
Abstract
The role of endogenous tissue-type plasminogen activator (tPA) in focal cerebral ischemic injury (FCII) after middle cerebral artery occlusion was studied using tPA gene-deficient (KO) mice and their wild-type (WT) littermates. The middle cerebral artery was occluded by thrombi induced by three different intensities of photochemical damage, a method that was newly introduced in mice. In both WT and KO mice, the intensity-dependent increase of FCII size was observed. The FCII size in tPA WT mice was smaller than in KO mice in cases of mild damage, whereas the FCII size was larger in WT mice than in KO mice in cases of severe damage. There was no difference in FCII size between WT and KO mice in cases of moderate damage. The number of microthrombi also increased with damage intensity in both WT and KO mice, but was less in WT mice at all intensities of damage. The results support the validity of the model of thrombotic occlusion by photochemical damage in mice, and suggest that endogenous tPA protects FCII through thrombolytic action on transient occlusion of middle cerebral artery with mild damage, but deteriorates on persistent occlusion with severe damage.
Tissue-type plasminogen activator (tPA) is a secretory serine protease that degrades fibrin through plasmin generation. Clinically, tPA is used as a thrombolytic agent. Its beneficial effect was shown in ischemic stroke patients treated within 3 hours after the onset of symptoms (NINDS group, 1995). However, the effect of tPA on CNS excitotoxicity has also been reported (Tsirka et al., 1995), suggesting that tPA may deteriorate cerebral infarction because excitotoxicity is thought to be essential for cerebral ischemic injury. Several lines of experiments conducted to confirm the role of tPA in cerebral infarction have been performed in animals, but results were controversial (Kilic et al., 1999; Klein et al., 1999; Nagai et al., 1999; Tabrizi et al., 1999; Wang et al., 1998).
In the present report, we induced focal cerebral ischemic injury (FCII) by middle cerebral artery (MCA) occlusion with thrombi induced by the local damage of vascular endothelial cells using a combination of infusion of Rose Bengal (RB), a photosensitive dye, and local photoillumination (Matsuno et al., 1993; Umemura et al., 1993). In this method, the area and intensity of vascular endothelial cell damage could be accurately controlled by RB dose and photoillumination intensity. Severe damage resulted in larger FCII size associated with later reperfusion of MCA and more microthrombi (Kawai et al., 1995; Kawano et al., 1998). This method has been used in rats, guinea pigs, and rabbits (Kawai et al., 1995; Umemura et al., 1993; Zhao et al., 2001), but not in mice. We confirmed the validity of this model in mice and studied the effect of tPA gene deficiency on FCII size.
MATERIALS AND METHODS
These studies were performed in tPA gene-deficient (KO) mice and their wild-type (WT) littermates (weight, 20 to 25 g). The mice were created by breeding pairs of heterozygotes, which were obtained by crossbreeding tPA KO and WT mice (gifts from the Center for Transgene Technology and Gene Therapy, KU Leuven, Belgium). Gene inactivation was obtained by homologous recombination in embryonic stem cells of the genes encoding tPA as previously described (Carmeliet et al., 1994). The genetic background of these mice was a mixture of 75% C57B1/6 and 25% SV129.
Animals were kept on a heating pad maintained at 37°C under halothane anesthesia. A catheter for infusion of RB was inserted in the left jugular vein. The skin between the left eye and left ear was incised and the temporal muscle was retracted. In the MCA region, a small opening (approximately 1.5 mm in diameter) was drilled, and the dura mater was kept intact. Irradiation with green light (wavelength, 540 nm) was directed by a 3-mm diameter optic fiber mounted on a micromanipulator (L-4887; Hamamatsu Photonics, Japan). The head of the optic fiber was placed on the opening in the skull base, and the surrounding tissue was shielded with shims to irradiate only the 1.5-mm diameter opening. Then, RB was injected through the catheter and the MCA was illuminated simultaneously. Infusion of RB was continued for 1 minute, whereas irradiation was continued for 30 minutes. We chose three different conditions: 10-mg/kg RB with 0.38 W/cm2 photoillumination to induce mild damage, 20-mg/kg RB with 1.0 W/cm photoillumination to induce moderate damage, and 20-mg/kg RB with 1.4 W/cm2 photoillumination to induce severe damage. Ten WT or KO mice were examined in each condition. After photoillumination, muscle and skin were replaced and the catheter was removed. Mice were killed 24 hours after illumination, and their brains were sectioned and stained with triphenyl tetrazolium chloride, which stains living tissue brick red (whereas the FCII remains white). The FCII area was measured by photography and then by planimetry. The data are presented as mean ± SD. Significance was determined using analysis of variance followed by the Fisher exact test.
