
Abstract
Growth factors promote cell growth and survival and protect the brain from developing injury after ischemia. In this article, the authors examined whether transforming growth factor-α (TGF-α) was protective in transient focal ischemia and whether alteration of cerebral circulation was involved. Rats received intraventricular TGF-α (50 ng, either split into 2 doses given 30 minutes before and 30 minutes after middle cerebral artery occlusion (MCAO), or 1 dose given 30 minutes after MCAO) or vehicle. Rats were subjected to 1-hour intraluminal MCAO and cerebral blood flow was recorded continuously by laser–Doppler flowmetry. Infarct volume was measured 1 and 4 days later. The effects of TGF-α on arterial tone were assessed in isolated rabbit basilar and common carotid arteries. Transforming growth factor-α before and after ischemia reduced infarct volume by 70% at 1 day and 50% at 4 days. Transforming growth factor-α given only after ischemia also did reduce infarct volume by 70% at 1 day and 80% at 4 days. The protective effect was more marked in cortex than in striatum. Transforming growth factor-α did not change cortical microvascular perfusion and did not modify arterial passive tone nor agonist-induced active tone. It can be concluded that TGF-α reduces infarct volume, even when the factor is exclusively administered at reperfusion, and that this effect is not mediated by changes in microvascular perfusion or cerebral arteries. It is therefore suggested that TGF-α has a protective effect against neuronal cell death after transient focal ischemia.
Transforming growth factor-α (TGF-α) is a 50-amino acid polypeptide that belongs to a family of mitogenic growth factors binding to the epidermal growth factor receptor (EGFR) (Massagué, 1983; Marquardt et al., 1984). Transforming growth factor-α and EGFR are expressed in the developing (Ma et al., 1992; Lazar and Blum, 1992; Ferrer et al., 1995; Kornblum et al., 1997, 1999, 2000) and adult brain, (Ferrer et al., 1995, 1996; Kornblum et al., 1995, 1997; Seroogy et al., 1991) where TGF-α is the most abundant ligand for EGFR (Lazar and Blum, 1992; Kaser et al., 1992; Seroogy et al., 1993). In the adult brain, TGF-α is expressed in neurons and glial cells of different regions, including the olfactory bulbs (Wilcox and Derynck, 1988), hypothalamus (Ma et al., 1994), cortex, and striatum (Kaser et al., 1992; Ferrer et al., 1995). Epidermal growth factor receptor is also widely expressed throughout the brain, and in adult mammals it is mainly expressed in neurons (Seroogy et al., 1993; 1994; Kornblum et al., 1995; Ferrer et al., 1996; Planas et al., 1998) and, to a lesser extent, astrocytes (Ma et al., 1994). The role of TGF-α and EGFR in neurons is intriguing, as they are usually involved in stimulating cell growth and proliferation (Derynck et al., 1987; Getchell et al., 2000). Indeed, TGF-α and EGFR are overexpressed in a variety of tumoral cells, which are subjected to autocrine growth stimulation (Derynck et al., 1987). However, the fact that EGFR is abundant in postmitotic cells suggests that activation of this receptor has neuronal effects other than promoting cell proliferation. Several lines of evidence support the view that TGF-α performs trophic actions on neurons. For instance, trophic effects have been shown in vitro in mesencephalic dopaminergic neurons (Alexi and Hefti, 1993; Engele and Schilling, 1996; Krieglstein and Unsicker, 1997), cortical neurons (Kornblum et al., 1999), and spinal motor neurons (Hanson et al., 1998). Furthermore, EGFR-null mice suffer neuronal degeneration in the olfactory bulb, piriform cortex, neocortex, and thalamus between postnatal days 5 and 8 (Kornblum et al., 1998).
Transforming growth factor-α acting through EGFR exerts a protective effect in permanent middle cerebral artery occlusion (MCAO) in rats (Justicia and Planas, 1999). In addition, administration of EGF protects the brain against global cerebral ischemia in the gerbil (Peng et al., 1998). Also, EGF increases survival of mesencephalic dopaminergic neurons subjected to an excitotoxic insult (Casper and Blum, 1995) and oxidative stress (Kinoshita et al., 1990; Maiese et al., 1993; Peng et al., 1998; Yamada et al., 1995). In this article, the authors examined whether TGF-α has protective effects in vivo against transient focal cerebral ischemia. The authors also explored possible effects of TGF-α on cerebral perfusion and vasoconstriction, which could mediate brain protection because TGF-α modifies blood flow and vasoconstriction in other vascular beds (Gan et al., 1987; Tepperman and Soper, 1994).
MATERIALS AND METHODS
Animals were handled in compliance with the Spanish legislation on “Protection of Animals used for Experimental and other Scientific Purposes,” and in accordance with the Directives of the European Community on this subject.
