
Abstract
Estrogens account for gender differences in the incidence and outcome of stroke, but it remains unclear to what extent neuroprotective effects of estrogens are because of parenchymal or vascular actions. Because reproductive steroids have vasoactive properties, the authors assessed the effects and mechanisms of action of 17-β-estradiol in rabbit isolated basilar artery. Cumulative doses of 17-β-estradiol (0.3 μmol/L to 0.1 mmol/L) induced concentration-dependent relaxation that was larger in basilar than carotid artery, in male than female basilar artery, and in KCl-precontracted than UTP-precontracted male basilar artery. Endothelium removal did not modify relaxation induced by 17-β-estradiol in basilar artery, whereas relaxation induced by acetylcholine (1 nmol/L to 0.1 mmol/L) was almost abolished. Neither the estrogen receptor antagonist ICI 182,780 (1 μmol/L), nor the protein synthesis inhibitor cycloheximide (1 μmol/L) affected 17-β-estradiol–induced relaxations. Relaxations induced by the K+ channel openers NS1619 and pinacidil in the same concentration range were greater and lower, respectively, when compared with relaxation to 17-β-estradiol, which was not significantly modified by incubation with the K+ channel blockers charybdotoxin (1 nmol/L and 0.1 μmol/L) or glibenclamide (10 nmol/L and 1 μmol/L). Preincubation with 17-β-estradiol (3 to 100 μmol/L) produced concentration-dependent inhibition of CaCl2-induced contraction, with less potency than the Ca2+ entry blocker nicardipine (0.01 to 10 nmol/L). The authors conclude that 17-β-estradiol induces endothelium-independent relaxation of cerebral arteries with tissue and gender selectivity. The relaxant effect is because of inhibition of extracellular Ca2+ influx to vascular smooth muscle, but activation of estrogen receptors, protein synthesis, or K+ efflux are not involved. Relatively high pharmacologic concentrations of 17-β-estradiol causing relaxation preclude acute vascular effects of physiologic circulating levels on the cerebral circulation.
Epidemiologic data indicate that women show less proneness to stroke than men during the reproductive years, but this gender-related protection is lost in the postmenopausal age, during which the mortality rate for stroke sufferers may be greater in women than in men (Wolf, 1990). However, the value of hormone replacement therapies in stroke prevention or outcome improvement has not been clearly established (Wilson et al., 1985; Finucane et al., 1993; Lafferty and Fiske, 1994; Pedersen et al., 1997; Petitti et al., 1998). 17-β-estradiol is increasingly considered an endogenous neuroprotective agent, and exogenous 17-β-estradiol produces neuroprotection against experimental stroke in both sexes (Simpkins et al., 1997; Dubal et al., 1998; Hawk et al., 1998; Toung et al., 1998; Rusa et al., 1999). More details on the role of reproductive steroids in stroke can be found in a recent review by Hurn and Macrae (2000).
Suggested mechanisms of estrogen-mediated neuroprotection include direct effects on neurones and glia (perfusion-independent mechanisms), such as prevention of oxidative damage (Kume-Kick and Rice, 1998), induction of neuroprotective protooncogene products (Alkayed et al., 1998), increase in transcription or activity of growth factors (Tanapat et al., 1999), and reduction of excitotoxic damage (Azcoitia et al., 1998). On the other hand, endogenous and exogenously administered 17-β-estradiol have been reported to preserve or improve recovery of blood flow in cerebral ischemia (Hurn et al., 1995; Pelligrino et al., 1998; Yang et al., 2000). Moreover, transdermal 17-β-estradiol induces reduction in the pulsatility index (that is, flow resistance) of the middle cerebral artery of postmenopausal women (Penotti et al., 1998). However, there also are reports indicating that neuroprotective effects of estrogens are unrelated to an influence on cerebral blood flow (Wang et al, 1999; Alkayed et al., 2000; Carswell et al., 2000). Therefore, whether neuroprotective effects of 17-β-estradiol may be related to its ability to reduce the severity of the ischemic insult through vascular mechanisms remains unclear.
