
Abstract
The balance between oxygen consumption and delivery in the rat brain after exposure to transient ischemia was quantitatively studied with single-spin echo T2-BOLD (blood oxygenation level–dependent) magnetic resonance imaging at 4.7 T. The rats were exposed to graded common carotid artery occlusions using a modification of the four-vessel model of Pulsinelli. T2, diffusion, and cerebral blood volume were quantified with magnetic resonance imaging, and CBF was measured with the hydrogen clearance method. A transient common carotid artery occlusion below the CBF value of approximately 20 mL·100 g−1·min−1 was needed to yield a T2 increase of 4.6 ± 1.2 milliseconds (approximately 9% of cerebral T2) and 6.8 ± 1.7 milliseconds (approximately 13% of cerebral T2) after 7 and 15 minutes of ischemia, respectively. Increases in CBF of 103 ± 75% and in cerebral blood volume of 29 ± 20% were detected in the reperfusion phase. These hemodynamic changes alone could account for only approximately one third of the T2 increase in luxury perfusion, suggesting that a substantial increase in blood oxygen saturation (resulting from reduced oxygen extraction by the brain) is needed to explain the magnetic resonance imaging observation.
Oxygen delivery and consumption are quantitatively closely matched in the living brain. However, this balance can be disturbed under both physiologic and pathologic conditions. It is well established that at the macroscopic scale, CBF increases in a disproportionate manner relative to the oxygen demand evoked by increased neuronal activity (Fox et al., 1988), leading to elevated oxygen saturation in the venous blood (Ogawa et al., 1993; van Zijl et al., 1998). This phenomenon has been demonstrated with invasive (Lund Madsen et al., 1995) and noninvasive techniques (Kwong et al., 1992; Oja et al., 1999a) and is the key prerequisite for BOLD MRI contrast (Ogawa et al., 1990) involving alterations in CBF, CBV, and CMRO2 (van Zijl et al., 1998). The BOLD effect is detected by T2 (Thulborn et al., 1982) or T2* MRI (Ogawa et al., 1990; Kwong et al., 1992), both of which are extensively used in functional studies of human brain.
Increasing interest has been placed on applying BOLD MRI under pathological conditions in the brain, such as during hypoxia (Turner et al., 1991; van Zijl et al., 1998), hypoperfusion (Gröhn et al., 2000; Kavec et al., 2001), and ischemia (De Crespigny et al., 1992; Roussel et al., 1995; Gröhn et al., 1998; Calamante et al., 1999; Grüne et al., 1999). Reduced cerebral perfusion is associated with adaptive changes in hemodynamics that are reflected in an increase in CBV or OER or both, as quantitatively detailed by positron emission tomography methodology (Heiss and Podreka, 1993). Under these conditions, reduced T2 or T2* identifies the parenchyma with increased deoxyhemoglobin concentration resulting from the mismatch between oxygen delivery and uptake. This datum is useful in the assessment of tissue viability (Gröhn et al., 1998; Lin et al., 2001). After a brief period of ischemia, an overshoot of CBF and CBV is generally observed (Hossmann, 1997). An excess amount of blood delivered to the brain is termed luxury perfusion, a condition that was observed in humans decades ago (Lassen, 1966). The data from animal models indicate that after ischemia, CMRO2 normalization is delayed (Snyder et al., 1975). During luxury perfusion, OER remains reduced for a short period after ischemia leading to an inevitable decrease in the deoxyhemoglobin fraction, thereby making it potentially accessible to BOLD MRI. Indeed, the data from animal model of transient ischemia have shown an increase in the T2*-weighted signal intensity (De Crespigny et al., 1992) on termination of vascular occlusion. This study indicates that BOLD MRI is capable of detecting luxury perfusion, yet the quantification of the contributions of CBV, CBF, and CMRO2 to the changes in the MRI relaxation coefficient has remained difficult to elucidate.
In the present work, we studied the temporal and quantitative behavior of single-spin echo T2 in the brain during and after brief episodes of graded vascular occlusions and ischemia. Our aim was to induce luxury perfusion in the rat brain and to study the ability of spin-echo T2-BOLD MRI to detect this response, and to separate the contributions of hemodynamic (CBF and CBV) or metabolic factors to the BOLD effect. The spin-echo BOLD model used for the data computations quantitatively describes the interrelations between cerebral T2 and physiologic variables (van Zijl et al., 1998; Gröhn et al., 2000; Kavec et al., 2001).