Focal cerebral ischemia in rats
Male Sprague–Dawley rats (body weight = 280 to 320 g; n = 76) obtained from Iffa-Credo (Lyon, France) were kept under a 12-hour light–dark cycle and were allowed free access to food and water. The day before ischemia, anesthesia was induced with 4% halothane (Zeneca Farms S.A., Porriño, Pontevedra, Spain) in a mixture of 70% N2O and 30% O2 through a face mask. Rats were placed in the prone position on a stereotaxic frame (Kopf Instruments, Tujunga, CA, U.S.A.) and anesthesia was maintained with 1.5% to 2% halothane. After a cranial midline skin incision, a stainless steel cannulae was implanted in the right lateral ventricle through a burr hole at the following coordinates (Paxinos and Watson, 1986): 0.3 mm posterior, 1.2 mm lateral, 3.2 mm ventral to bregma. In a group of rats, a truncated 21G needle was implanted in a burr hole to measure cortical perfusion. The needle was placed at the following coordinates: 2.0 mm posterior, 3.5 mm right to bregma. Both the cannulae and truncated needle were fixed to the skull with two miniature screws and dental cement. The wound was sutured and the animal was allowed to recover for 24 hours.
Focal cerebral ischemia was produced by transient intraluminal occlusion of the MCA, as previously reported (Soriano et al., 1997). Briefly, rats were anesthetized with halothane as above. After tracheal intubation for controlled ventilation, anesthesia was maintained with 1% to 1.5% halothane. The flexible plastic tip (0.5 mm in diameter, PF319; Perimed, Stockholm, Sweden) of a master laser–Doppler probe (PF418; Perimed) was introduced through the cranial 21G needle until the tip of the probe touched the brain surface, thus measuring the fronto-parietal cortical perfusion (CP) in the territory of the MCA with a laser–Doppler flowmeter (PF4001 Master, Perimed). The left femoral artery was cannulated to monitor mean arterial blood pressure (MABP), and body temperature was maintained at 37.5°C with a heating blanket connected to a rectal probe. The right common, external, and internal carotid arteries were identified through a ventral cervical midline incision. After electrocoagulation and cutting of its branches, the external carotid artery was ligated and cut 2 mm distal to the bifurcation. The pterygopalatine artery was ligated, and microclips were placed across both the common and internal carotid arteries. A 2.6-cm length of 3–0 monofilament nylon suture heat-blunted at the tip was introduced into the external carotid artery through a puncture. After tightening a silk suture around the external carotid artery and the intraluminal nylon suture to prevent bleeding, the microclips were removed. Then the nylon suture was gently advanced into the internal carotid artery and circle of Willis until the origin of the MCA was reached, at approximately 22 mm from the carotid bifurcation. This was indicated by slight resistance to nylon suture advance and sudden decrease in CP as measured by the laser–Doppler probe. Both common carotid arteries were clamped. The clip on the left common carotid artery was released after 50 minutes. Ten minutes later, the filament was gently removed and the clip on the right common carotid artery was released. After surgery, rats were allowed to recover spontaneous breathing and were kept in their cages with free access to food and water.
After 2 different experimental dosing regimens, 50 ng TGF-α was administered in the lateral ventricle. In the first study, the dose of TGF-α was split in 2 administrations (25 ng in 5 μL of vehicle each, infused over a 15-minute period) 30 minutes before and 30 minutes after MCAO (n = 15), as similar dosing reduces infarct volume after permanent focal ischemia (Justicia and Planas, 1999). Control subjects received the same volume of vehicle (phosphate-buffered saline [PBS]) twice (n = 17), and infarct volume was evaluated at 1 or 4 days postischemia. In the second study, the pretreatment was omitted and 50 ng TGF-α (n = 13) or PBS (n = 10) was given as 1 single injection 30 minutes after ischemia, and infarct volume was measured 1 or 4 days later. Rats were killed while under halothane anesthesia, and the brains were rapidly frozen and kept at −20°C. Twenty-micrometer coronal brain sections were obtained with a cryostat. Sections were collected every 1 mm and were stained with cresyl violet. The infarcted area was identified macroscopically by pallor and microscopically by structural disorganization and histologic signs of neuronal and tissular damage, as defined by Garcia and coworkers (1993). Infarct area was measured in each stained section with an image analyzing system (AIM Image Research, Canada). Infarct volume was calculated by integrating data from all sections. Measurements were performed by one of the authors while being blinded to the treatment. Statistical analyzes were performed by means of the two-tailed, unpaired, Student's t-test to compare results from TGF-α–infused animals and the corresponding controls regarding hemodynamic data (CP and MABP) and infarct volume data. Infarct volume was determined in control and TGF-α groups either at 1 or 4 days postischemia. Then the effects of 2 factors, treatment (TGF-α vs. controls) and postischemic time (1 and 4 days), on infarct size were tested by two-way analysis of variance. This was followed by the Fisher's least significant differences test as a post hoc analysis for group comparisons. P < 0.05 was considered significant.
