
Abstract
The present study assessed the role of PARP [poly(adenosine diphosphate-ribose) polymerase] activation in experimental pneumococcal meningitis. Mice with a targeted disruption of the PARP1 gene were protected against meningitis-associated central nervous system complications including blood-brain barrier breaching and increase in intracranial pressure. This beneficial effect was paralleled by a significant reduction in meningeal inflammation, as evidenced by significantly lower cerebrospinal fluid leukocyte counts and interleukin-1β, −6, and tumor necrosis factor-α concentrations in the brain (compared with infected wild-type mice). The reduction in inflammation and central nervous system complications was associated with an improved clinical status of infected, PARP1-deficient mice. A similar protective effect was achieved by PARP inhibition using 3-aminobenzamide, the pharmacologic efficacy of which was confirmed by a marked attenuation of meningitis-induced poly(ADP)ribose formation. When the rat brain-derived endothelial cell line GP8.3 was cocultured with macrophages, exposure to pneumococci induced endothelial cell death and was paralleled by PARP activation and a reduction in the oxidized form of cellular nicotinamide adenine dinucleotide content. Treatment with 3-aminobenzamide significantly attenuated cellular nicotinamide adenine dinucleotide depletion and pneumococci-induced cytotoxicity. Thus, PARP activation seems to play a crucial role in the development of meningitis-associated central nervous system complications and pneumococci-induced endothelial injury. Inhibitors of PARP activation could provide a potential therapy of acute bacterial meningitis.
Reactive oxygen species (ROS) and reactive nitrogen intermediates (RNI) are terminal mediators of brain damage in bacterial meningitis (Koedel and Pfister, 1999a). Both ROS and RNI can be cytotoxic through a number of independent mechanisms. Their cytotoxic effects comprise inactivation of a variety of enzymes, damage to membrane ion transporters, and initiation of lipid peroxidation. Besides these modes of action, ROS and RNI produce a complex variety of lesions in the DNA that give rise to single- and double-strand breaks (Lieber, 1998; Halliwell, 1999). The chromatin-associated zinc finger protein, poly[adenosine diphosphate (ADP)]-ribose) polymerase-1 (PARP1), catalyzes the transfer of the ADP-ribose moiety from its substrate, the oxidized form of nicotinamide adenine dinucleotide (NAD+), to diverse nuclear acceptor proteins, and is activated by these DNA strand breaks (Ikejima et al., 1990; Weinfeld et al., 1997). Extensive PARP1 activation can initiate an energy-consuming futile intracellular cycle, leading to rapid depletion of cellular stores of NAD+ and adenosine triphosphate, which results in cell dysfunction and necrotic-type cell death (Ha and Snyder, 2000). This cytotoxic pathway was observed in vitro in cells exposed either to various oxygen-centered free radicals or to peroxynitrite (Heller et al., 1995; Szabo et al., 1996). The causal role of PARP1 in pathology was also observed in experimental disease models using mice with an inactivation of the PARP1 gene. Pronounced protection was observed in cerebral and myocardial ischemia-reperfusion injury (Endres et al., 1997; Pieper et al., 2000b), hemorrhagic shock (Liaudet et al., 2000), and diabetic pancreatic β-cell destruction (Burkart et al., 1999; Pieper et al., 1999). Furthermore, PARP1 was suggested to be involved in experimental arthritis (Szabo et al., 1998), colitis (Jijon et al., 2000), and endotoxic shock (Oliver et al., 1999). However, the role of PARP1 in central nervous system (CNS) infection is not completely unknown.
In bacterial meningitis, an unfavorable clinical outcome of the disease is mainly due to CNS complications (Pfister et al., 1993). The major CNS complications causing brain injury comprise cerebrovascular alterations, brain edema, hydrocephalus, and increased intracranial pressure (ICP). Although there is substantial evidence that ROS and RNI are central mediators of meningitis-associated CNS complications (Koedel et al., 1995; Buster et al., 1995; Leib et al., 1996; Kastenbauer et al., 1999), the mechanisms underlying ROS- and RNI-induced brain injury in meningitis are poorly defined.
To examine the possible involvement of PARP1 activation in pneumococcal meningitis, we studied the inflammatory host response, development of CNS complications, and clinical status in PARP1-deficient mice. We also investigated the effect of the PARP inhibitor 3-aminobenzamide on meningitis-associated pathophysiologic alterations in a rat model and on pneumococci-induced endothelial cell injury. The data suggest a central role of PARP in the initiation of meningeal inflammation and development of CNS complications in acute bacterial meningitis.
MATERIALS AND METHODS
Animals
Experiments were conducted in male PARP1 −/− and wild-type litter mates consisting of a mixed genetic background of 129SV and C57BL/6 (kindly provided by Prof. A. Fontana). These mice stem from colonies that were established from breeding pairs originally derived from Prof. Wang (C57BL/6@IARC; International Agency for Research on Cancer, Lyon, France) (Wang et al., 1995). The PARP-inhibitor study was performed in male Wistar rats (Charles River Wiga GmbH, Sulzfeld, Germany). All experiments were approved by the Government of Upper Bavaria.
Mouse model of pneumococcal meningitis
Meningitis was induced by a transcutaneous injection of 15 μL 107 colony-forming units per milliliter of Streptococcus pneumoniae strain type 3 into the cisterna magna during short-term anesthesia with halothane. Mice were weighed, put into cages, and allowed to wake. Mice were clinically evaluated 24 hours after infection. The clinical score comprises the following criteria: presence of tremor and piloerection, vigilance, beam-balancing test, and postural-reflex test according to Bederson et al. (1986). Thereafter, the body temperature was measured with a rectal probe, mice were weighed, and mice were anesthetized by an intraperitoneal injection of ketamine/xylazine. To evaluate blood-brain barrier (BBB) permeability, 0.5 mL 2% Evans blue (EB) was administered intraperitoneally. A catheter was inserted into the cisterna magna via a burr hole in the occipital bone to measure ICP and to determine cerebrospinal fluid (CSF) leukocyte counts. Forty-five minutes after EB application, mice were deeply anesthetized with thiopental and perfused transcardially with ice-cold phosphate-buffered saline (PBS). Brains were extracted and rapidly frozen.
Determination of Evans blue levels in mouse brain homogenates
Blood-brain barrier integrity was investigated using the EB extravasation method (Lu et al., 1997). Frozen brain sections were homogenized, centrifuged, and 0.5 mL formamide was added to the resulting pellets. The samples were placed into a thermomixer for 48 hours at 37°C and centrifuged. The absorbance of the supernatant and a standard curve were measured spectrophotometrically at 650 nm. The EB concentration was expressed as ng/mg brain protein.
Measurement of myeloperoxidase concentration in mouse brain homogenates
Brain myeloperoxidase concentration, a marker for leukocyte infiltration, was determined as described previously (Koedel et al., 1999).
Immunoassays for the murine cytokines interleukin-1β, −6, and tumor necrosis factor-α
Immunoreactive interleukin-1β, −6, and TNF-α were determined using commercially available EIA kits (R&D Systems GmbH, Wiesbaden-Nordenstadt, FRG). Briefly, frozen brain sections were homogenized in sample buffer (10 mmol/L Hepes, pH 7.9; 10 mmol/L potassium chloride; 1.5 mmol/L magnesium chloride; and a mixture of protease inhibitors including phenyl-methylsulfonyl fluoride, aprotinin, leupeptin, and pepstatin A). The homogenates were centrifuged and 50 μL supernatant was used for each determination. Immunoreactive interleukin-1β, −6, and TNF-α concentrations were expressed as pg/mg brain protein.