MATERIALS AND METHODS
Experimental
A modification of the four-vessel model modified from Puslinelli et al. (1982) was used for graded occlusion of CCAs (Gröhn et al., 2000) and induction of ischemia. Briefly, male Wistar rats (250–350 g) were initially anesthetized with 0.8% to 1.5% halothane in a 70:30 nitrous oxide-to-oxygen ratio, and both vertebral arteries were occluded by electrocoagulation. Three days later, rats were reanesthetized with intraperitoneal urethane (1.2–1.5 g/kg) for MRI and CBF determination. All CBF measurements were carried out in the magnet bore simultaneously with the quantification of MRI variables, so that T2 BOLD and the trace of the diffusion tensor (Dav = 1/3 Trace D) were determined from one rat group without a contrast-agent injection (n = 17) and Dav and CBV were determined from a second group injected with an iron-based contrast agent (n = 6; see below). The CCAs were exposed and surrounded with silicon-coated snares joined to a controllable screw device, as previously described in detail (Gröhn et al., 2000). The device allowed for graded or complete occlusion of CCAs and release while the animal was inside the magnet. Femoral arteries were cannulated for blood pressure measurements (Cardiocap II; Datex, Helsinki, Finland) and sampling for blood gas analyses (i-Stat Corp., East Windsor, NJ, U.S.A.). Core temperature was monitored and maintained at approximately 37°C with a heating blanket during all experiments. Two to four sets of parametric control MRI scans were acquired before 15-minute graded CCA occlusion (n = 10) or 7-minute complete ischemia (n = 13). In the animals exposed to graded CCA occlusion during which there was no decrease in the water diffusion coefficient, a 15-minute complete occlusion of CCAs was induced after a 30-minute recovery period. After the release of CCA occlusion, MRI variables were monitored for 90 minutes. All animal experiments were approved by the Ethical Committee of the National Laboratory Animal Center, University of Kuopio.
Determination of cerebral blood flow
Cerebral blood flow was quantified using the hydrogen-clearance method, as described previously (Gröhn et al., 2000). Briefly, two 1-mm platinum/iridium electrodes were inserted 1 to 1.3 mm into both sides of the cortex 1.5 mm caudal to the bregma and 3 mm lateral from the midline. Silver/silver chloride reference electrodes were inserted subcutaneously in the flank. A clearance curve was recorded after loading with a 10/65/25 mixture of hydrogen/nitrogen/oxygen and was analyzed using the initial-slope approach (Olesen et al., 1971).
Magnetic resonance imaging
All MRI experiments were carried out at 4.7 T using a horizontal magnet (Magnex Scientific, Abdington, U.K.) equipped with actively shielded gradients (Magnex) interfaced either to an s.m.i.s. (Surrey Medical Imaging Systems, Guildford, U.K.) or to a Varian UnityInova (Varian, Palo Alto, CA, U.S.A.) console. A surface coil (diameter, 27 mm) was used for signal transmission and detection. A coronal imaging slice (slice thickness, 2 mm) was chosen according to T1-weighted pilot images so that the center of the slice was 4.1 mm from the surface of the brain.
A set of T2 images was acquired using a Hahn single-spin echo sequence with repetition time (TR) of 1,500 milliseconds and echo times (TE) in the sequence of 20, 50, 110, and 80 milliseconds. In six animals exposed to 7-minute ischemia, CBV was quantified using 6 mg/kg intravenous superparamagnetic iron oxide particles (AMI-227, Guerbet, France) (Payen et al., 1998) and a spin-echo sequence with TEs of 12, 20, 40, and 58 milliseconds. An adiabatic BIR-4 refocusing pulse was used to minimize pulse imperfections in signal transmission with a surface coil. Images for Dav were acquired using the identical sequence (TR, 1,500 milliseconds; TE, 80 milliseconds) with diffusion sensitizing bipolar gradients around the refocusing pulse with b-values of approximately 0, 500, and 1000 s/mm2. A field of view of 40 mm with a data matrix of 128×64 or 128×32 (both zero-filled to 128×128 before Fourier transformation) were used in the graded occlusion 15-minute ischemia and 7-minute ischemia experiments, respectively. These data-acquisition conditions led to temporal resolutions of 10 minutes for each absolute T2 and 4 minutes for Dav in the graded occlusion 15-minute ischemia group, and 5 and 2 minutes in 7-minute ischemia group, respectively.
Data analysis
Absolute T2 and Dav images were computed on a pixel-by-pixel basis by fitting the data to a single-exponential as a function of TE or b-values, respectively. Parietotemporal cortices (25 pixels) were analyzed for parametric MRI images so that hemispheric data were used independently. The signal-to-noise ratios in the Hahn single-echo images used for computations of absolute T2 images ranged from 112.5 ± 10.2 (TE, 20 milliseconds) to 18.3 ± 3.5 (TE, 110 milliseconds), thus enabling accurate quantification of T2. All cerebral data from graded occlusions were incorporated into the data set (n = 33), whereas MRI data from hemispheres showing greater than 12% (2 SD of control values) (Minematsu et al., 1992) reduction of Dav during CCA occlusions were included to the 7-minute (n = 16) and 15-minute ischemia (n = 14) data sets.
Changes in CBV were determined using the established linear relation between the relaxation rate change (ΔR2, R2 = 1/T2) and intravascular AMI-227 concentration, C(t) (Payen et al., 1998).
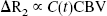
Hematocrit was assumed to remain constant. The relaxation rate change was obtained from T2 values before and after injection of AMI-227 as follows:
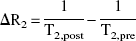
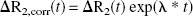
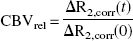
T2-BOLD theory
A theory quantitatively combining spin-echo BOLD changes with physiologic parameters including CBF, CMRO2, CBV, and OER (van Zijl et al., 1998; Ulatowski et al., 1999) has been successfully applied to several hemodynamic challenges, including acute hypoperfusion and ischemia (Gröhn et al., 2000; Kavec et al., 2001), hypoxia (van Zijl et al., 1998), and hypercapnia (Ulatowski et al., 1999). The theory provides a framework to estimate hemodynamic changes in the acute phase of cerebral ischemia (Gröhn et al., 2000). In the following section, the key equations and quantitative assumptions of the theory that are relevant to the current experimental settings are summarized.