Isolated rabbit arteries
Male New Zealand White rabbits (Technology Transferring Center, Polytechnic University of Valencia, Valencia, Spain) (body weight = 2.5 to 3 kg, n = 5) were killed by injection of 25 mg/kg sodium thiopental (Braun Medical Rubí, Barcelona, Spain) and 1.5 mL of 10 mmol/L KCl solution through the ear vein. The whole brain, including the brainstem, was removed, and the basilar artery was dissected free. A midline throat incision provided access to common carotid arteries, one of which was dissected free. Four 3-mm-long segments of basilar artery and four 4-mm-long segments of carotid artery were obtained. For recording of isometric tension, segments were mounted in an organ bath by using tungsten wires (89 μm in diameter) for basilar arteries and stainless steel wires (207 μm in diameter) for carotid arteries. Two pins were introduced through the arterial lumen; one was fixed to a stationary support and the other was connected to a strain gauge (Universal Transducing Cell UC3; Gould Statham, Oxnard, CA, U.S.A.). Isometric tension was amplified (Hewlett-Packard 8805D; San Diego, CA, U.S.A.) and recorded (Omniscribe D5237–5; Houston Instrument, Gistel, Belgium). Each organ bath contained 5 mL Ringer-Locke solution at 37°C and was bubbled with a 95% O2 and 5% CO2 mixture to give a pH of 7.3 to 7.4. Previously determined optimal resting tensions of 0.5 g and 2 g were applied to basilar and carotid arterial segments, respectively, which were then allowed to equilibrate for 30 to 60 minutes before starting the experiments.
The contractile capacity of each arterial segment was assessed by exposure to 50 mmol/L KCl Ringer-Lock solution. Basilar arteries contracting less than 0.5 g and carotid arteries contracting less than 2 g were discarded. After washing out the KCl several times until resting tension was recovered, the effects of TGF-α on passive tone and agonist-mediated contraction were tested. Basilar arteries were contracted 3 times with uridine 5′-triphosphate (UTP, 0.1 mmol/L). Between the first and second contraction arteries were preincubated (10 minutes) with TGF-α (5 nmol/L), which was washed out between the second and third contraction to test the reversibility of possible effects of TGF-α. As a control, basilar arteries were run in parallel without preincubation with TGF-α. For comparison, a similar protocol was performed in carotid arteries, except for the agonist, which was 5-hydroxytryptamine (5-HT, 10 μmol/L). The second and third responses to agonists were expressed as percentage of the first contraction in the same arterial segment. In another set of experiments, basilar arteries were precontracted with UTP (0.1 mmol/L), and the effects of cumulative concentrations of TGF-α (1, 5, and 10 nmol/L) on the maintained active tone were assayed. After TGF-α, cumulative concentrations of acetylcholine (Ach, 0.1, 1, and 10 μmol/L) were added to check on relaxant ability and functional integrity of the endothelium in the arterial segments. Student's t-test was used for statistical analysis of differences between control and TGF-α-treated arteries. P < 0.05 was considered significant.
Drugs and solutions
Human recombinant TGF-α (Oncogene Science Calbiochem-Novabiochem International, Nottingham, U.S.A.) was prepared following the instructions of the manufacturer. The lyophilized product was first dissolved in 10 mmol/L acetic acid up to a concentration of 1 μg/μL and stored at −20°C. The working solution was prepared by diluting this stock solution with 0.1 mol/L PBS to the concentration of 5 ng/μL, yielding pH 7 to 7.5. UTP, 5-HT, and Ach were dissolved and diluted in saline solution (0.9% NaCl). The Ringer-Locke solution had the following composition (mmol/L): NaCl 120, KCl 5.4, CaCl2 2.2, MgCl2 1.0, NaHCO3 25, and glucose 5.6. In KCl-depolarizing solution, NaCl was replaced by an equimolar amount of KCl. All products and reagents, unless otherwise stated, were from Sigma-Aldrich Química (Spain).
RESULTS
In vivo neuroprotective effects of TGF-α
Body temperature and MABP were continuously recorded during surgery. Mean body temperature (mean ± SEM) was 37.5°C ± 0.1°C in both groups—control subjects and rats receiving TGF-α. Mean arterial blood pressure (mean ± SEM) was 104.0 ± 1.9 in control subjects and 100.4 ± 2.5 mm Hg in rats treated with TGF-α. No significant differences were found between the groups. Blood pH, Pco2, Po2, and glucose were not significantly different before and after TGF-α treatment (mean ± SEM before TGF-α: pH = 7.49 ± 0.01, Pco2 = 32 ± 3 mm Hg, Po2 = 228 ± 10 mm Hg, and glucose = 85 ± 10 mg/dL; after TGF-α: pH = 7.42 ± 0.03, Pco2 = 39 ± 4 mm Hg, Po2 = 188 ± 17 mm Hg, and glucose = 81 ± 11 mg/dL). Mortality rates and the number of rats per group not developing infarct after ischemia are shown in Table 1. Infarct volume was measured at 1 (n = 7 control subjects, n = 7 TGF-α) and 4 days (n = 10 control subjects, n = 8 TGF-α) after MCAO with treatment before and after ischemia. Transforming growth factor-α protected the brain in focal ischemia by reducing infarct volume (Fig. 1A and 1), as assessed with two-way analysis of variance by treatment and time (Ftreatment (1, 27) = 12.8, P < 0.001), at 1 (P < 0.05) and 4 days postischemia (P < 0.05). Infarct volumes (mean ± SEM; in mm3) were 135 ± 31 (n = 7) and 39 ± 9 (n = 7) at 1 day, and 195 ± 21 (n = 10) and 93 ± 39 (n = 8) at 4 days for control and TGF-α groups. The effect of time evaluation of infarct volume was not significant.