Experimental groups in the mouse model
The following experimental groups were investigated: wild-type mice injected intracisternally with 15 μl PBS (n = 6), wild-type mice injected intracisternally with S pneumoniae (n = 9), and PARP1-deficient mice injected intracisternally with S pneumoniae (n = 9).
Rat model of pneumococcal meningitis
A well-characterized rat model of pneumococcal meningitis that also permits the study of cerebrovascular alterations was used in this study (Koedel et al., 2000). Briefly, meningitis was induced by intracisternally injection of 150 μL 107 colony-forming units/mL S pneumoniae. Twenty-four hours later, rats were clinically evaluated as described previously (Koedel et al., 2000). Thereafter, rats were anesthetized, tracheotomized, and artificially ventilated. A catheter was inserted into the left femoral artery for continuous monitoring of mean arterial blood pressure. The left femoral vein was cannulated for EB administration to evaluate BBB permeability (Paul et al., 1998). A catheter was inserted into the cisterna magna for continuous ICP monitoring and to determine CSF leukocyte counts and CSF interleukin-6 concentrations. Regional cerebral blood flow (rCBF) was measured by laser-Doppler flowmetry. For measurement of carbon dioxide reactivity of cerebral vessels, hypercapnia was produced with 10% carbon dioxide, 21% oxygen, and the balance of nitrogen as described previously (Koedel et al., 2000). The autoregulatory capacity of cerebral vessels was studied by increasing mean arterial blood pressure by the intravenous infusion of norepinephrine as described by Koedel et al. (2000).
TUNEL staining
For the detection of DNA damage in situ, frozen brain sections containing lateral ventricles and hippocampal tissue were analyzed using a commercially available in situ cell death detection kit (Roche Molecular Biochemicals, Mannheim, Germany).
Experimental groups in the rat model
The following experimental groups were investigated: (1) rats injected intracisternally with 150 μL PBS (n = 8), (2) untreated rats injected intracisternally with S pneumoniae (n = 8), (3) infected rats treated intraperitoneally with the PARP inhibitor 3-aminobenzamide (30 mg/kg) 6 and 18 hours after infection (n = 6).
Western blot analysis
To determine the expression of platelet endothelial cell adhesion molecule (PECAM)-1 and occludin, and the formation of ADP-ribose polymers during pneumococcal meningitis, brain protein extracts were subjected to gel electrophoresis on 4% to 12% NuPage Tris-Bis gels; for Western blot analysis of brain PARP1 expression, 4% to 12% NuPage Tris-Glycine gels were used for separation of total cellular proteins (both from Novex, Frankfurt, Germany). After transfer of proteins on PVDF membranes, primary incubations were carried out with a 1:2000 dilution of the polyclonal anti-PECAM-1 antibody H-300, a 1:1000 dilution of the polyclonal anti-PARP antibody H-250 (both from Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.), a 1:2000 dilution of the mouse monoclonal antibody to poly(ADP-ribose) 10H (Alexis Deutschland GmbH, Grünberg, Germany), or a 1:1000 dilution of a polyclonal anti-occludin antibody (Zymed Laboratories, San Francisco, CA, U.S.A.). Bound primary antibody was detected using a peroxidase-conjugated antibody against immunoglobulin G (1:2,000 dilution; Sigma Chemicals, Deisenhofen, Germany) and a chemiluminescent substrate (Amersham Pharmacia Biotech, Freiburg, Germany).
Cell culture experiments
The simian virus 40 large T-immortalized rat brain-derived endothelial cell line GP8.3 (a gift by Dr. J. Greenwood, London, UK) was cultured in Ham F10 medium supplemented with 20% fetal calf serum, 80 μg/mL heparin, 7.5 μg/mL endothelial cell growth supplement, 0.5 μg/mL ascorbic acid, 2 mmol/L glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin (assay medium). The murine macrophagelike cell line RAW 264.7 (a gift by Prof. Dr. F. Krombach, Munich, Germany) was kept in RPMI 1640 supplemented with 10% FCS, 2 mmol/L glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. For experiments, GP8.3 endothelial cells were seeded onto 24-well, collagen type I-coated culture dishes (Becton Dickinson, Lincoln Park, NJ, U.S.A.) at a density of 1 × 105 cells per well, and cultured for 72 hours until GP8.3 cells formed a confluent monolayer. At that point, the medium was replaced with the Ham F10 assay medium and the various stimuli (see Experimental groups in vitro). For coculture experiments, RAW 264.7 cells were seeded onto cell culture inserts (3-μm pore size; Becton Dickinson) at a density of 1 × 104 cells per well, and cultured for 72 hours. Inserts were then placed into wells containing confluent GP8.3 cells, Ham F10 assay medium, and the various stimuli.
Measurement of cellular NAD+ content
At 24 hours after stimulation, GP8.3 endothelial cells were extracted with 500 μL ice-cold 0.4 mol/L perchloric acid. After neutralization with 80 μL 2.5 mol/L potassium bicarbonate, cellular debris was removed by centrifugation. The supernatants were assayed for NAD+ using a modification of the colorimetric method (Bernofsky and Swan, 1973) in which the reduced form of nicotinamide adenine dinucleotide (NADH) produced by enzymatic cycling with alcohol dehydrogenase reduces MTT [3-(4,5-dimethylthiazo-2-yl)-2,5-diphenyltetrazoliumbromide] to formazan.
MTT colorimetric assay
Cell viability was assessed by the mitochondrial-dependent reduction of MTT to formazan as described previously (Denizot and Lang, 1986).
Propidium iodide staining
The GP8.3 endothelial cells were stained with propidium iodide as described by Barres et al. (1992).
Immunocytochemical staining for ADP-ribose polymers and L-nitrotyrosine
To examine whether pneumococcal stimulation causes ADP-ribose polymer formation in vitro, cells were fixed with absolute ethanol. To allow for PARP activity, cell ghosts were incubated in 100 mmol/L Tris-hydrochloride (pH 8) containing 10 mmol/L magnesium chloride, 1mmol/L dithiothreitol, and 400 μmol/L NAD+ at 30°C for 2 hours (Kupper et al., 1996). Thereafter, cells were incubated with a 1:50 dilution of the monoclonal antibody 10H directed against poly-(ADP-ribose). Immune complexes were detected by subsequent incubation with a fluorescein isothiocyanate conjugated-labeled sheep antimouse immunoglobulin G (diluted 1:200; Sigma Chemicals).
For the detection of L-nitrotyrosinelike immunoreactivity, endothelial cells were fixed with paraformaldehyde and incubated with rabbit polyclonal antinitrotyrosine immunoglobulin G (1:100 dilution, Upstate Biotechnology, Lake Placid, NY, U.S.A.). The immunofluorescence was accomplished with a 1:200 dilution of a fluorescein isothiocyanate conjugated-labeled goat antirabbit immunoglobulin G (Sigma Chemicals).
Nitroblue tetrazolium staining technique
Superoxide generation was determined using the nitroblue tetrazolium staining technique as described (Koedel and Pfister, 1999b).
Measurement of nitrite/nitrate concentrations
Nitric oxide production was assessed by measuring nitrite/nitrate using a commercially available colorimetric assay kit (Calbiochem-Novabiochem, San Diego, CA, U.S.A.).