An MRI voxel contains tissue and a mixture of blood vessels with different oxygenation statuses (i.e., arterioles, capillaries, and venules). Therefore, the contribution of both blood and tissue R2 has to be accounted for in calculating relaxation effects in perfused tissue (parenchyma). Because slow exchange between microvessels and tissue is expected to prevail in ischemia if the blood-brain barrier is intact, the signal attenuation in a spin-echo experiment can be expressed as
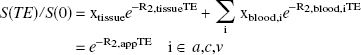
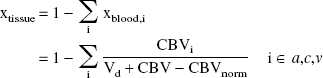
The R2 in blood can be derived as a volume-weighted sum of the relaxation rates in different compartments (van Zijl et al., 1998) as follows:
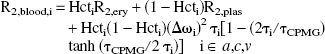
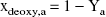
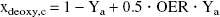
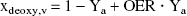
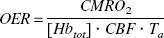
Experimental data from cerebral ischemia studies show that the R2 of the tissue remains unaltered after a brief period of ischemia (Knight et al., 1994; Li et al., 2000). The relaxation rate for the rat cerebral tissue (R2,tissue) at 4.7 T was determined from R2 data (Results), assuming an OER of 0.36 under normal physiologic conditions. In the urethane-anesthetized Wistar rat cortex, CBVnorm is 4.77 ± 0.13% (Sandor et al., 1986) and Vd is 820 μL/g (Schwab et al., 1997). A normal physiologic cerebral microvascular hematocrit value of 0.4 was used in the computations. In the present animal model, the arteries were occluded at the level of CCAs and vertebral arteries, thus lacking the arterial branching sequence before the occlusion site; therefore, this assumption is justified (Gröhn et al., 2000).
RESULTS
Magnetic resonance imaging and hemodynamic responses after graded occlusions
Blood pressure, core temperature, and blood gases were within the normal range during experiments (Table 1).
Physiologic variables before, during, and after occlusion
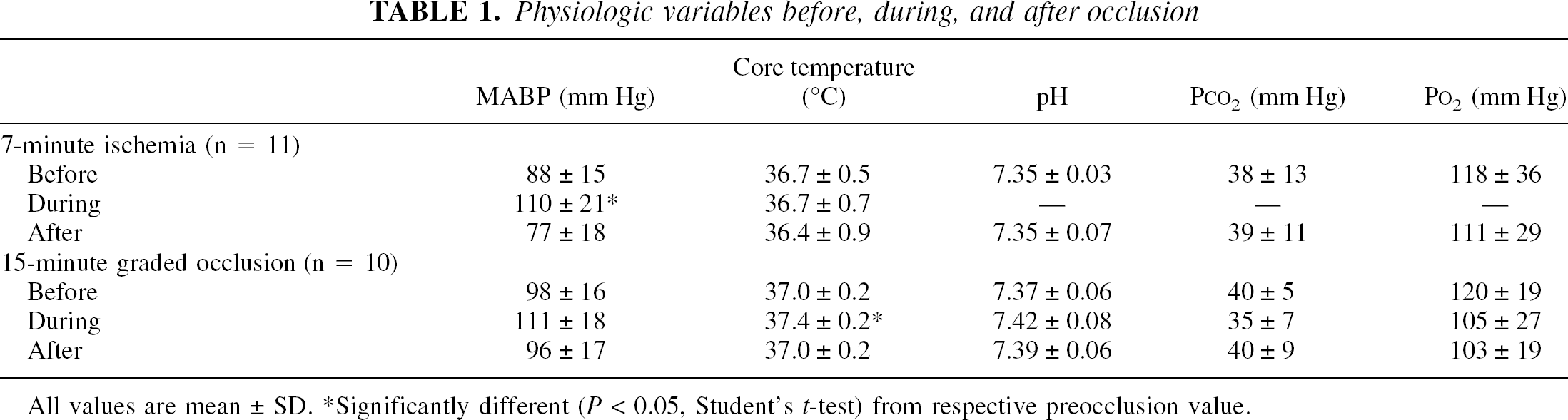
All values are mean ± SD.
Significantly different (P < 0.05, Student's t-test) from respective preocclusion value.