Classic genomic effects of estrogenic steroids are mediated by activation of nuclear receptors and subsequent transcription and translation processes. However, estrogens also show nongenomic, acute effects on vascular tone (Kitazawa et al., 1997), most of which have been described in coronary arteries (Jiang et al., 1991, 1992; White et al., 1995). Because of the relevance of estrogens in stroke (Hurn and Macrae, 2000), the authors studied the effects and mechanisms of action of 17-β-estradiol on vascular tone of isolated cerebral arteries. For this purpose, the authors assessed the vasoactive effects of 17-β-estradiol in rabbit basilar artery and, for comparison, in carotid artery. Once relaxant effects of 17-β-estradiol were evidenced, influence of gender and nature of active tone on relaxation were investigated. The authors also assessed the importance of functional endothelium and the involvement of estrogen receptor stimulation and protein synthesis activation in 17-β-estradiol–induced relaxation. Finally, the involvement of K+ efflux and Ca2+ influx modulation in the relaxant effects of 17-β-estradiol were investigated.
MATERIALS AND METHODS
Experiments were conducted in compliance with the Spanish legislation on “Protection of Animals used for Experimental and other Scientific Purposes” and in accordance with the Directives of the European Community on this subject.
Tissue preparation
Thirty-six male and five female New Zealand White rabbits (Technology Transferring Center, Polytechnic University of Valencia, Spain), weighing 2.5 to 3 kg, were killed by injection of 25 mg/kg sodium thiopental (Tiobarbital, B Braun Medical, Jaén, Spain) and 1.5 mL of 10 mmol/L KCl solution through the ear vein. The whole brain, including the brainstem, was removed and the basilar artery was dissected free. A midline throat incision provided access to common carotid arteries, one of which was dissected free. Four 3-mm-long segments of basilar artery and four 4-mm-long segments of carotid artery were obtained. Some segments of basilar artery were mechanically devoid of endothelium by gentle rubbing with a stainless steel rod introduced through the arterial lumen. For isometric tension recording, segments were mounted in an organ bath by using tungsten wires (89 μm in diameter) for basilar arteries and stainless steel wires (700 μm in diameter) for carotid arteries. Two pins were introduced through the arterial lumen. One pin was fixed to a stationary support, whereas the other pin was connected to a strain gauge (Universal Transducing Cell UC3, Gould Statham, Oxnard, CA, U.S.A.). Isometric tension was conveniently amplified (Hewlett-Packard 8805D, San Diego, CA, U.S.A.) and recorded (Omniscribe D5237-5, Houston Instrument, Gistel, Belgium). Each organ bath contained 5 mL Ringer-Locke solution at 37†C and bubbled with a 95% O2 and 5% CO2 mixture to give a pH of 7.3 to 7.4. Previously determined optimal resting tensions of 0.5 g and 2 g were applied to basilar and carotid arterial segments, respectively. Then the segments were allowed to equilibrate for 30 to 60 minutes before starting the experiments.
Experimental procedure
The contractile capacity of every arterial segment was assessed by exposure to 50 mmol/L KCl Ringer-Locke solution. Basilar arteries contracting less than 0.5 g and carotid arteries contracting less than 2 g were discarded. Cumulative concentration-response curves to 17-β-estradiol (0.3 μmol/L to 0.1 mmol/L) were obtained in arteries precontracted with 50 mmol/L KCl or with 0.1 mmol/L UTP. To assess the role of the endothelium in the effects of 17-β-estradiol, responses were elicited in some rubbed, KCl-precontracted basilar arteries. The functional state of the endothelium was verified by challenge of UTP-precontracted arteries with acetylcholine (1 nmol/L to 0.1 mmol/L). The involvement of estrogen receptors and protein synthesis activation in the effects of 17-β-estradiol were assessed by incubating some basilar arteries with ICI 182,780 (1 μmol/L) and cycloheximide (1 μmol/L), respectively. The role of K+ efflux modulation in the effects of 17-β-estradiol was studied, on one hand, by comparing the relaxant effects of the estrogen with those of two K+ channel openers— the selective Ca2+-activated K+ channel (KCa) opener NS1619 (0.3 μmol/L − 0.1 mmol/L) and the selective ATP-dependent K+ channel (KATP) opener pinacidil (0.3 μmol/L − 0.1 mmol/L). On the other hand, concentration-response curves to 17-β-estradiol were obtained after incubation of the basilar arteries with two K+ channel blockers—the selective Ca2+-activated K+ channel (KCa) blocker charybdotoxin (1 and 100 nmol/L) and the selective ATP-dependent K+ channel (KATP) blocker glibenclamide (0.01 and 1 μmol/L). Finally, the ability of 17-β-estradiol to modulate Ca2+ entry was assessed by obtaining concentration-response curves to CaCl2 (10 μmol/L to 10 mmol/L) in the absence or presence of different concentrations of the estrogen (3, 10, 30, and 100 μmol/L). For this purpose, basilar arteries were washed 3 times (10-minute intervals) with Ca2+ free medium containing 1 mmol/L ethylene glycol-bis-[β-aminoethyl ether] N,N,N′,N′-tetraacetic acid (EGTA). Then, arteries were stimulated with Ca2+ free, 50 mmol/L KCl medium (with or without 17-β-estradiol) and cumulative concentrations of CaCl2 were added. For comparison, different concentrations of the L-type Ca2+ channel blocker nicardipine (0.01, 0.1, 1, and 10 nmol/L), rather than estrogen, were assayed in a separate series of experiments.
