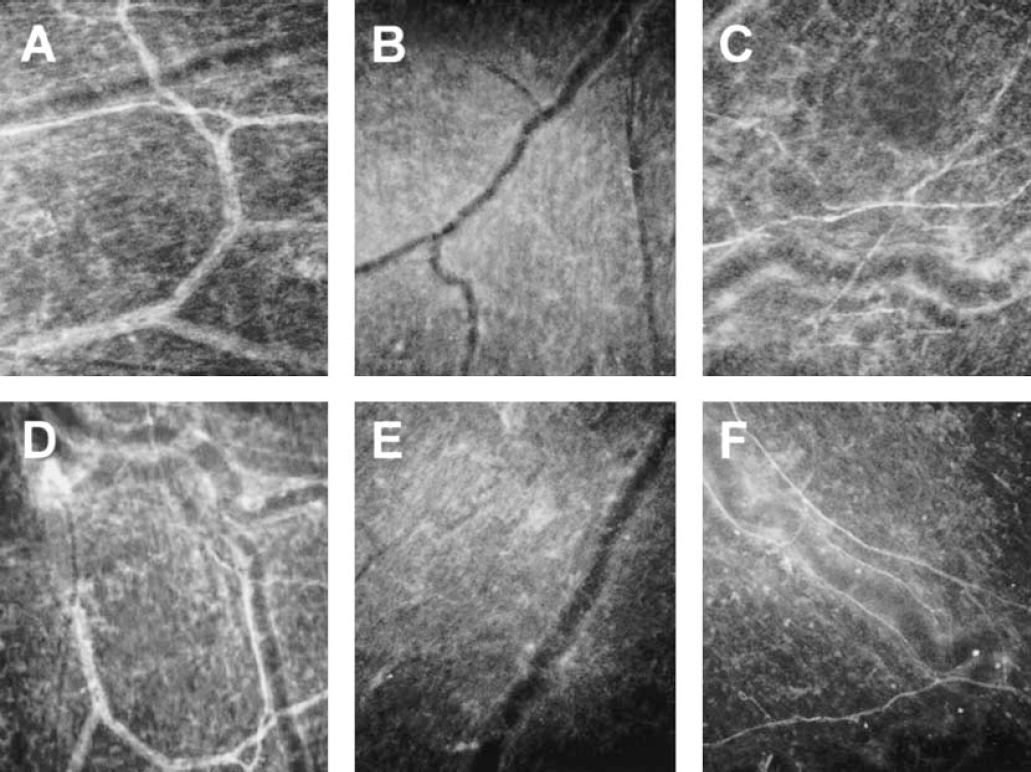
Immunochemistry results for calcitonin gene-related peptide
Tumor necrosis factor-α bioactivity in rat cerebrospinal fluid.
At 6 hours after intracisternal instillation of PCW, TNF-α bioactivity in the CSF of rats receiving intravenous saline was equivalent to 899 ± 447 pg/mL compared to 115 ± 61 pg/mL in naratriptan-treated meningitis (P = 0.045).
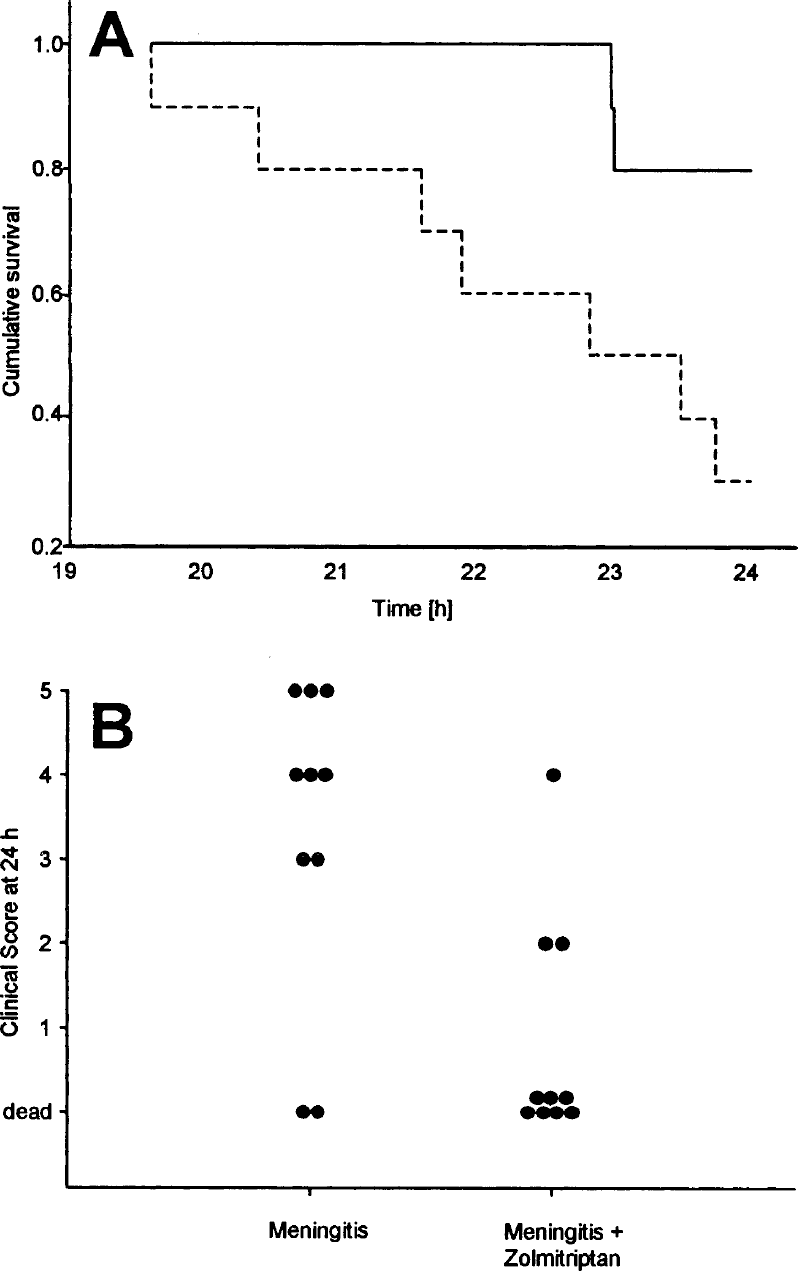
Mouse experiments
At 24 hours after inoculation, 7 out of 10 mice with untreated meningitis had died compared to 2 out of 10 mice with zolmitriptan-treated meningitis. Kaplan-Meier survival curves for these animals are shown in Fig. 5A. This difference in survival was statistically significant (P = 0.02, log-rank test). The clinical score at 24 hours (Fig. 5B) was 0.8 ± 1.4 in untreated meningitis compared to 3.3 ± 1.9 in zolmitriptan-treated meningitis (P < 0.01, Mann-Whitney U test). Mean CSF bacterial titers were 7 × 108 cfu/mL (range, 4 × 107 –3 × 109) in animals receiving vehicle IP compared to 3 × 109 cfu/mL (range, 3 × 107 −2 × 1010) in animals given zolmitriptan IP (P = 0.931, Mann-Whitney U test). Similar effects were observed with naratriptan treatment (data not shown).
Calcitonin gene-related peptide concentrations in human cerebrospinal fluid samples