Cerebral blood flow in the cerebral cortex was 65 ± 24 mL·100 g−1·min−1 and the control value of Dav was 0.70 ± 0.04 × 10-3 mm2/s. Cerebral T2 as determined with the data matrixes of 128 × 64 or 128 × 32 was 50.3 ± 1.3 or 52.2 ± 1.6 milliseconds (P < 0.01), respectively. The postocclusion cerebral T2 is plotted as a function of CBF during 15 minutes of graded occlusion (Fig. 1A). It is evident that T2 was prolonged only when CBF had decreased below a threshold of 30% of normal, corresponding to an average flow of approximately 20 mL·100 g−1·min−1. Interestingly, this value was close to the CBF level below which a drop of more than 0.15 × 10-3 mm2/s in Dav has been observed in the same animal model system (Gröhn et al., 2000). Previous studies have shown that the expression of ischemic energy failure occurs at this CBF threshold (Busza et al., 1992; Kohno et al., 1995); thus, in the present case it is likely that ischemia developed in those brains in which CBF decreased below approximately 20 mL·100 g−1·min−1. The CBF determined during graded occlusion showed no correlation to reperfusion CBF measured within 15 minutes of the release of CCA occlusions (Fig. 1B). Interestingly, in some animals substantial CBF responses were detected even after mild reductions in flow that occurred during occlusion; in these animals, reperfusion AT2 was less than 1.5 milliseconds.
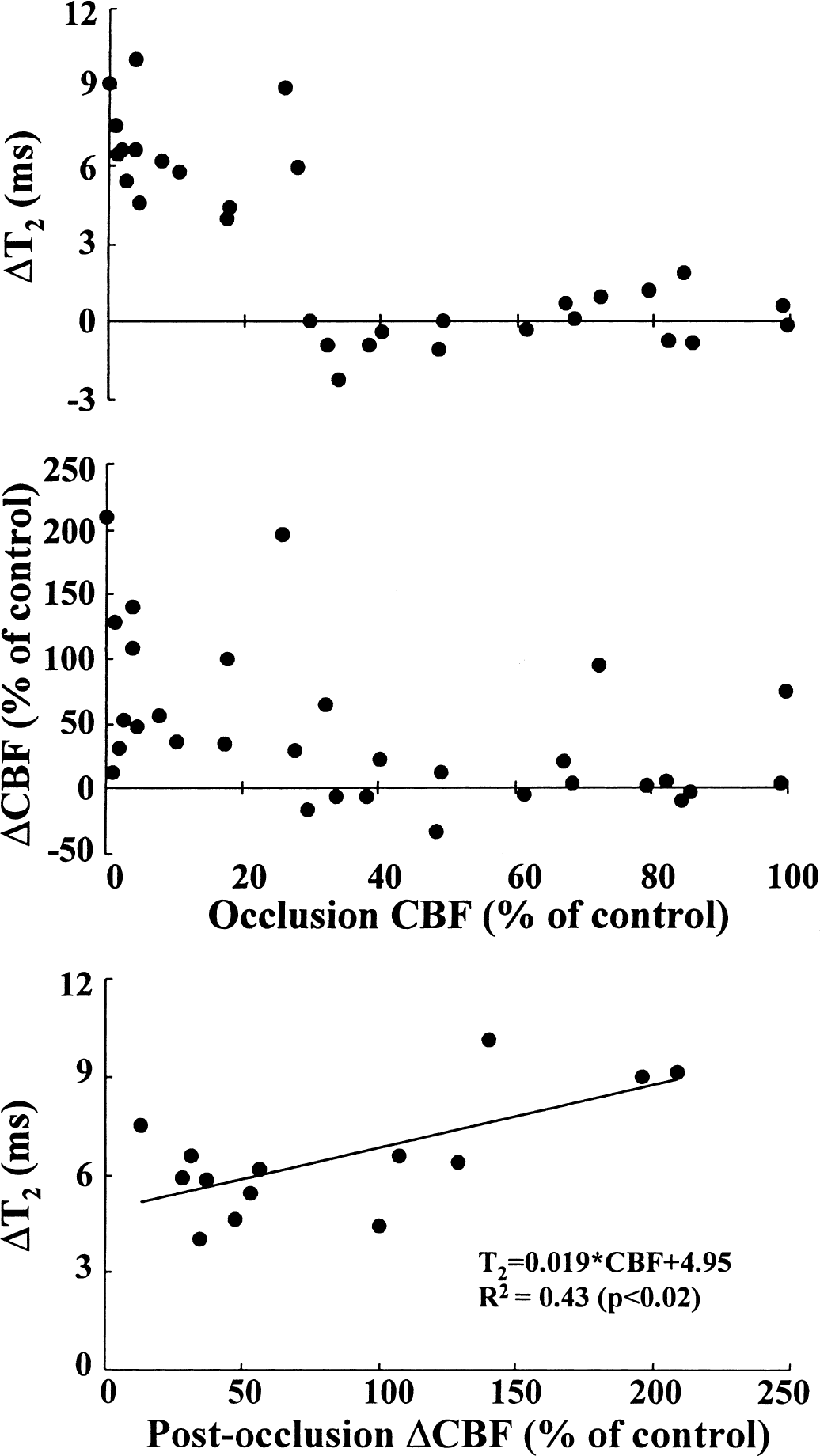
Postocclusion cerebral ΔT2
A plot of postischemic ΔT2 as a function of postischemic CBF change after 15 minutes of ischemia showed a correlation (T2 = 0.02 × CBF + 4.95, R2 = 0.43, P < 0.02) with a shallow slope between the two variables (Fig. 1C).