Data analysis
Relaxant responses elicited by 17-β-estradiol, acetylcholine, NS1619, and pinacidil were expressed as percentage of the active tone achieved with KCl or UTP. Contractile responses to CaCl2 were expressed as percentage of previous response to KCl before Ca2+ washing, and data were fitted with a sigmoidal curve. Maximum effect (Emax) and half-maximal effective drug concentration (EC50) were calculated for each concentration-response curve. The pEC50 was calculated as the negative logarithm to base 10 of the EC50 for statistical analysis. These pharmacologic parameters are “apparent” for 17-β-estradiol and K+ channel openers because no real Emax was detected at the greatest concentrations used. One-way analysis of variance followed by Dunnett's multiple comparison test was used to compare the effects of 17-β-estradiol in the groups of basilar arteries from female rabbits, carotid arteries, UTP-precontracted arteries, rubbed arteries, ICI 182,780, cycloheximide, K+ channel agonists, and K+ channel antagonists with the effects of the estrogen in a control group of basilar arteries from male rabbits. Student's unpaired t-test was used to compare the effects of acetylcholine in intact and rubbed arteries. Finally, one-way analysis of variance followed by Student-Newman-Keuls multiple comparisons test was used to compare the effects of CaCl2 in 17-β-estradiol- and nicardipine-treated arteries, at the different concentrations used, with the effects of CaCl2 in their respective control groups. P < 0.05 was considered significant.
Drugs and solutions
17-β-estradiol, pinacidil, and glibenclamide were dissolved and diluted in dimethyl sulfoxide. ICI 182,780, cycloheximide, and NS1619 were dissolved and diluted in ethanol. Total dimethyl sulfoxide or ethanol added to the organ bath for preincubations or concentration-response curves was, at the most, 0.59% v/v and did not affect arterial tone. UTP, acetylcholine, and charybdotoxin were dissolved and diluted in saline solution (0.9% NaCl). Nicardipine was dissolved in bidistilled water and diluted in saline solution. All drugs were from RBI-Sigma-Aldrich Química (Madrid, Spain), except for ICI 182,780 (kindly provided by AstraZeneca, Madrid, Spain), cycloheximide (Calbiochem, Bad Soden, Germany), and nicardipine (kindly provided by Ferrer Internacional, Barcelona, Spain). The Ringer-Locke solution had the following composition (mmol/L): NaCl 120, KCl 5.4, CaCl2 2.2, MgCl2 1.0, NaHCO3 25, and glucose 5.6. In Ca2+ free medium, CaCl2 was omitted from the composition, and, when indicated, EGTA was added. In KCl-depolarizing solution, NaCl was replaced by an equimolar amount of KCl.
RESULTS
Cumulative doses of 17-β-estradiol (0.3 μmol/L to 0.1 mmol/L) induced concentration-dependent relaxation of active tone in rabbit basilar artery (Fig. 1). The highest relaxation was obtained in basilar arteries from male rabbits and precontracted by depolarization with 50 mmol/L KCl (apparent pEC50 = 4.65 ± 0.04, and Emax = 77 ± 3% of active tone). By comparison, relaxations were significantly less in carotid arteries (apparent pEC50 = 4.37 ± 0.02, P < 0.01; and Emax = 28 ± 2%, P < 0.01), in basilar arteries from female rabbits (apparent pEC50 = 4.72 ± 0.07, P < 0.01; and Emax = 59 ± 7%, P < 0.01), and in basilar arteries precontracted with 0.1 mmol/L UTP (apparent pEC50 = 4.40 ± 0.05, P < 0.01; and Emax = 47 ± 7%, P < 0.01).