In another set of experiments, six WT or KO mice representing the three conditions were perfused with 10% neutral buffered formalin 3 hours after photoillumination. Their brains were used for fibrin immunostaining to count the number of microthrombi, which contain fibrin in the infarct area. Briefly, brains were removed, embedded in paraffin, and sectioned into 5-μm slices. Fibrin(ogen) was stained via a two-step procedure with a rabbit antihuman fibrinogen antibody (Dako, Kyoto, Japan) diluted 1:50, followed by peroxidase-conjugated goat antirabbit immunoglobulin G antibody (Dako Envision kit, Dako, Japan). Peroxidase activity was visualized with diaminobenzidine (DAB Substrate kit; Nichirei, Tokyo, Japan). After staining, the number of microthrombi in the FCII area in the sections was counted in three individual fields of the microscope at 100x magnification. The numbers of microthrombi-rich animals (i.e., animals with an average of more than 10 microthrombi in the three fields) were compared between WT and KO mice, or among animals in each condition. The statistical significance of the difference in the ratio of microthrombi-rich animals between groups was determined using the Mann-Whitney test.
RESULTS
Physiologic variables
Physiologic variables recorded in WT and KO mice (n = 5 in each group) after the operation were within normal range (pH = 7.4 ± 0.07 vs. 7.4 ± 0.05, P
Focal cerebral ischemic injury size
The FCII size of WT or KO mice in each damage condition is shown in Fig. 1. In WT mice, FCII size (0.83 ± 1.5 mm for mild damage, 4.0 ± 2.0 mm for moderate damage, and 14 ± 6.4 mm for severe damage) increased with the intensity of damage. The difference in FCII size between moderate and severe MCA damage was statistically significant (P < 0.001). These data were similar to previous data obtained in different species (Kawano et al., 1998), showing the validity of this model in mice. In KO mice, increasing FCII size (4.6 ± 4.2 mm for mild damage, 5.8 ± 3.1 mm for moderate damage, and 8.2 ± 2.5 mm for severe damage) was also dependent on the intensity of damage, though these differences were not statistically significant (P = 0.48 between mild and moderate damage; P = 0.15 between moderate and severe damage). Compared with WT mice, FCII size in KO mice was significantly (P < 0.05) larger in cases of mild damage. However, FCII size in KO mice was significantly (P < 0.001) smaller than that in WT mice with severe damage. Cerebral infarct sizes were not different in mice with moderate damage.