Experimental groups in vitro
The following experimental groups were investigated: GP8.3 endothelial cells cultured alone or with the RAW 264.7 macrophages, stimulated with 108 colony-forming units/mL heat-killed pneumococci, (1) untreated, (2) treated with 0.5 mmol/L 3-aminobenzamide, or (3) treated with 5 mmol/L 3-aminobenzamide. The GP 8.3 cells cultured alone or with RAW 264.7 in the Ham F10 assay medium without pneumococci, served as negative controls.
Statistical analysis
For statistical analysis, data obtained 24 hours after pneumococcal challenge (both in vitro and in vivo) were compared by one-way analysis of variance and Scheffes test. Data are given as mean ± SD.
RESULTS
Activation of PARP during experimental pneumococcal meningitis
In Western blot analysis, PARP1 protein was absent in brain homogenates from PARP1-deficient mice. Brain PARP1 protein expression did not differ between PBS-injected and pneumococci-infected wild-type mice (at 24 hours after intracisternally injection;Fig. 1A). Furthermore, no immunoreactive species corresponding to the molecular size of PARP1 cleavage products (especially the 85-kDa fragment) were detectable.
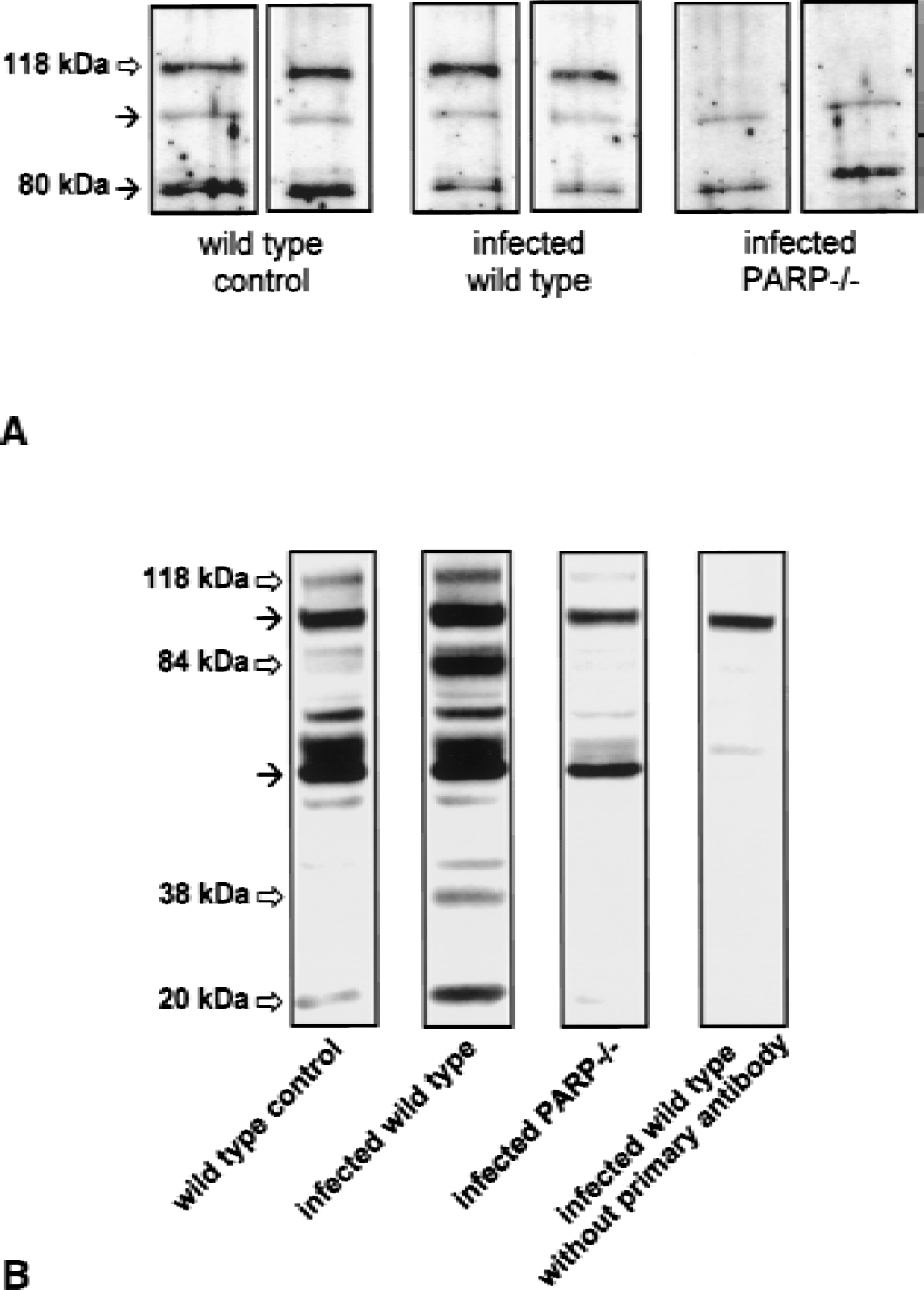
Western blot analysis of PARP [poly(adenosine diphosphate-ribose) polymerase] expression
Adenosine diphosphate-ribose polymer formation is a marker for PARP catalytic activity (Kawamitsu et al., 1984). To determine whether pneumococcal meningitis induces poly(ADP ribosyl)ation, mouse and rat brain homogenates were subjected to immunoblot analysis with monoclonal antibodies to PAR. The extent of poly(ADP ribosyl)ated proteins was increased in mouse (Fig. 1B) and rat brains (data not shown) 24 hours after pneumococcal inoculation compared with uninfected control animals. The array of poly(ADP ribosyl)ated proteins was consistent with recent studies in which several (ADP ribose) protein acceptors in the MW 20,000- to 120,000-Da range were identified by immunoblotting of cell and tissue extracts (Simbulan-Rosenthal et al., 1998; Szabados et al., 1999). Both targeted disruption of the PARP1 gene (Fig. 1B) and pharmacologic PARP inhibition with 3-aminobenzamide (data not shown) were associated with no or nearly absent immunoreactvity for poly(ADP ribose).
Effect of PARP1 depletion and PARP inhibition on the clinical status
Clinical examination revealed a similar degree of disease in all infected wild-type mice as evidenced by piloerection, lethargy, and impaired motor functions (parameters of the clinical score), and loss of body weight (−13.3% ± 2.8%) and hypothermia (34.6 °C ± 0.7 °C). Infected PARP1-deficient mice had significantly improved clinical scores (Fig. 2A), and showed less of a reduction in body weight (−8.2% ± 2.9%) and body temperature (36.8 °C ± 0.4 °C). None of the PBS-injected control mice exhibited any signs of disease within the observation period. No significant change in body weight (+0.9% ± 3.0%) was observed. Body temperatures (36.7 °C ± 0.7 °C) were within the normal range.