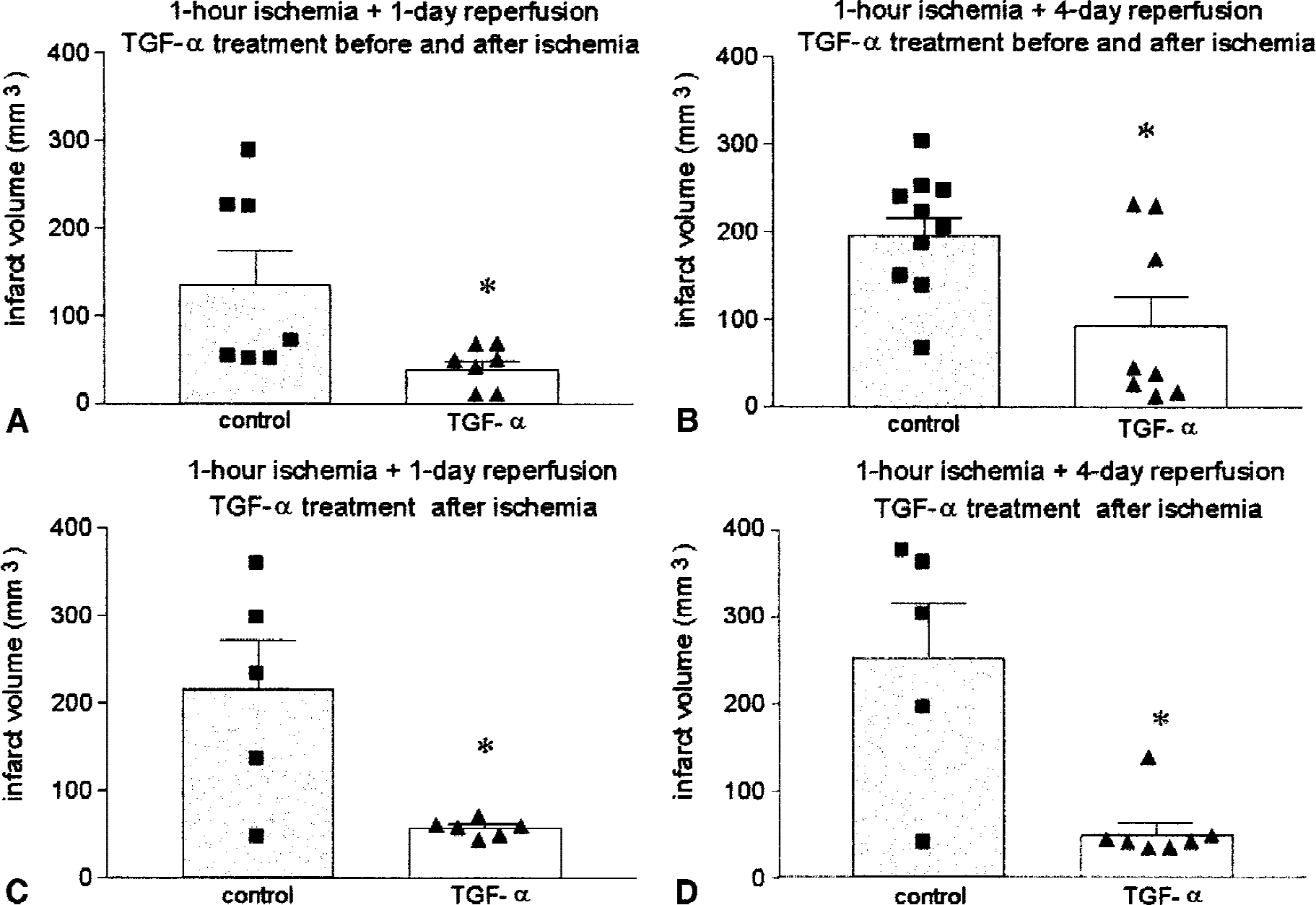
Infarct volume (mm3) after middle cerebral artery occlusion (MCAO) in controls (vehicle administered) and rats receiving 50 ng TGF-α either in 2 administrations
Number of rats per group, mortality rate, and rats without brain infarct
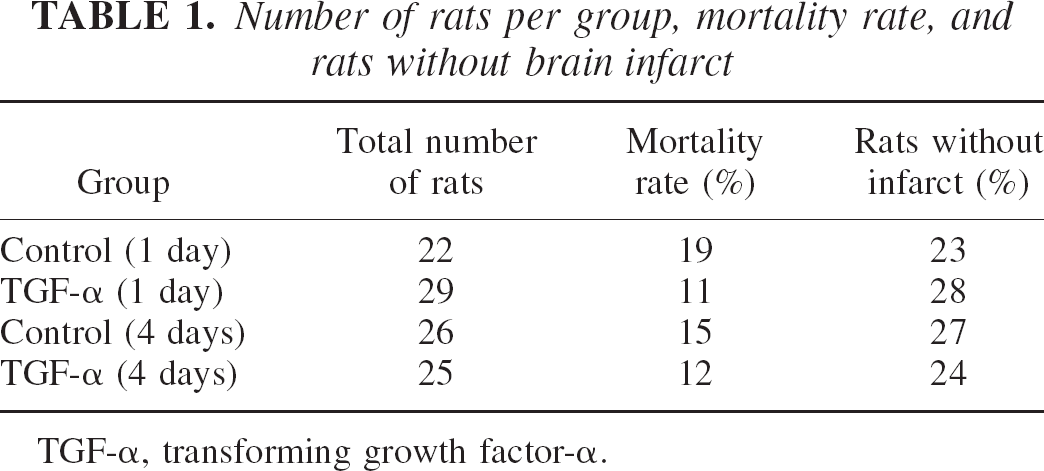
TGF-α, transforming growth factor-α.