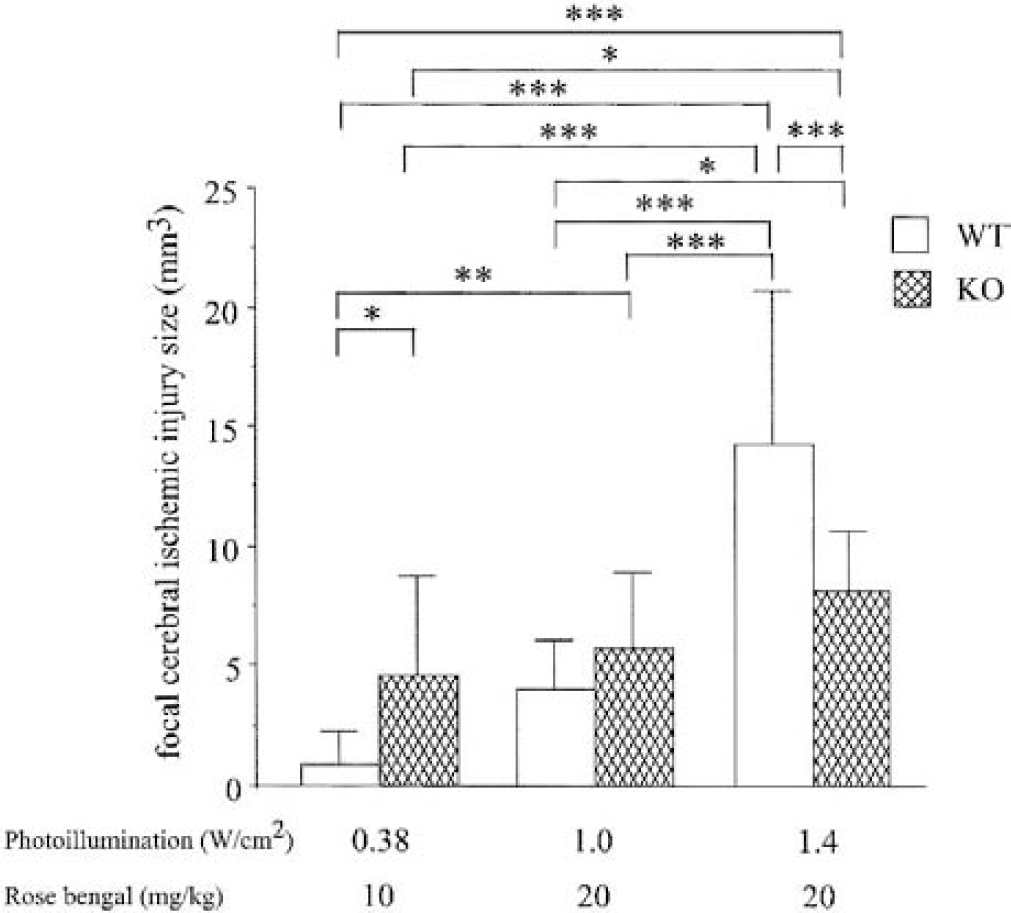
Cerebral infarct size of tissue-type plasminogen activator (tPA)–knockout mice (KO) and their wild-type (WT) littermates in various degrees of middle cerebral artery damage. Intensity of photoillumination and Rose Bengal (RB) dose are shown in the bottom. In both WT and KO mice, intensity of a damage-dependent increase of cerebral infarct size was observed. In mild damage induced by 0.38 W/cm2 light and 10-mg/kg RB, WT mice had smaller infarct sizes than KO mice. However, in severe damage induced by a combination of 1.4 W/cm2 light and 20-mg/kg RB, WT mice had larger cerebral infarct sizes than KO mice. In moderate damage induced by 1.0 W/cm2 light and 20-mg /kg RB, there was no significant difference in these infarct size. Data represent mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001.
Microthrombi
Photographs of brain sections of the FCII area are shown in Fig. 2. In both WT and KO mice, the number of microthrombi was larger in cases of severe damage (Figs. 2A and 2B) than in cases of mild damage (Figs. 2C and 2D). The KO mice had more microthrombi (Figs. 2B and 2D) than WT mice (Figs. 2A and 2C) in each degree of damage. The numbers of microthrombi-rich animals showing more than 10 microthrombi in the microscopic field are shown in Table 1. In WT mice, the number of microthrombi-rich animals was associated with the intensity of damage. The number of microthrombi-rich mice with mild damage (1 of 6) was significantly lower than that of mice with severe damage (5 of 6), whereas the number of microthrombi-rich mice with moderate damage was intermediate (4 of 6). The changes in microthrombi in tPA KO mice (5 of 6 in cases of mild and moderate damage, and 6 of 6 in cases of severe damage) were similar to WT mice, but these differences were not significant. In cases of mild damage, the number of microthrombi-rich mice was significantly higher in KO mice compared with WT mice. The number of microthrombi in KO mice in cases of moderate or severe damage was also higher than that in WT mice, but the differences between WT and in KO mice were not significant.