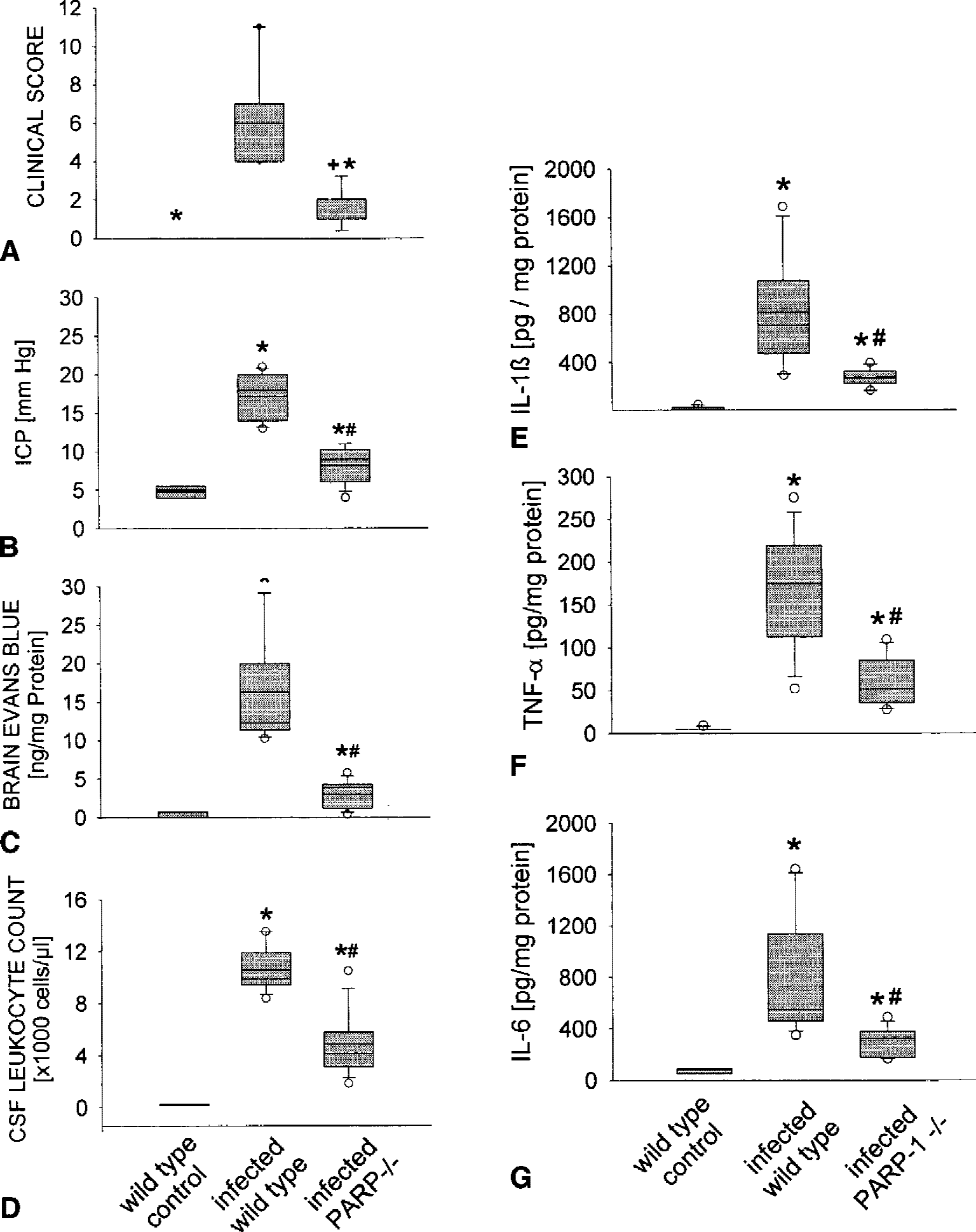
Effect of PARP1 depletion on the clinical status, central nervous system complications, and the host immune response in experimental pneumococcal meningitis.
Infected rats also showed typical signs of meningitis. Treatment with 3-aminobenzamide significantly improved the clinical scores of infected rats compared with those of untreated, infected rats (Table 1).
Effect of PARP inhibition on meningitis-associated intracranial complications
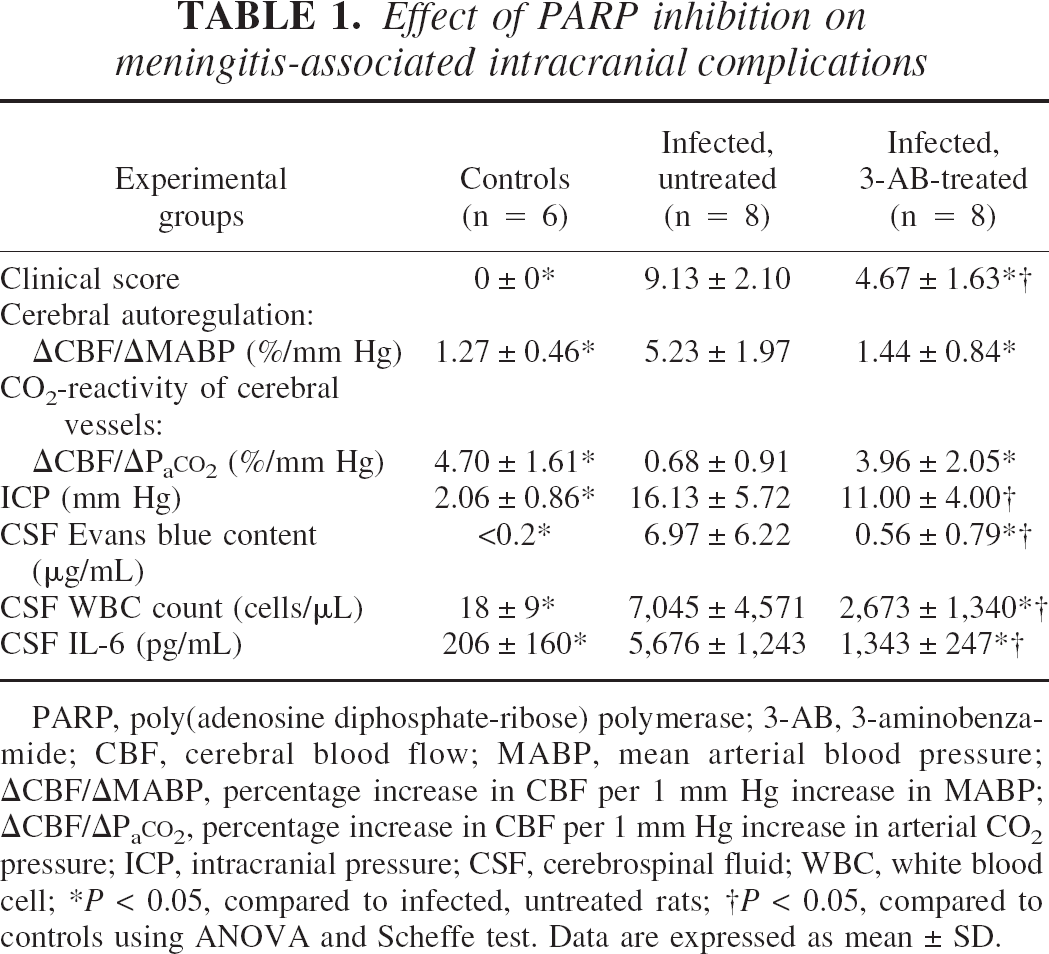
PARP, poly(adenosine diphosphate-ribose) polymerase; 3-AB, 3-aminobenzamide; CBF, cerebral blood flow; MABP, mean arterial blood pressure; ΔCBF/ΔMABP, percentage increase in CBF per 1 mm Hg increase in MABP; ΔCBF/ΔPaCO2, percentage increase in CBF per 1 mm Hg increase in arterial CO2 pressure; ICP, intracranial pressure; CSF, cerebrospinal fluid; WBC, white blood cell;
P < 0.05, compared to infected, untreated rats;
P < 0.05, compared to controls using ANOVA and Scheffe test. Data are expressed as mean ± SD.