Transforming growth factor-α given as 1 single dose at 30 minutes after ischemia, without preischemic treatment, also reduced infarct volumes at 1 and 4 days after MCAO (Fig. 1C and 1). Measures of infarct volume (mean ± SEM; in mm3) were 216 ± 56 (n = 5) and 57 ± 4 (n = 6) for control and TGF-α groups, respectively, at 1 day, and 250 ± 63 (n = 5) and 48 ± 14 (n = 7) at 4 days. This single treatment after ischemia did also effectively reduce infarct volume at 1 day (P < 0.05) and 4 days (P < 0.005). Two-way analysis of variance by treatment and time showed a global significant effect of postischemic TGF-α treatment (Ftreatment (1, 18) = 19.5, P < 0.001), but not of the time of study (1 or 4 days). Moreover, a clear difference in the group variances was observed (Fig. 1C and 1), which turned out to be highly significant (F4, 5 =169, P < 0.0001 at 1 day and F4, 6 = 14, P < 0.005 at 4 days). Subsequent data analysis using the unpaired t-test with Welch's correction, which does not assume equal variances, confirmed that TGF-α significantly reduced mean infarct volume (P < 0.05). Decrease of group variability was mainly attributed to the strong reduction of cortical infarction in the rats treated with TGF-α (Fig. 2). Indeed, in the group of rats receiving pretreatment with TGF-α, reduction of cortical infarct size was seen at 1 (Fig. 2A) and 4 days (Fig. 2B) (P < 0.05). After administration of TGF-α after ischemia, cortical infarct volume also was reduced at 1 (P < 0.05) and 4 days (P < 0.005) (Fig. 2C and 2). Also, striatal infarct volume showed a small decrease after TGF-α, which was significant (P < 0.005) in the group that received TGF-α after ischemia and that was studied at 4 days (Fig. 2D).
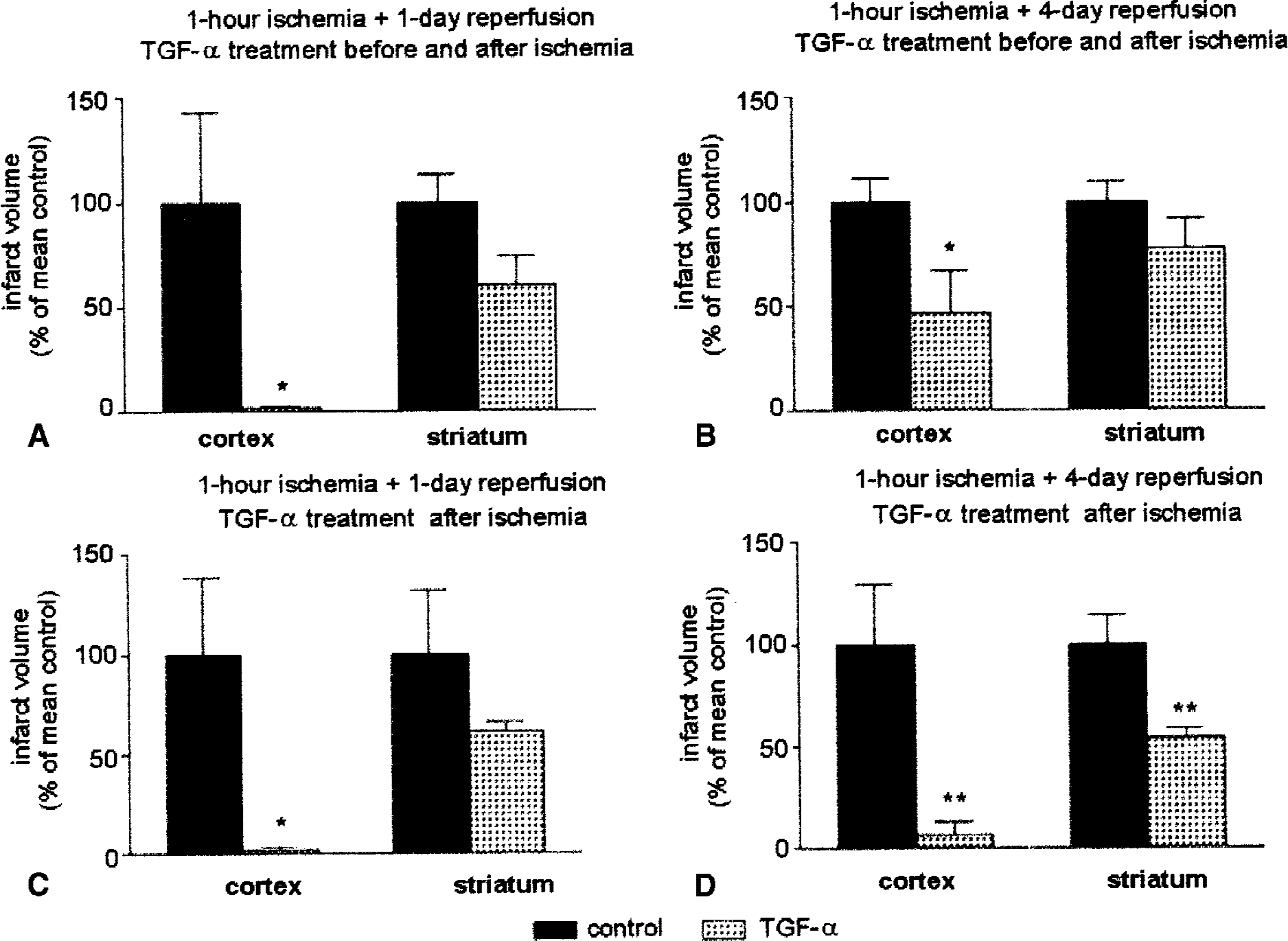
Effect of 50 ng transforming growth factor-α (TGF-α) on infarct volume in cortex and striatum after treatment before and after ischemia
Hemodynamic effects of TGF-α