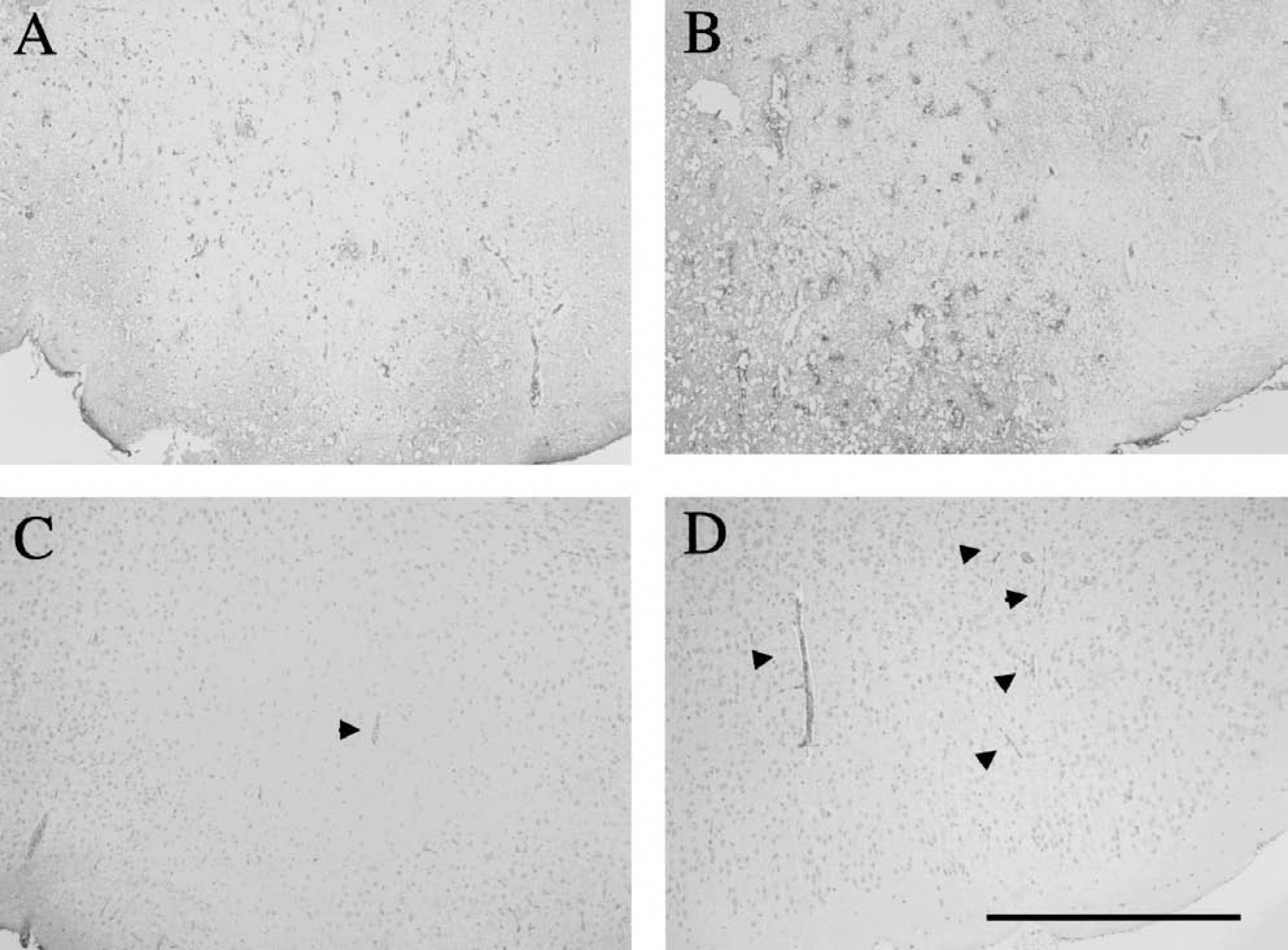
Fibrin(ogen) immunohistochemistry in the focal cerebral ischemic injury (FCII) area of wild-type (WT) and tissue-type plasminogen activator (tPA)–knockout (KO) mice 3 hours after photoillumination. In severe damage, the number of microthrombi was larger than in mild damage in both mice, but KO mice
Microthrombi in area of focal cerebral ischemic injury in wild-type or tPA-knockout mice

Data represent number of microthombi-rich animals (animals with more than 10 microthrombi in average of 3 different fields in area of focal cerebral ischemic injury under microscope in 100x magnification) per number of animals to be studied.