The CGRP concentration in controls was 27.1 ± 1.7 (range, 25.3–31.74) pg/mL. The concentration of CGRP in CSF of patients with bacterial meningitis was 67.9 ± 87.5 (range, 29.2–266.1) pg/mL (Fig. 6). The difference between the groups was statistically significant (P < 0.01, Mann-Whitney U test).
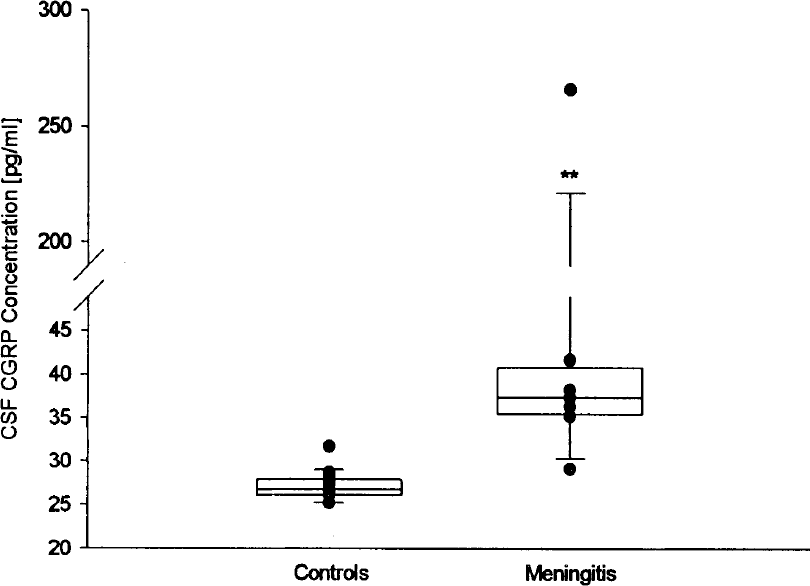
The calcitonin gene-related peptide (CGRP) concentrations in the cerebrospinal fluid (CSF) of patients with bacterial meningitis (n = 10) are significantly higher than those found in controls. ** P = 0.001 (Mann-Whitney U test).
DISCUSSION
We have shown for the first time that pharmacologic intervention with triptans has potent antiinflammatory effects in bacterial meningitis in the rat. Zolmitriptan and naratriptan significantly reduced the influx of leukocytes into the CSF as well as the increase of regional cerebral blood flow and ICP and the formation of brain edema. Triptans also significantly improved survival and clinical score at 24 hours in mice infected intrathecally with live pneumococci. Our experiments demonstrate that in the early phase of bacterial meningitis, inflammation mediated by the trigeminovascular system contributes to bacterially induced host responses.