Cortical perfusion was assessed by laser–Doppler flowmetry in a group of rats given TGF-α (n = 6) and vehicle (PBS, n = 6). Basal values, expressed in arbitrary perfusion units (PU), were not significantly different in TGF-α and PBS-treated rats (82 ± 24 PU, and 137 ± 43 PU, respectively;P = 0.29). Preischemic infusion of TGF-α (25 ng/15 min) or its vehicle did not affect baseline or MABP. Ischemia decreased CP to less than 30% of baseline, without changes in MABP. Administration of TGF-α before ischemia did not significantly alter CP values during the 1-hour episode of transient ischemia or at reperfusion upon filament withdrawal. During the ischemic insult, CP was 28% ± 10% of baseline in TGF-α–treated rats, and 27% ± 1% in PBS-treated rats. At reperfusion, CP recovered to 101% ± 26% of baseline in TGF-α–treated rats, and to 115% ± 3% in PBS-treated rats. Likewise, the second infusion of TGF-α (25 ng/15 min) 30 minutes after the transient episode of ischemia did not change CP or MABP at reperfusion.
Effects of TGF-α in isolated arteries
Exposure to 50 mmol/L KCl depolarizing medium induced arterial contractions, which amounted to 1085 ± 435 mg in basilar artery, and 4688 ± 1638 mg in carotid artery. Control contractions induced by UTP (0.1 mmol/L) in basilar artery were 88% ± 39% of the response to KCl, and those induced by 5-HT (10 μmol/L) in carotid artery were 127% ± 11% of the response to KCl. On its own, TGF-α (5 nmol/L) did not modify the passive arterial tone of basilar or carotid arteries. Moreover, as shown in Table 2, preincubation with TGF-α (5 nmol/L) did not modify the contractile effects of UTP in basilar artery. Contractile effects of 5-HT were not significantly modified in carotid arteries by TGF-α preincubation. After washing out TGF-α, responses to UTP in basilar arteries, and to 5-HT in carotid arteries were the same in treated and untreated arterial segments. Cumulative concentrations of TGF-α (1, 5, and 10 nmol/L) did not significantly modify the active tone induced in basilar artery by precontraction with UTP (0.1 mmol/L). Relaxant ability and functional integrity of the endothelium were shown by significant (P < 0.01) reductions in active tone induced by Ach (1 and 10 μmol/L) (Fig. 3).