Magnetic resonance imaging and hemodynamic responses during and after ischemia
T2 increase (i.e., a positive BOLD response during reperfusion) was observed only in the brains in which the ischemic CBF threshold was crossed during occlusion, a finding that is suggestive of the physiological mechanisms underlying this phenomenon. We therefore studied more carefully the temporal and quantitative interrelations of T2, Dav, CBF and CBVrel in the animals exposed to 7 minutes of global ischemia. It has been shown that cerebral functions and cerebral metabolism can fully recover from ischemia of this duration (Alger et al., 1989). A decrease in T2 (Fig. 2A) signifying negative BOLD effect (−5.8 ± 3.4 milliseconds) was detected in acute ischemia (Gröhn et al., 1998, 2000). Dav had decreased by −0.18 ± 0.09×10−3 mm2/s before the release of the occlusion (Fig. 2C). As expected from previous results (Fig. 1A), an increase in T2 by 4.6 ± 1.2 milliseconds (P < 0.01) was observed after removal of occlusion, and T2 returned to the control level within 10 minutes of reperfusion (Fig. 2A). After reperfusion, Dav had normalized in the first time point of recovery by 6 minutes, and remained at the control level for the entire observation period (Fig. 2C).
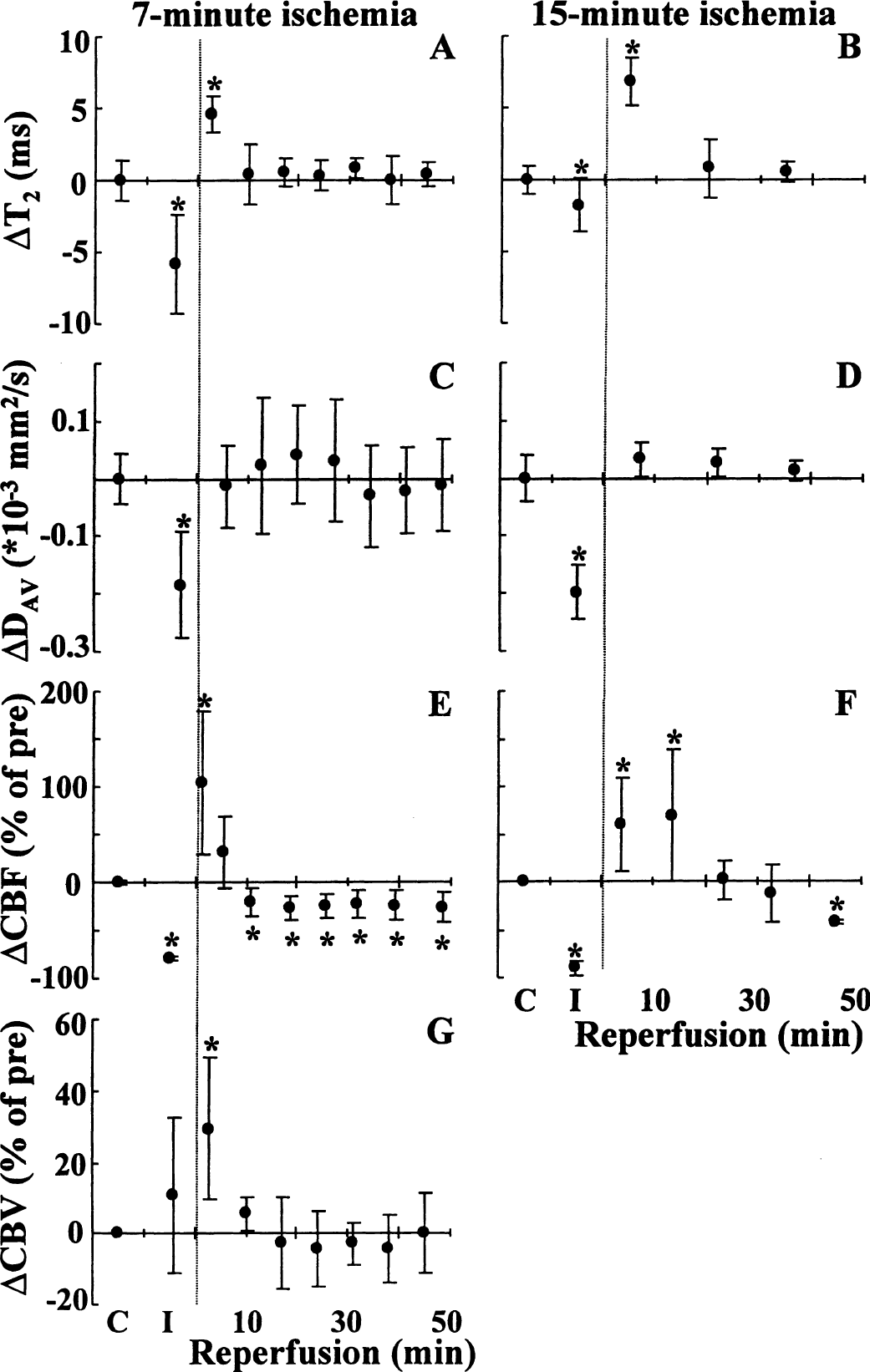
ΔT2 (
In the animals exposed to 15 minutes of ischemia after graded CCA occlusion, a T2 decrease of −1.7 ± 1.8 milliseconds (P < 0.05 compared with the preischemic value) was determined (Fig. 2B). The degree of Dav reduction during ischemia (−0.20 ± 0.05 × 10-3 mm2/s) was as severe in these rats as in the 7-minute ischemia group. A T2 increase of 6.8 ± 1.7 milliseconds (P < 0.01) was observed in the first reperfused time point, followed by normalization to preischemic values.