Using two animal models, we investigated the effect of triptan treatment on different aspects of bacterial meningitis. In particular, intrathecal infection of mice with live encapsulated pneumococci allowed us to study the effects of triptans in untreated meningitis, whereas PCW-induced meningitis in rats was used to model the host response caused by bacterial lysis. Antibiotic-induced lysis of pneumococci triggers the release of PCW. The concentration of cell walls, which consist mainly of teichoic acid (Höltje and Tomasz, 1976), is related to the clinical outcome in human pneumococcal meningitis (Schneider et al., 1999).
Two findings argue that the antiinflammatory effects observed in our rat experiments are related to typical agonist action of the triptans at serotonergic receptors on perivascular nerve fibers. First, triptan treatment inhibited the depletion of CGRP and SP from these fibers during meningitis as demonstrated by immunochemistry. Second, the antiinflammatory effects were reversed to a large extent by coapplication of 5HT1B and 5HT1D receptor antagonists. The fact that inflammation was not restored completely may be explained by additional action of triptans on other serotonin receptors, such as the 5HT1F receptor (Martin et al., 1997).
All of the antiinflammatory effects of triptans that were observed during our experiments may be linked to the inhibition of neuropeptide release. Both SP and CGRP have been shown to promote the activation of neutrophils and microvascular endothelial cells, which is required for the invasion of neutrophils into the CSF space. In detail, SP and CGRP upregulate the expression of the β2-integrins CD11 and CD18 on neutrophils and promote their adherence to endothelial cells in vitro (Zimmerman et al., 1992). Furthermore, SP enhances the production of leukotrienes, TNF-α secretion, and regulates LFA-1 and ICAM-1 (Saban et al., 1997). Activating effects on human neutrophils exceeding those of SP were also demonstrated for CGRP (Richter et al., 1992). Endothelial cells may be activated through signaling by compounds of the bacterial cell wall (Freyer et al., 1999), but they are also a source as well as a target of proinflammatory cytokines (Freyer et al., 1999). Both CGRP and SP stimulate the production and release of these cytokines from immune competent cells (Sakuta et al., 1995). In our study, we found a decrease of TNF-α in the CSF of animals with triptan-treated meningitis. Induction of the adhesion molecules ICAM-1 and VCAM-1 on the endothelial surface by SP was previously demonstrated (Quinlan et al., 1998). Thus, reduced CSF leukocyte counts in triptan-treated meningitis may be related to decreased release of neuropeptides otherwise promoting endothelial and leukocyte activation. Additional inhibition might be mediated by direct action of triptans on endothelial 5HT1B receptors (Riad et al., 1998).
Decreased endothelial activation may exert a protective effect on the blood—brain barrier by reducing the production of inducible NO (Freyer et al., 1999), a mediator of trigeminal nociceptive transmission (Hoskin et al., 1999). Since activation of the vascular endothelium is a prerequisite for the transcytosis of pneumococci through cerebral endothelial cells (Ring et al., 1998), triptans might theoretically also decrease bacterial invasion into the CNS. These effects could not be studied in our mouse model due to the intrathecal mode of infection. On the other hand, presence of identical bacterial concentrations supports the conclusion that improved survival and clinical score of the triptan-treated mice was related to influences on the inflammatory host response rather than to bacterial mechanisms.
Reduction of rCBF increase in response to triptans may reflect vasoconstriction mediated by 5HT1B receptors on perivascular myocytes. However, no significant effect of triptans on rCBF was demonstrated in nonmeningitic animals, and earlier studies have shown that the TVS is not essential for CBF autoregulation under physiologic conditions (Sakas et al., 1989;Vraamark et al., 1998). More likely, reduced hyperemia again is an effect of reduced levels of vasoactive neuropeptides. This mechanism is in keeping with the findings after chronic nasociliary nerve sectioning in PCW-induced meningitis (Weber et al., 1996).
Increased ICP in the acute phase of bacterial meningitis may be attributed to at least three mechanisms (Quagliarello and Scheld, 1992). First, blood—brain barrier disruption causes vasogenic edema due to extravasation of plasma compounds and hyperemia. Second, cytotoxic compounds released from activated leukocytes, astrocytes (Freyer et al., 1996), microglia (Draheim et al., 1999), endothelial cells (Freyer et al., 1999), and bacteria cause cytotoxic brain edema. Finally, the increased ICP may be due to an increase of CSF outflow resistance (Scheld et al., 1980). Lower ICP and reduced brain edema in the triptan group may thus result from attenuated hyperemia as well as better preservation of the blood—brain barrier secondary to reduced leukocyte recruitment.
Relevance of these experimental findings for bacterial meningitis in humans is suggested by the clinically observed activation of the trigeminovascular system, and by our demonstration of significantly increased CGRP concentrations in the CSF of patients with bacterial meningitis. Our results may open a new approach in the design of pharmacologic interventions to treat pain and reduce life-threatening intracranial complications during the early phase of bacterial meningitis.