Effect of PARP1 depletion and PARP inhibition on meningitis-associated central nervous system complications
Pneumococcal infection caused a significant increase in ICP and BBB permeability, as indicated by detectable EB concentrations in mouse brain and rat CSF (Figs. 2B and 2C; Table 1). Infected PARP1-deficient mice showed a significantly attenuated increase in ICP and in brain Evans blue concentrations. Treatment with 3-aminobenzamide did not lead to a significant reduction in ICP but significantly attenuated EB extravasation into the CSF, indicating a protective effect on BBB integrity (Table 1).
To study the autoregulatory capacity of cerebral vessels, mean arterial blood pressure was slowly increased by the intravenous infusion of norepinephrine, and rCBF was recorded in control and infected rats. In control rats, rCBF was maintained relatively constant when mean arterial blood pressure was increased by intravenous norepinephrine (Table 1). In infected rats, a dramatic increase in rCBF occurred during norepinephrine-induced arterial hypertension, indicating an impairment of cerebrovascular autoregulation. Treatment with 3-aminobenzamide prevented the meningitis-associated loss of cerebrovascular autoregulation. Carbon dioxide reactivity of cerebral vessels was assessed by producing hypercapnia with 10% carbon dioxide, 21% oxygen, and a balance of nitrogen. In control rats, there was an increase of rCBF of 4.7 ± 1.6% per 1 mm Hg increase in arterial carbon dioxide pressure, indicating an intact hypercapnic reactivity of cerebral vessels. Twenty-four hours after pneumococcal infection, the rCBF response to hypercapnia was markedly reduced in infected rats. Similar to the rCBF response to hypertension, treatment with 3-aminobenzamide prevented the pneumococci-induced loss of the hypercapnic reactivity of cerebral vessels (Table 1).
To assess meningitis-associated vascular injury in vivo, we determined brain protein levels of PECAM-1 and occludin by Western immunoblotting using mouse-brain homogenates. Recent work implicated the use of PECAM-1 expression in regional vascular beds as an indicator of endothelial cell surface area (Eppihimer et al., 1998; Ogunshola et al., 2000; Wiessner et al., 2001). Occludin expression was shown to be an appropriate marker for the tight junction permeability properties of endothelial cells in different tissues (Wachtel et al., 1999; Bolton et al., 1998; Wang et al., 2001). Twenty-four hours after pneumococcal infection, infected wild-type mice displayed a significant decrease in brain PECAM-1 protein levels compared with that of uninfected controls (Fig. 3). Western blot analysis of occludin expression showed a slight (but not significant) reduction of brain occludin levels in mice subjected to pneumococcal meningitis (Fig. 3). In addition, a band with a size of approximately 50-kDa, apparently a proteolytic fragment of occludin, became visible in brain homogenates from infected wild-type mice (Wachtel et al., 1999). Infected PARP1-deficient mice exhibited higher levels of PECAM-1 protein expression and a lower extent of occludin proteolysis (Fig. 3).
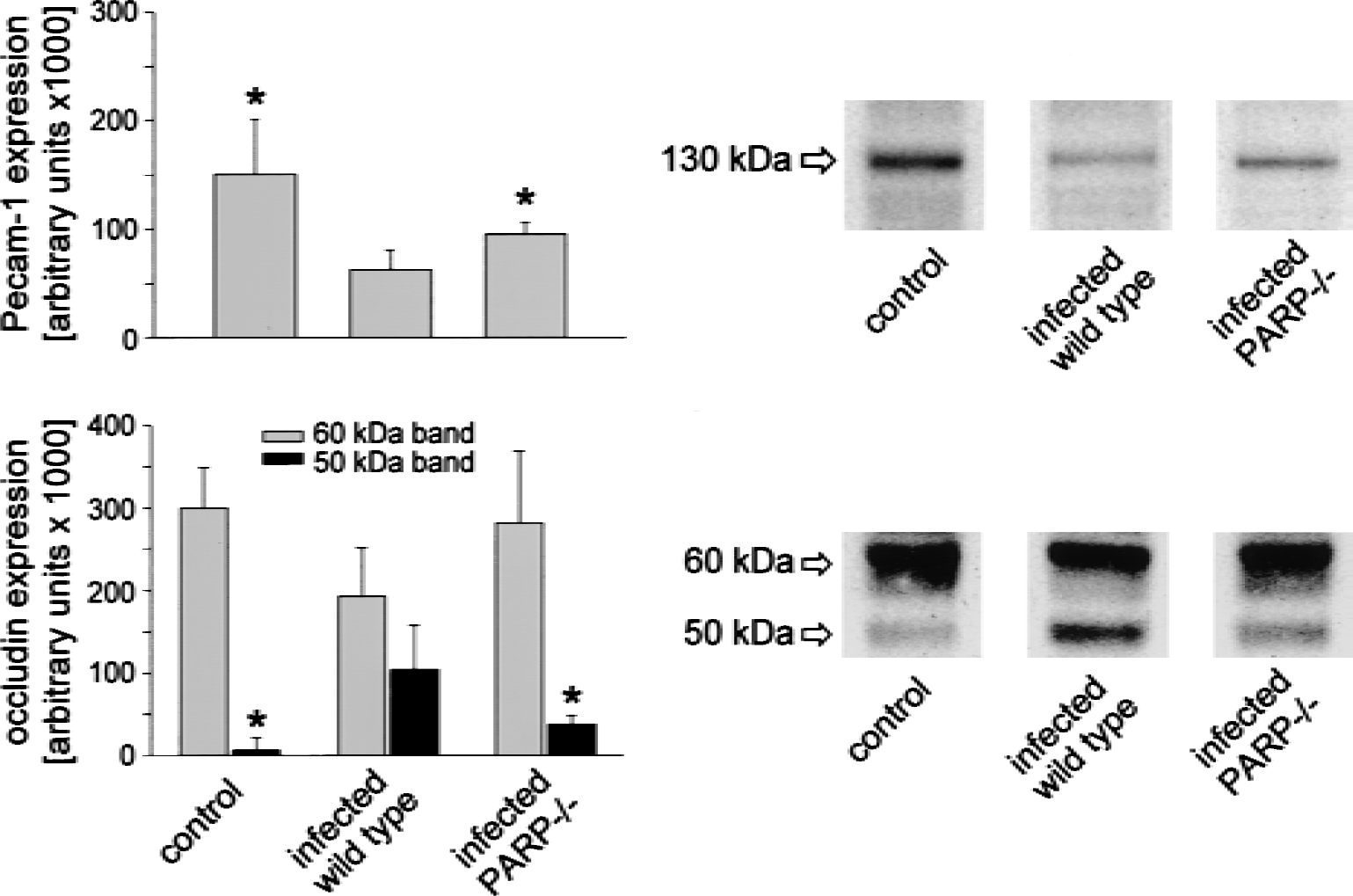
Western blot analysis of platelet endothelial cell adhesion molecule (PECAM-1) and occludin expression in mouse brain homogenates. Twenty-four hours after pneumococcal infection, infected wild-type mice displayed a significant decrease in brain PECAM-1 protein levels compared with uninfected controls (upper graphs). In addition, a proteolytic fragment of occludin (molecular size, ≈ 50 kDa) became detectable in brain homogenates from infected wild-type mice (lower graphs). Infected PARP-deficient mice exhibited higher levels of PECAM-1 protein expression and a lower extent of occludin proteolysis. Western blot analysis was performed on brain protein extracts of five randomly selected animals of each experimental group investigated, and one representative example for each group was shown. Blots were scanned, and optical densities were determined using a computer-imaging analysis system (VisitronSystems GmbH, Puchheim, Germany). Intensity values are given as arbitrary units. * P < 0.05 compared with infected wild-type mice.