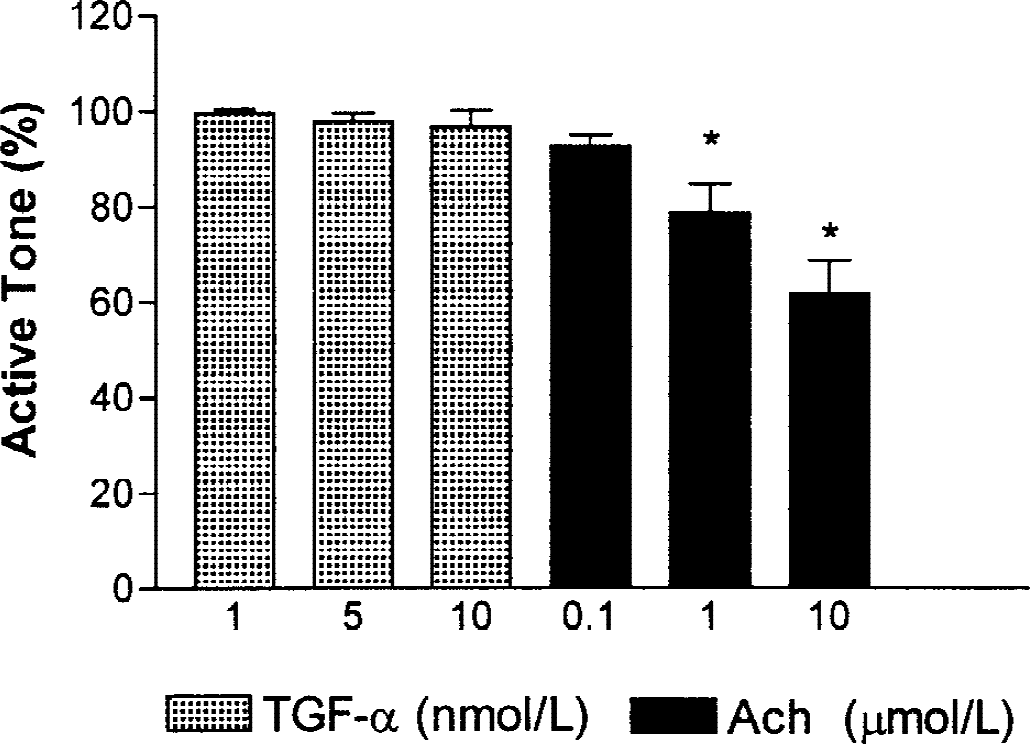
Effects of cumulative concentrations of transforming growth factor-α (TGF-α; 1, 5, and 10 nmol/L, n = 4) and acetylcholine (Ach; 0.1, 1, and 10 μmol/L, n = 10) on active tone of rabbit isolated artery precontracted with uridine 5′-triphosphate (0.1 mmol/L). Values (mean ± SEM) are expressed as percentage of the initial active tone. * P < 0.01.
Effects of transforming growth factor-α oncontraction of isolated arteries

Data are expressed as percentage of the first agonist-induced contraction in the same arterial segment before TGF-α incubation. Values are mean ± SEM from n arterial segments. UTP, uridine 5′-triphosphate; 5-HT, 5-hydroxytryptamine; TGF-α, transforming growth factor-α.
DISCUSSION
The current data show that TGF-α reduces infarct volume in transient MCAO in the rat. The results suggest that TGF-α has a protective effect against transient focal cerebral ischemia, which agrees with the authors' previous study showing a protective effect in permanent focal cerebral ischemia (Justicia and Planas, 1999) and protection against excitotoxic striatal lesion (Alexi et al., 1997). Although early reperfusion of the ischemic tissue (that is, thrombolysis) is a therapeutic strategy in cerebral ischemia (Overgaard, 1994), reperfusion also has some damaging effects, which differentiate the pathophysiology of irreversible and reversible cerebral ischemia (McAuley, 1995). The authors now show that the protective effect of TGF-α is not overcome by reperfusion injury.
In this study, the authors found that acute treatment with TGF-α before and after ischemia reduced (70%) infarct volume at 1-day recirculation. Then, an additional group of rats was studied at 4 days postischemia to assess whether TGF-α retarded ischemic brain damage rather than exerting a protective effect. Rats received TGF-α according to the same dosing regimen as above, then infarct volume was evaluated at 4 days. Again, the results showed a significant reduction (53%) of infarct volume by TGF-α, thus indicating that the effect of TGF-α was maintained and that it favored brain protection. Furthermore, administering TGF-α only after ischemia did also reduce infarct volume (73% at 1 day and 80% at 4 days) to an extent similar to that including treatment before ischemia, thus adding some further interest to any hypothetical therapeutic value of this treatment. In addition, TGF-α decreased the associated group variability by homogenizing the response to ischemia in relation to the control group. Although rather large, the variability found in controls (45% coefficient of variation of infarct volume at 4 days) is comparable with that reported in several studies of focal ischemia (McAuley, 1995). From the current results, it is obvious that future research for any putative therapeutic use of TGF-α, together with assessing functional recovery, should address finding out which is the window for effective treatment after ischemia, the most appropriate doses, and the administration route.