Bilateral CCA occlusion caused a severe CBF decrease of more than 80% (Figs. 2E and 2F). Cerebral blood flow increased by 103 ± 75% within 5 minutes of reperfusion after 7-minute ischemia. A moderate cerebral hypoperfusion developed as CBF decreased to a level that was approximately 20% lower than that determined before ischemia (Fig. 2E). In the rats exposed to 15 minutes of ischemia, CBF was increased by 84 ± 64% after 10 minutes of recovery, followed by a delayed hypoperfusion (Fig. 2F). During 7-minute ischemia, CBVrel was increased by 11 ± 22%, and by the first time point of reperfusion had increased by 29 ± 20%. The subsequent time points during recovery showed values at the control level (Fig. 2G). Using the established relation between CBF and CBV (Grubb et al., 1974), the CBF increase of 103% (Fig. 2E) would correspond to a CBV increase of 30% (Fig. 2G), which is in good agreement with the measured value.
T2 simulations
The theoretical framework (van Zijl et al., 1998) provides a sound foundation to estimate the contributions of hemodynamic or metabolic variables on T2 both under physiologic conditions and during reduced CBF. Prolonged parenchymal T2 in the postischemic brain may result from increased microvascular CBV (due to long T2 of blood) or inhibition of CMRO2 (due to reduced OER and xdeoxy,i). We have simulated parenchymal T2 using different CBVrel values as a function OER (Fig. 3). During ischemia, these computations showed that at an OER of 0.9, a value determined during reduced cerebral perfusion (Heiss and Podreka, 1993), the negative intravascular BOLD effect should have been curtailed T2 by 1.3 milliseconds. This value was smaller than the ΔT2 measured at 2.5 minutes of ischemia, but close to that measured at 5 minutes.
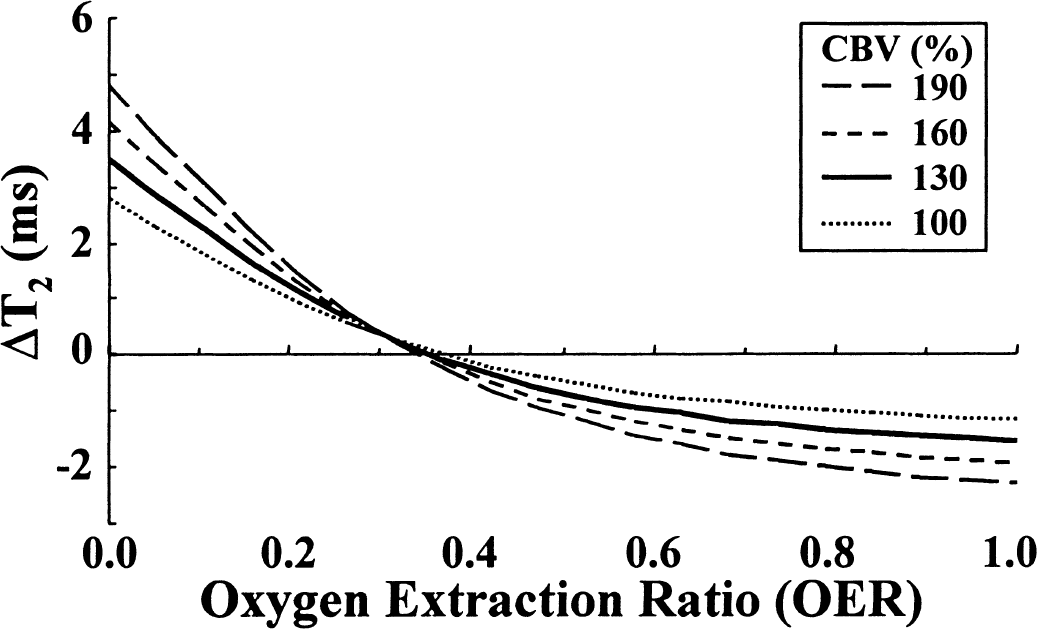
Parenchymal ΔT2 as a function of OER at different CBVs. Changes in parenchymal T2 as a function of OER were computed as described in Materials and Methods.