Effect of PARP1 depletion and PARP inhibition on the host immune response
Pneumococcal infection led to a massive leukocyte infiltration into the subarachnoid space in wild-type mice and in untreated, infected rats. Targeted disruption of the PARP1 gene was associated with a significant reduction in CSF pleocytosis compared with that of infected wild-type mice (Fig 2D). In accordance with this finding, brain myeloperoxidase concentrations were significantly lower in infected PARP-deficient mice than in infected wild-type mice (1.4 ± 1.1 vs. 4.1 ± 1.3 U/g protein in infected wild-type mice and 0.4 ± 0.2 U/g protein in wild-type controls). Likewise, treatment with 3-aminobenzamide significantly reduced CSF pleocytosis compared with that of untreated infected rats (Table 1).
To further characterize the role of PARP1 in the pneumococci-induced host immune response within the CNS, mouse-brain homogenates obtained either from wild-type mice or PARP1-deficient mice were analyzed for their expression of the proinflammatory cytokines interleukin-1β, −6, and TNF-α. Twenty-four hours after pneumococcal inoculation, a significant increase in brain protein concentrations of all these cytokines was detectable in infected wild-type mice compared with controls (Figs. 2E, 2F, and 2G). Targeted disruption of the PARP1 gene was associated with significantly diminished brain levels of interleukin-1β, −6, and TNF-α. In accordance with these findings, increased levels of interleukin-6 were observed in rat CSF 24 hours after pneumococcal infection. Treatment with 3-aminobenzamide caused a marked reduction of CSF interleukin-6 concentrations (Table 1).
Determination of meningitis-associated brain injury
Because DNA damage is implicated as an essential step in PARP activation the presence of DNA strand breakage was investigated by the TUNEL (terminal deoxynucleotidyl transferase-mediated 2′-deoxyuridine 5′-triphosphate-biotin nick end labeling) method. It is worthwhile to note that the TUNEL assay detects free 3′-hydroxyl ends due to single- or double-strand breaks (Willingham, 1999; Stadelmann and Lassmann, 2000). Both, single- and double-strand breaks have been shown to activate PARP with almost equal efficiency (Ikejima et al., 1990; Weinfeld et al., 1997).
All TUNEL-positive cells were detected within the inflammatory infiltrates in the subachnoid and ventricular spaces. This process contrasts the findings in the brain parenchyma, which was devoid of TUNEL-positive cells (Fig. 4).
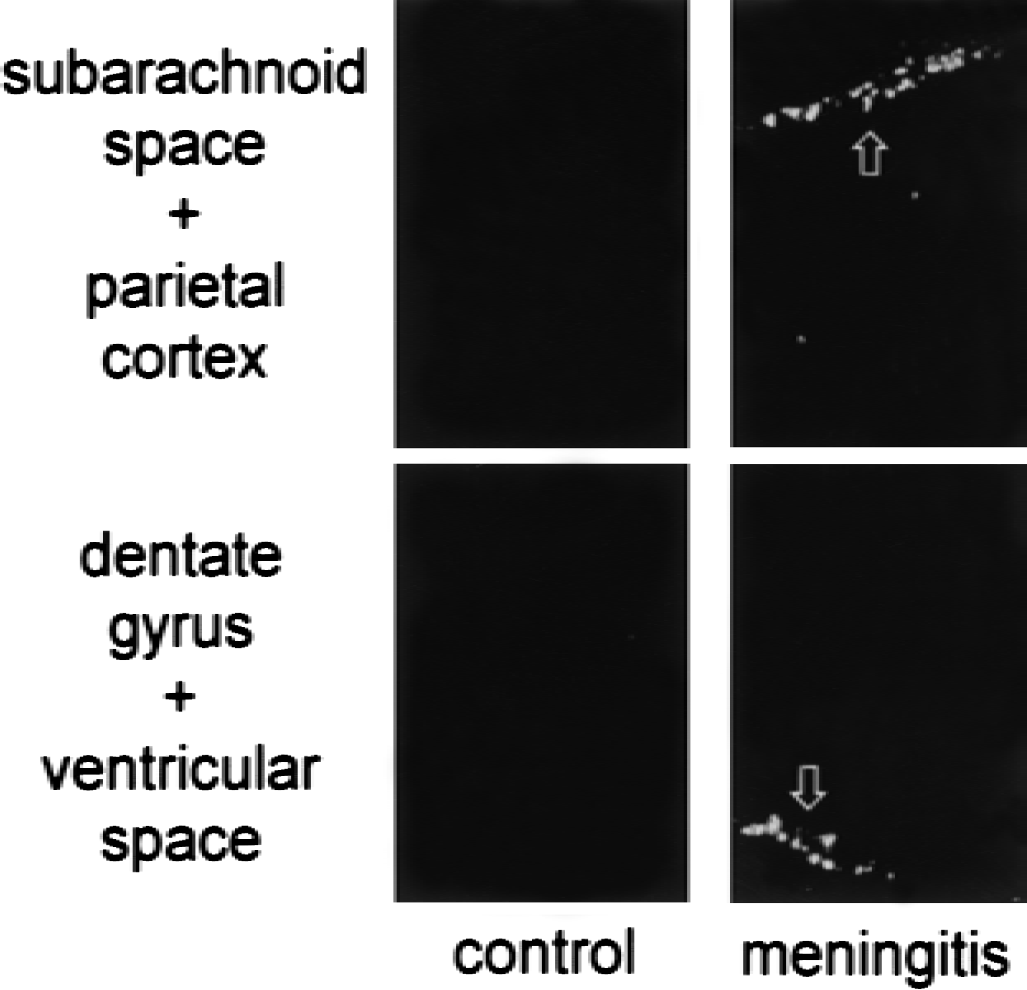
TUNEL staining of brain sections from wild-type mice injected either with phosphate-buffered saline or pneumococci (representative examples). The TUNEL-positive cells were detected within the inflammatory infiltrates in the subarachnoid and ventricular spaces, but not within the brain parenchyma.
Effect of 3-aminobenzamide on pneumococci-induced endothelial cell injury in vitro
Because 3-aminobenzamide treatment was protective against the development of meningitis-associated endothelial dysfunction, additional experiments were performed to assess the role of PARP activation in pneumococci-induced injury of brain endothelial cells. When GP8.3 endothelial cells were exposed to heat-killed pneumococci, no or only mild cytotoxicity could be observed. However, when endothelial cells were cocultured with macrophages, pneumococci induced a significant reduction in endothelial cell viability, as assessed by mitochondrial MTT reduction (Fig 5A) and counting of nuclei stained with propidium iodide (55 ± 10 vs. 98 ± 13 cells/microscopic field in controls, P < 0.05;Fig. 5C). Pneumococci-induced cytotoxicity was paralleled by a marked reduction in cellular NAD+ content (Fig. 5B) and a positive staining of GP8.3 endothelial cells for the PARP reaction product poly(ADP-ribose) (Fig. 5C). In addition, exposure to pneumococci resulted in superoxide dismutase-inhibitable nitroblue tetrazolium staining reduction, which indicates superoxide production (data not shown), in increased nitrite/nitrate concentrations in the cell culture supernatant (30.6 ± 7.1 vs. 12.4 ± 5. 9 μmol/L in controls, P < 0.05), and a prominent immunostaining for L-nitrotyrosine (Fig. 5C). These latter findings support the concept of a central role of oxidants in both the so-called PARP cell-suicide hypothesis (Szabo, 1998) and pneumococci-induced cell injury (Koedel and Pfister, 1999b).