After finding this infarct volume reduction by TGF-α, the authors were concerned whether TGF-α had a direct neuroprotective effect or rather a hemodynamic effect that reduced the severity of ischemia at the time of MCAO and shortly after reperfusion, an effect that is known for several drugs. For instance, the NO donor spermine/NO is a cerebral vasodilator (Salom et al., 1998) that reduces infarct size after focal cerebral ischemia by improving intraischemic brain perfusion (Salom et al., 2000). The authors explored the effects of TGF-α on cerebral circulation, as previous data show that TGF-α stimulates femoral arterial blood flow (Gan et al., 1987) and gastric mucosal blood flow (Tepperman and Soper, 1994) in vivo, and it is a potent inhibitor of the contractile responses of coronary arterial strips to various smooth muscle agonists in vitro (Gan et al., 1987). On the one hand, the authors studied the effect of intraventricular administration of TGF-α on CP by means of laser–Doppler flowmetry in the rat, and on the other hand, the authors examined the effect of TGF-α on the arterial tone of basilar and carotid arteries isolated from rabbits. Transforming growth factor-α had no effect on the cerebral circulation as: (1) there were no systemic vascular effects because MABP was not modified by intraventricular TGF-α infusion; (2) there were no local vascular effects because neither passive tone nor agonist-induced active tone were modified by TGF-α in cerebral arteries (that is, basilar artery) or in arteries supplying the cerebrovascular tree (that is, common carotid artery); and, (3) there was no pre-, intra-, or postischemic change in CP in response to TGF-α infusion, which does not support the possibility of local microcirculatory changes through an effect of intraventricular TGF-α on brain metabolism. Thus, it is unlikely that the protective effect of TGF-α against transient focal cerebral ischemia is because of systemic or local effects leading to a reduction in the severity of the ischemic insult. There is considerable variation between species in the vascular response to certain growth factors of the EGF family (Gan et al., 1986). Therefore, it is feasible that the vascular effects of human recombinant TGF-α reported in a canine model (Gan et al., 1987) are not necessarily the same in other animals, such as rat and rabbit, which were studied here. Moreover, differences in sensitivity to TGF-α of distinct vascular beds cannot be ruled out. The reported in vivo effects of TGF-α on blood flow were found after peripheral administration (Gan et al., 1987; Tepperman and Soper, 1994). Injection of TGF-α into the circulation causes accumulation in brain vasculature rather than entering the brain parenchyma, as it hardly crosses the blood–brain barrier (Pan et al., 1999). Therefore, the effect of circulating TGF-α on blood flow might differ from that of TGF-α administered intraventricularly, which would exert actions upon various neural cell types.
The neuroprotective effect of TGF-α in brain is mediated through the EGFR (Justicia and Planas, 1999). Cellular targets for exogenous TGF-α include neurons and astroglia, although astroglia is considered as the main cellular target of TGF-α (Engele and Schilling, 1996; Mazzoni and Kenigsberg, 1996; Kornblum et al., 1998, 1999; Ma et al., 1994, 1999). Indeed, TGF-α induces astrogliosis (Rabchevsky et al., 1998), and the EGFR is up-regulated in glia after brain injury, such as that arising from hypothalamic lesions (Junier et al., 1993) and focal ischemia (Planas et al., 1998). Ligand-binding to the EGFR causes receptor dimerization, tyrosine-phosphorylation, and activation of a number of intracellular signaling pathways, such as that of the mitogen-activated protein kinases, (Moghal and Sterneberg, 1999; Kornblum et al., 1999) and STAT proteins (Darnell et al., 1994; Ruff-Jamison et al., 1994; Grandis et al., 1998). Activation of these intracellular signalling pathways may be crucial for transducing the extracellular TGF-α signal to the nucleus and inducing a cellular response involved in brain protection against an ischemic insult. Recently, the authors showed nuclear translocation of STAT-1 and STAT-3 in the ischemic cortex after administration of TGF-α in the rat, thus indicating that this pathway is involved in TGF-α signalling (Justicia et al., 2000).
In the current study, the protective effect of TGF-α was more marked in cortex than striatum. Selective protection of brain cortex versus striatum in cerebral ischemia has been reported for several substances, such as estradiol (Dubal et al., 1998). The reason why the cortex, compared with the striatum, is a better target for TGF-α (after intraventricular administration) remains unclear. However, the fact that EGFR is expressed in cortical and striatal cells (Planas et al., 1998), whereas protection is greater in cortex, suggests that differences might be accounted for by features such as the intrinsic local lesion severity probably conditioned by the severity of local blood flow reduction and diffusion after intraventricular administration of TGF-α. However, the participation of other factors cannot be excluded—for example, the dependence on selective growth factor for neuronal survival in different brain regions. Also, a recent article showed that protection achieved by administration of glial cell line-derived neurotrophic factor in the intrastriatal 6-hydroxydopamine lesion model depends on the site of administration of the trophic factor (Kirik et al., 2000). This latter study supports the interesting view that trophic factor neuroprotection requires a direct action on terminal innervation. The cortex sends dense projections to the ipsilateral striatum (Paxinos, 1985), therefore in the current model, TGF-α will be accessible to nerve terminals in the striatum arising from the cortex. In contrast, it is likely that the access of nerve ends arising from the striatum to TGF-α will be more limited, as striatal projections essentially innervate the globus pallidus, entopeduncular nucleus, and substantia nigra (Paxinos, 1985), which are distant from the lateral ventricle. The possibility that TGF-α might exert a trophic action locally in nerve terminals, by either a direct action or by causing microenvironmental changes, and that this effect might contribute to protecting neuronal cell degeneration needs to be explored further.
In summary, the current results demonstrate that TGF-α reduces infarct volume at 1 and 4 days after transient MCAO, an effect that is not attributable to changes in microvascular perfusion, and that does not require preischemic administration. It is therefore suggested that TGF-α exerts a neuroprotective effect in temporary focal cerebral ischemia.
Footnotes
Acknowledgments:
The authors thank Ms. M.C. Tirados and Ms. M.C. Máñez for technical assistance.