Using the CBV and CBF data (Fig. 2) as physiologic constraints, the calculations of parenchymal T2 during postocclusion hyperemia at unchanged CMRO2 gave a net T2 increase of 1.6 milliseconds, which is approximately 30% of the actual value determined (Fig. 2A). However, this figure is virtually identical to the 1.9-millisecond value derived from the ΔT2 versus ΔCBF plot (Fig. 1C) for the 100% CBF increase. Clearly, additional factors to those provided by CBF and CBV are needed to explain the experimentally determined T2. Potential contributing factors include the reduced OER due to depressed CMRO2. Using the measured postischemic hemodynamic data, an OER of approximately 0.05 should prevail to account for a postischemic T2 increase of 4 to 5 milliseconds (Fig. 3). Considering the quantified CBF and CBV and using Eq. 9, CMRO2 would not be restored to a level of more than 5% to 10% of the control level when T2 increased by 4.6 ± 1.2 milliseconds (Fig. 2A), as determined in the early moments of reperfusion. Using the data in Fig. 3 we can estimate that in those animals showing a CBF increase after mild occlusions without BOLD (Fig. 1B), the hemodynamic changes during a steady CMRO2 would result in a T2 change of 1 to 1.5 milliseconds. This value confirms the good quantitative agreement between the theoretical and experimental observations.
DISCUSSION
The present study shows that the positive BOLD effect appears only when the CBF threshold of approximately 20 mL·100 g−1·min−1 is crossed during a brief vascular occlusion, which sheds light on important physiologic mechanisms for the MRI detection of luxury perfusion (Turner et al., 1991; De Crespigny et al., 1992). This value is close to the previously reported CBF threshold for a large diffusion decrease (Busza et al., 1992; Kohno et al., 1995; Gröhn et al., 2000) and energy failure (Crockard et al., 1987). Therefore, the observed CBF dependence of the T2 effect supports the concept that reduced energy metabolism is needed for postischemic BOLD measurements. The adaptive changes in CBF and CBV during “hyperemia” can explain only a fraction of the early T2 change, and the main body of this effect of luxury perfusion is attributable to the reduced OER due to depression of CMRO2 in the early moments of recovery, leading to low xdeoxy in the (micro)vasculature.
Both T2 (van Zijl et al., 1998; Oja et al., 1999a; Ulatowski et al., 1999; Golay et al., 2001) and T2* (Ogawa et al., 1993; Lin et al., 1997; Davis et al., 1998; Kim et al., 1999) BOLD MRI can be used to quantify cerebral hemodynamics and metabolism, rendering these techniques powerful tools for physiology studies. The calibration procedures for the T2 BOLD are more straightforward than those for T2*, because the former method evaluates intravascular blood oxygenation-dependent relaxation changes and the exchange of water between the vasculature and tissue proceeds at a slow exchange rate relative to the MRI time scale (van Zijl et al., 1998; Golay et al., 2001). The contributions from field inhomogeneities independent of BOLD and the substantial extravascular effects on T2* have hampered the quantification of physiologic variables in absolute terms with this MRI method, given that venous oxygen saturation has been quantified by phase images using T2* (Haacke et al., 1997). Using the calibration data for blood as a function of oxygenation and hematocrit, the microvascular CBV (van Zijl et al., 1998; Ulatowski et al., 1999) and OER (Oja et al., 1999a; Golay et al., 2001) figures have been derived from cerebral T2 data that have clearly agreed with those acquired by positron emission tomography techniques.
Hemodynamic responses after an ischemic episode have recently been reviewed (Hossmann, 1997). It is a common observation that after a global ischemia, a two-step response consisting of a brief hyperemia followed by a hypoperfusion is detected (Lassen, 1966l Snyder et al., 1975; Todd et al., 1986; Hossmann, 1997). In hyperemia, CBF increases by 50% to 150% and CBV by 20% to 100% (Todd et al., 1986). The flow-volume changes result from near-complete relaxation of vessel tone caused by accumulation of vasodilating agents (Hossmann, 1997). After hyperemia, a long-lasting period of hypoperfusion occurs with reduced CBF (Snyder et al., 1975; Todd et al., 1986; Hossmann, 1997) and normal or slightly reduced CBV (Todd et al., 1986). The magnitude and duration of CBF and CBV changes in the present study both were similar to those reported after ischemic periods of equivalent duration (Todd et al., 1986; Allen et al., 1988).
Established interrelations between CBF thresholds for energy failure and extensive diffusion decline starts at approximately 20 mL·100 g−1·min−1 (Busza et al., 1992; Gröhn et al., 2000). Below this value, the full biochemical picture of ischemia is expressed with breakdown of adenylates, acidification, loss of ionic homeostasis, and the release of numerous vasoactive substances such as adenosine from the cells. These factors, together with increased extracellular [K+] and [H+], clearly evoke an almost completely “relaxed” tone of the cerebral vasculature that leads to a substantial increase in vessel diameter (Hossmann, 1997). Because CMRO2 becomes severely inhibited in ischemia (Ljunggren et al., 1974), it may take several minutes after the restoration of CBF to fully restore oxygen uptake. Indeed, it has been shown in a canine model of 15-minute cerebral ischemia that substantial nonoxidative glucose metabolism occurs for approximately 30 minutes of reperfusion (Snyder et al., 1975). Consistent with this observation, Allen et al. (1988) reported that after 15 minutes of complete forebrain ischemia in gerbils, lactate (as detected by 1H nuclear magnetic resonance spectroscopy) remained higher than 3 mmol/kg for longer than 25 minutes. 31P nuclear magnetic resonance spectroscopy studies have shown a time lag of several minutes in ATP recovery after global forebrain ischemia (Alger et al., 1989; Allen et al., 1992). Furthermore, venous oxygen saturation remains high for at least 5 minutes of global forebrain ischemia (Snyder et al., 1975).