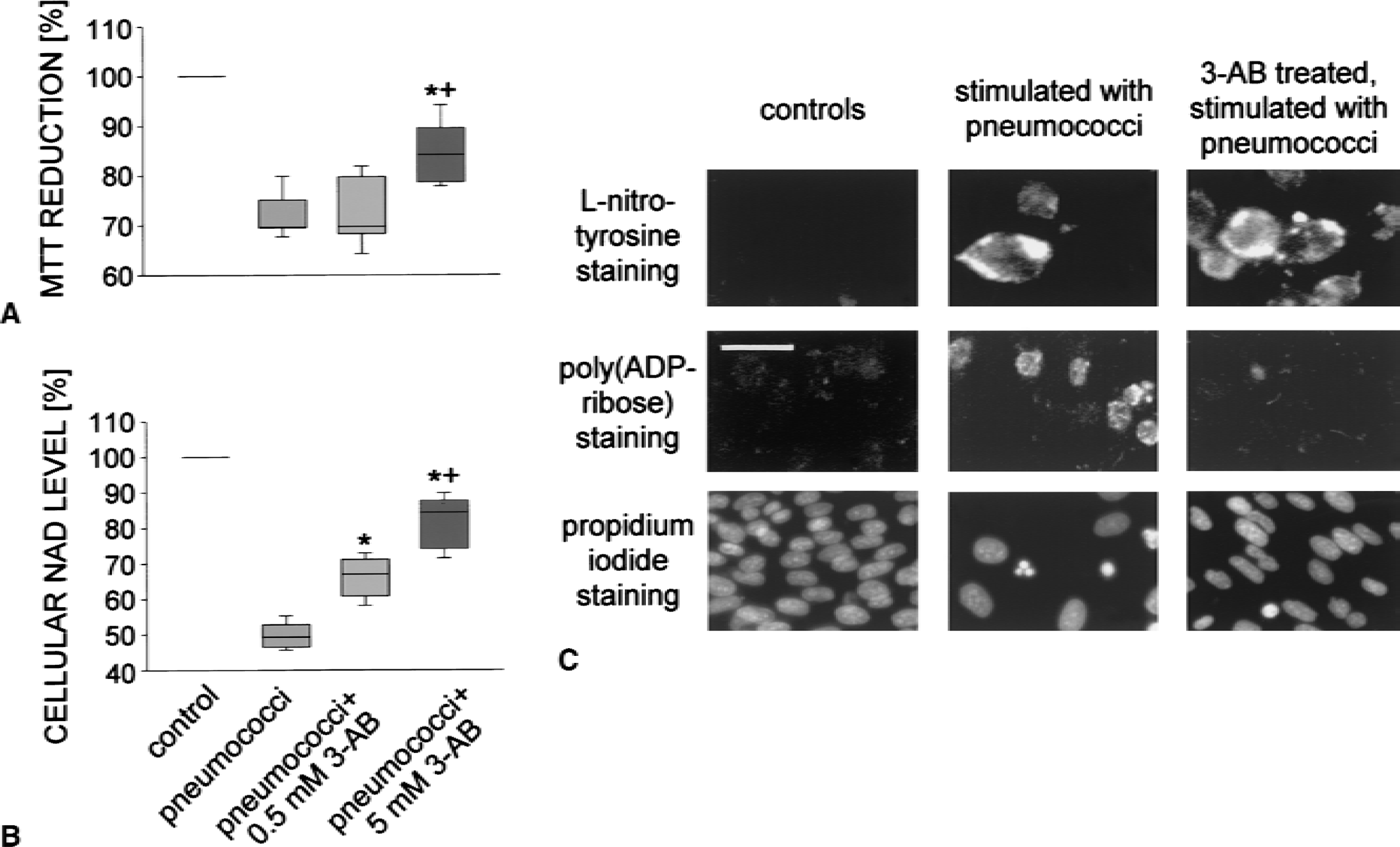
When rat brain-derived endothelial cells were cocultured with RAW 264.7 macrophages, exposure to pneumococci led to a significant cellular loss, as assessed by the colorimetric MTT assay
When testing the effect of 3-aminobenzamide on pneumococci-induced cytotoxicity in our coculture system, an approximately 50% reduction in cell death was found (Fig. 5A; 77 ± 9 cells/microscopic filed in the coculture system exposed to pneumococci and treated with 5 mmol/L 3-aminobenzamide, P < 0.05). This effect was associated with the near absence of ADP-ribose polymer formation in GP8.3 endothelial cells and a significant increase in cellular NAD+ content (Fig. 5B). However, 3-aminobenzamide did not alter pneumococci-induced production of superoxide, nitric oxide/nitrite (30.3 ± 3.3 and 26.8 ± 7.4 μmol/L in supernatants of pneumococci-stimulated cocultures treated with 0.5 and 5 mmol/L 3-aminobenzamide, respectively;P not significant), and peroxynitrite (Fig. 5C), arguing against the role of a radical-scavenging side effect in the protective activity of 3-aminobenzamide.
DISCUSSION
Our findings show that (1) PARP is activated during experimental pneumococcal meningitis and in brain-derived endothelial cells exposed to S pneumoniae; (2) genetic disruption of the PARP1 gene provides profound protection against the development of meningitis-associated CNS complications, resulting in an improved clinical status of infected animals; (3) the alleviation of CNS complications is associated with an attenuated inflammatory host response; and (4) pharmacologic PARP inhibition also improves the clinical course of pneumococcal meningitis. These results provide compelling evidence for a crucial role of PARP activation in brain pathophysiology during bacterial meningitis, and suggest that pharmacologic inhibition of PARP may be a promising approach for adjuvant therapy of this inflammatory disease.
During the past decade, evidence accumulated that free radicals and oxidants are critical mediators in the pathogenesis of bacterial meningitis (Koedel and Pfister, 1999a). Free radicals and oxidants may contribute to brain injury during bacterial meningitis through a variety of independent mechanisms. One of these pathways involves the induction of DNA strand breakage and subsequent PARP activation, thus initiating an energy-consuming intracellular cycle that can ultimately result in cellular energy depletion and death (Ha and Snyder, 2000). This mechanism was shown to act as an effector of oxidant-induced tissue damage in septic shock (Szabo et al., 1997a; Kuhnle et al., 1999), ischemia-reperfusion injury (Endres et al., 1997; Pieper et al., 2000b), and streptozotocin-induced diabetes (Burkart et al., 1999; Pieper et al., 1999). However, we observed PARP activation in our animal models of pneumococcal meningitis, as evidenced by ADP-ribose polymer formation in the absence of noteworthy DNA damage. When using the TUNEL method to detect DNA strand breaks, only a few positive cells in the inflammatory infiltrate but not in brain parenchyma were detected. Thus, DNA strand breakage may provide a PARP-activation signal in CNS-infiltrating leukocytes, but not in the brain endothelium or in associated brain astrocytes that provide the functional unit to maintain BBB integrity. Therefore, mechanisms other than DNA strand breakage may be involved in PARP activation in bacterial meningitis. This suggestion is supported by a recent report that showed a fast activation of PARP in primary rat brain cortical neurons that was evoked by1,4,5 triphosphate-Ca2+ mobilization not involving DNA breaks. The PARP was identified as a novel downstream target for phospholipase C (Homburg et al., 2000). The suggestion that PARP activation is not determined solely by DNA damage is also supported by the observation of incongruenties in localization of basal in vivo poly(ADP ribosyl)ation and DNA strand breaks in various tissues including kidney, stomach, and pancreas (Pieper et al., 2000a).