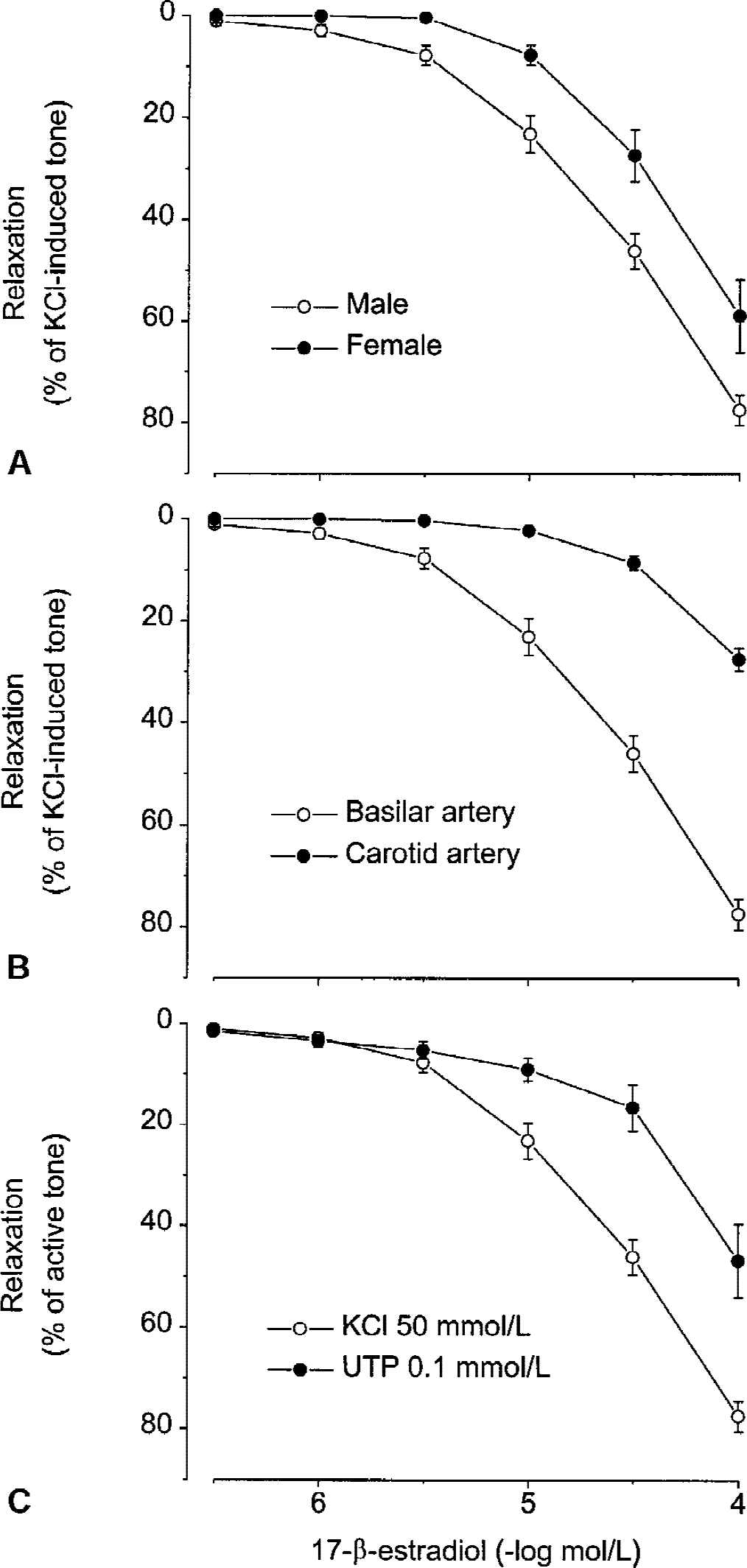
Influence of gender, arterial tissue, and nature of the agent eliciting active tone on the relaxant effects of 17-β-estradiol:
Mechanical rubbing of the endothelium did not significantly modify relaxation induced by 17-β-estradiol in basilar arteries from male rabbits (Fig. 2A), whereas, in the same arterial segments, relaxation induced by acetylcholine (1 nmol/L to 0.1 mmol/L) was almost completely abolished (Fig. 2B).