P <0.05 versus tPA KO mice in mild damage; †P <0.05 versus wild-type (WT) mice in severe damage.
tPA, tissue-type plasminogen activator; KO, knockout.
DISCUSSION
In the present study, the intensity of damage was dependent on the increase of FCII in both WT and KO mice, which indicates that this thrombosis model can also be applied to mice. However, compared with WT mice, KO mice showed larger FCII in cases of mild damage and smaller FCII in cases of severe damage. These results suggest that endogenous tPA protects against cerebral ischemic injury in cases of mild damage, but that this protective effect deteriorates when MCA damage is more severe.
Both FCII and the number of microthrombi in WT mice were associated with the intensity of MCA damage, a finding that is consistent with those obtained in studies of other rodents (Kawai et al., 1995; Kawano et al., 1998; Takamatsu et al., 2000). These observations support the validity of this thrombosis model in mice. The pathogenic process of arterial occlusion in humans is different from that in experimental animals. In most patients, the cerebral artery is spontaneously recanalized in the early phase of cerebral infarction. The event is frequently followed by rethrombosis. However, in the other models of focal ischemia, such as nylon thread insertion and ligation of the MCA, the MCA is not occluded by thrombi. In contrast, thrombotic occlusion of the MCA by photochemical damage mimics recanalization followed by rethrombosis in guinea pigs and primates (Kawano et al., 1998; Takamatsu et al., 2000). The application of this model to mice enabled us to study the molecular mechanisms of ischemic stroke by using gene-transferred mice.
In mild damage, KO mice had larger FCII sizes than WT mice, which was in line with the finding of a larger number of microthrombi in KO mice. If we consider the evidence that a reduction in the number of microthrombi by treatment with an anticoagulant agent reduced FCII size in this model (Kawai et al., 1995), we speculate that endogenous tPA reduces FCII size through microthrombi degradation by thrombolytic action in cases of mild damage. Spontaneous fibrinolytic activity in tPA-deficient mice was reduced compared with WT mice (Carmeliet et al., 1994). This result strongly indicates that reduction of spontaneous fibrinolysis is associated with the increased number of fibrin clots in tPA-deficient mice. However, KO mice had smaller FCII sizes in severe damage, though the number of microthrombi was similar to that of WT mice. The absence of differences in the number of microthrombi between KO and WT mice may suggest that the occlusion persisted long enough to cause the maximum FCII, even though endogenous tPA is exited in cases of severe damage. Under such conditions, the toxic effect of tPA, such as stimulation of N-methyl-
The present results suggest that earlier thrombolytic therapy with exogenous tPA may be still beneficial in ischemic stroke patients. But if the thrombus is too rigid and resistant to be dissolved or if tPA treatment is not administered early enough, tPA may deteriorate ischemic stroke. This prediction was confirmed in an animal model (Nagai et al., 2001) and is in line with the clinical observations of a tPA trial (NINDS group, 1995). Therefore, the late timing of tPA treatment in patients with ischemic stroke should receive more attention. Furthermore, the suppression of tPA activity might be beneficial for patients with persistent occlusion (Vivien and Buisson, 2000; Yepes et al., 2000).