New enzymes other than PARP1 with the capability to catalyze the formation of ADP-ribose polymers were identified, including PARP 2, V PARP, and tankyrase. Although limited data are available regarding the overall contribution of PARP-like enzymes to poly(ADP ribosyl)ation reactions in vivo, it appears from analysis of PARP1-deficient mice that poly(ADP ribosyl)ation in the brain almost exclusively reflects PARP1 (Pieper et al., 2000a). Consistent with this observation, we demonstrated here that no or only low levels of poly(ADP ribosyl)ated proteins were detectable by immunoblot analysis in brain homogenates from infected PARP1-deficient mice. Thus, PARP1 presumably mediates ADP-ribose polymer formation during pneumococcal meningitis.
In the present study, targeted disruption of the PARP1 gene was associated with an improved clinical status of infected mice. This outcome benefit was related to an alleviation of meningitis-associated CNS complications, which are considered to be major predictive factors of an unfavorable clinical outcome of bacterial meningitis (Pfister et al., 1993). Thus, PARP1-deficient mice had significantly lower ICP values than infected wild-type mice. Studies in animal models identified vasogenic brain edema, provoked by a breakdown of the BBB, as the predominant cause of the increase in ICP in bacterial meningitis (Quagliarello et al., 1986). According to this concept, the attenuated increase in ICP observed in infected PARP1-deficient mice was associated with reduced brain Evans blue concentrations, indicating a lesser degree of BBB disruption (compared with that of infected wild-type mice). This finding is consistent with previous results that show a significant attenuation of hemorrhagic shock- and reperfusion injury-induced intestinal epithelial hyperpermeability by depletion of the PARP1 gene and pharmacologic PARP inhibition using 3-aminobenzamide (Liaudet et al., 2000; Cuzzocrea et al., 1997). As seen in PARP1 knockout mice, a marked reduction of the meningitis-associated increase in BBB permeability was also observed when infected rats were treated with 3-aminobenzamide. In addition, we demonstrated that inhibition of PARP by 3-aminobenzamide was protective against the development of endothelial dysfunction in bacterial meningitis, as evidenced by the maintenance of the cerebrovascular reactivity to hypertension and hypercapnia. This observation was further strengthened by Western blot analysis of brain PECAM-1 and occludin expression, which suggested a protective effect of PARP1 deficiency against the meningitis-associated loss of PECAM-1 expression and occludin proteolysis—two indicators for vascular injury. Our in vivo observation complies with recent ex vivo studies that showed an improved endothelium-dependent relaxant response to acetylcholine in aortic rings isolated from rats subjected either to splanchnic artery occlusion and reperfusion or to endotoxic shock when treated with the PARP inhibitor 3-aminobenzamide (Cuzzocrea et al., 1997; Szabo et al., 1997a). In these experimental models, the development of endothelial dysfunction was attributed to the overproduction of oxidants that induce PARP activation, thus provoking depletion of cellular energy stores and, finally, endothelial cell death. This concept also seems valid for meningitis-associated endothelial dysfunction. Thus, our cell culture experiments showed that in the presence of macrophage exposure of brain-derived endothelial cells to S pneumoniae, oxidants were produced, ADP-ribose polymer was formed, cellular NAD+ was depleted, and in cell death occurred. The PARP inhibition by 3-aminobenzamide interfered with poly(ADP ribosyl)ation and NAD+, but not with ROS and RNI production.
During the past decade, a substantial body of work has implicated that CNS complications occur as a consequence of an exaggerated inflammatory host response to bacterial products. We demonstrated here that the improved clinical status and the reduction in CNS complication by genetic PARP1 depletion and pharmacologic PARP inhibition was accompanied by an attenuated leukocyte infiltration into the CNS. Accordingly, outside the CNS, inhibition of PARP reduced leukocyte recruitment in carrageenan- and zymosan-triggered models of local inflammation (Szabo et al., 1997b), arthritis (Szabo et al., 1998), peritonitis (Cuzzocrea et al., 1999), and after ischemia-reperfusion injury (Chatterjee et al., 2000). Furthermore, leukocyte accumulation in the lung after hemorrhagic shock (Liaudet et al., 2000) and into the jeopardized tissue after myocardial reperfusion injury (Zingarelli et al., 1998) was markedly lessened in PARP1-deficient mice. The mechanisms by which the absence of functional PARP or PARP inhibition protects against leukocyte infiltration is thought to be through decreased expression of endothelial cell adhesion molecules and proinflammatory cytokines (Szabo et al., 1997b; Yang et al., 2000; Oliver et al., 1999). According to this concept, we observed that the suppression of leukocyte infiltration was associated with reduced brain concentrations of interleukin-1β, −6, and TNF-α in infected PARP1-deficient mice, and with reduced CSF interleukin-6 levels in infected rats treated with 3-aminobenzamide. Similarly, Jijon et al. (2000) reported that pharmacologic PARP inhibition resulted in a significant decrease of TNF-α and interferon-γ secretion in experimental colitis. Furthermore, an almost complete abrogation of the accumulation of TNF-α and interferon-γ in the serum was observed in PARP1-deficient mice subjected to endotoxic shock (Oliver et al., 1999). Recent studies showed a functional association between PARP1 and nuclear factor (NF)-κB, a key regulatory molecule in inflammation (Hassa and Hottiger, 1999; Oliver et al., 1999).
Through this association, PARP1 may regulate NF-κB–dependent transcription and, thus, the synthesis of inflammatory mediators (Oliver et al., 1999). Previous data from our group showed that NF-κB activation plays a fundamental role in the induction, aggravation, and perpetuation of the inflammatory host response during pneumococcal meningitis (Koedel et al., 2000). Because the genes for interleukin-1β, −6, and TNF-α are under the transcriptional control of NF-κB (Barnes and Karin, 1997), it is conceivable that the suppression of meningitis-induced interleukin-1β, −6, and TNF-α synthesis in PARP1-deficient mice is due to the decreased ability of this genotype to transactivate through NF-κB.
In summary, PARP1 deficiency and PARP inhibition provided a profound advantage of the host to control the BBB integrity and the inflammatory process in bacterial meningitis. Meningitis-associated CNS complications and the symptoms in diseased animals are thereby mitigated. Thus, pharmacologic PARP inhibition holds promise for the adjunctive therapy of bacterial meningitis.
Footnotes
Acknowledgments:
The authors thank Dr. Beatrice Grabein and Ms. Maindok from the Max-von-Pettenkofer Institute for Hygiene and Microbiology for the pneumococcal preparation, and Ms. J. Benson for copyediting the manuscript.