The assignment of parenchymal T2 changes to the intravascular BOLD effect using the present procedure necessitates an unchanged R2,tissue. It is well established that residual flow is needed for water accumulation to the brain under flow-compromised conditions (Crockard et al., 1980). Crockard et al. (1980) have shown that 60 minutes of one-sided carotid occlusion in gerbils with a CBF less than 20 mL·100 g−1·min−1 results in a decrease in the specific gravity, an index of edema. In a baboon model of transient MCA occlusion, the brain water content remained unchanged during 30 minutes of ischemia and 60 minutes of reperfusion (Bell et al., 1985). Therefore, water accumulation to the flow-compromised brain is a time-dependent phenomenon. Previous MRI studies have shown that cortical T2, a marker of tissue water content, remains unchanged for the first 60 to 90 minutes of ischemia induced by permanent (Knight et al., 1994) or transient MCA occlusion (Li et al., 2000). In view of these data, it reasonable to assume that tissue water content is unaffected during 15 minutes of ischemia induced by the modified Pulsinelli (Pulsinelli et al., 1982) model. Furthermore, T2 shortening on induction of focal ischemia (Gröhn et al., 1998; Calamante et al., 1999) has been completely explained by the intravascular BOLD effect (Gröhn et al., 2000; Kavec et al., 2001). Recently, a quantitative MRI method has been reported to overcome the effects of R2,tissue on the BOLD signal in long-lasting ischemia (Grüne et al., 1999). This method uses R2 and R2* as determined by CPMG multiecho and gradient-echo MRI, respectively. In this way, one can then subtract the tissue relaxation changes sensed by R2 from the absolute R2* to obtain the plain BOLD effect. This method has also been applied to human stroke patients for the determination of CMRO2 changes by MRI (An and Lin, 2000). It should be noted, however, that the method can only provide changes in physiologic variables through BOLD because the interrelations between R2* and physiologic variables needed for absolute quantification of physiology have not yet been established.
In the present model of cerebrovascular occlusions, a good quantitative match between the T2 values obtained by the model and experiments is evident at 5 minutes of ischemia; however, at an earlier time point, the experimental T2 values tend to be shorter than expected (Figs. 2A and 2B). Although the exact reason for this discrepancy is not known, it appears to be related to partial “voluming” with macrovasculature (Oja et al., 1999b; Golay et al., 2001). Because venous blood in the ischemic brain has negligible oxygen saturation, T2 would have been approximately 10 milliseconds; thus, even a 10% volume fraction by large veins in the average MRI voxel (Oja et al., 1999b) would explain our observation of a short T2 in early ischemia. This situation is feasible by taking into account the spatial resolution used (128 × 32 versus 128 × 64 matrix in 7-minute and 15-minute ischemia groups, respectively) and the time-dependent changes in ischemic hemodynamics (Kettunen et al., 2001).
The key question to be addressed is whether the CBF and CBV increases in postischemic hyperemia alone could account for the observed T2 BOLD effect. Our data unequivocally point to a key contribution of reduced OER as a result of depressed CMRO2 to the T2 BOLD in luxury perfusion. The methods used for quantification of CBF and T2 have distinct temporal resolutions, making it difficult to obtain time-matched data. The associated CBV change with the CBF increase explains only one third of the T2 increase if CMRO2 proceeds at control rate directly from the start of reperfusion. For instance, we compute that after 7 minutes of ischemia, a reduction of OER to 0.05 is needed to reconcile the observed BOLD effect of approximately 5 milliseconds. Under these conditions, the possible partial voluming by venous macrovasculature is not harmful to the T2 data because the low oxygen extraction and the increased washout due to the high CBF would make the oxygenation of venous blood similar to that in the arteriole bed (Snyder et al., 1975). A reduction of maximal amount of oxygen extracted in the capillary bed leads to an altered T2 versus OER curve (Fig. 3) to the right (Gröhn et al., 2000), which will inevitably promote the BOLD response in luxury perfusion. In this instance it should be noted that OER versus parenchymal T2 response curves are magnetic-field dependent (Gröhn et al., 2000). For example, at 1.5 T CBV changes begin to increase T2 at higher OER values than at 4.7 T. Consequently, T2 BOLD changes on luxury perfusion would appear after milder OER reductions at 1.5 T in quantities equivalent to 4.7 T, though their relative magnitude would be smaller because of the inherently longer cerebral T2 values in this field.
To conclude, we have shown that significantly increased T2 is observed after a short period of global ischemia. The observed T2 changes are consistent with a previous report of hyperintensity in T2*-weighted images after short periods of ischemia at 2.0 T (De Crespigny et al., 1992). In the absence of water accumulation, a large part of the parenchymal T2 BOLD effect can be ascribed to the reduced CMRO2, with minor contributions from increased CBF and CBV.
Footnotes
Acknowledgments:
The authors thank Mr. Kari Mauranen for his help with the statistical analyses and Ms. Niina Kuhmonen for her expert technical assistance.