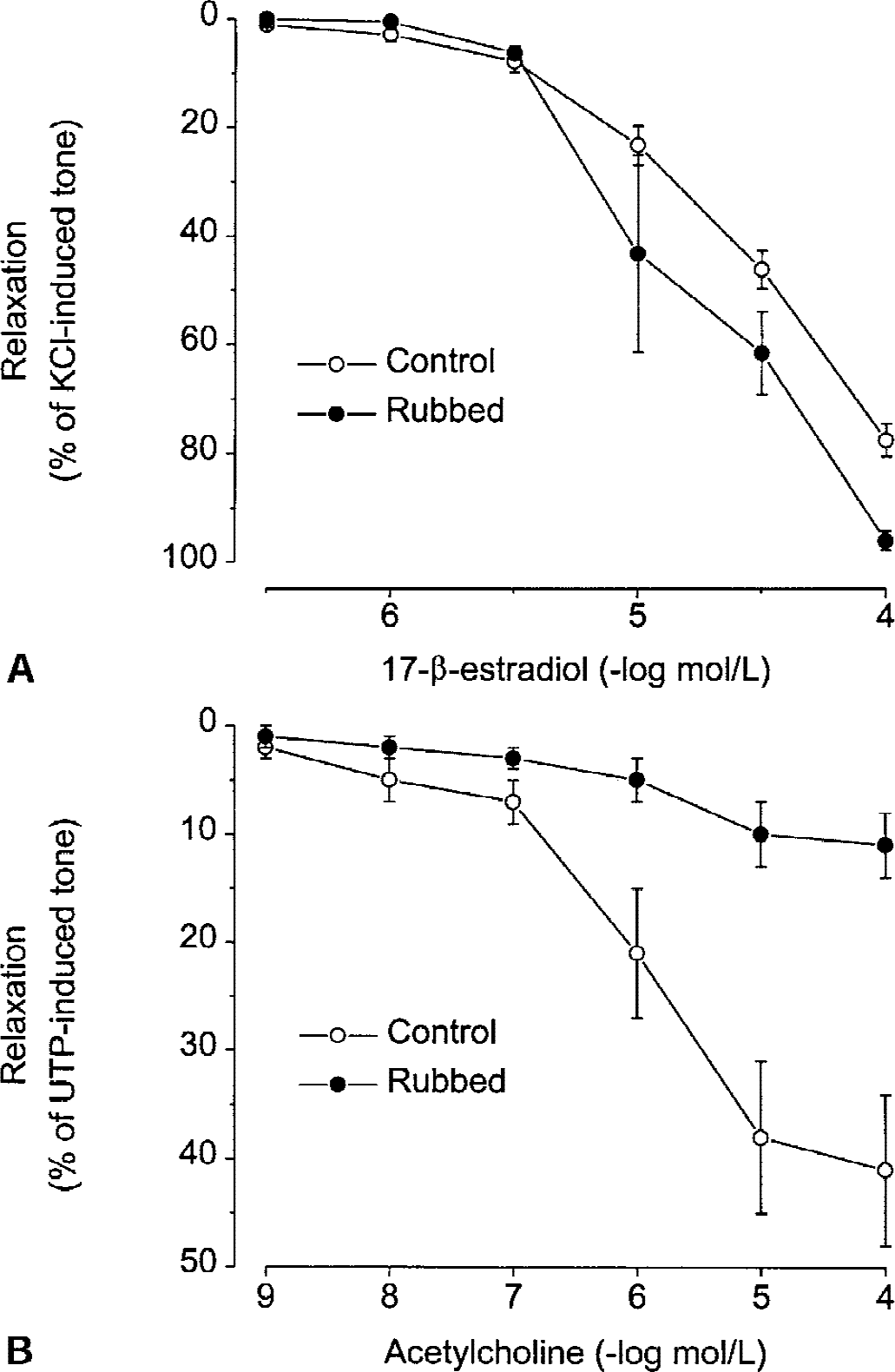
Effects of endothelium removal by mechanical rubbing of the rabbit basilar artery intima on
As shown in Fig. 3, relaxation to 17-β-estradiol was not significantly modified by incubation of basilar arteries from male rabbits with the estrogen receptor antagonist ICI 182,780 (1 μmol/L) or the protein synthesis inhibitor cycloheximide (1 μmol/L).
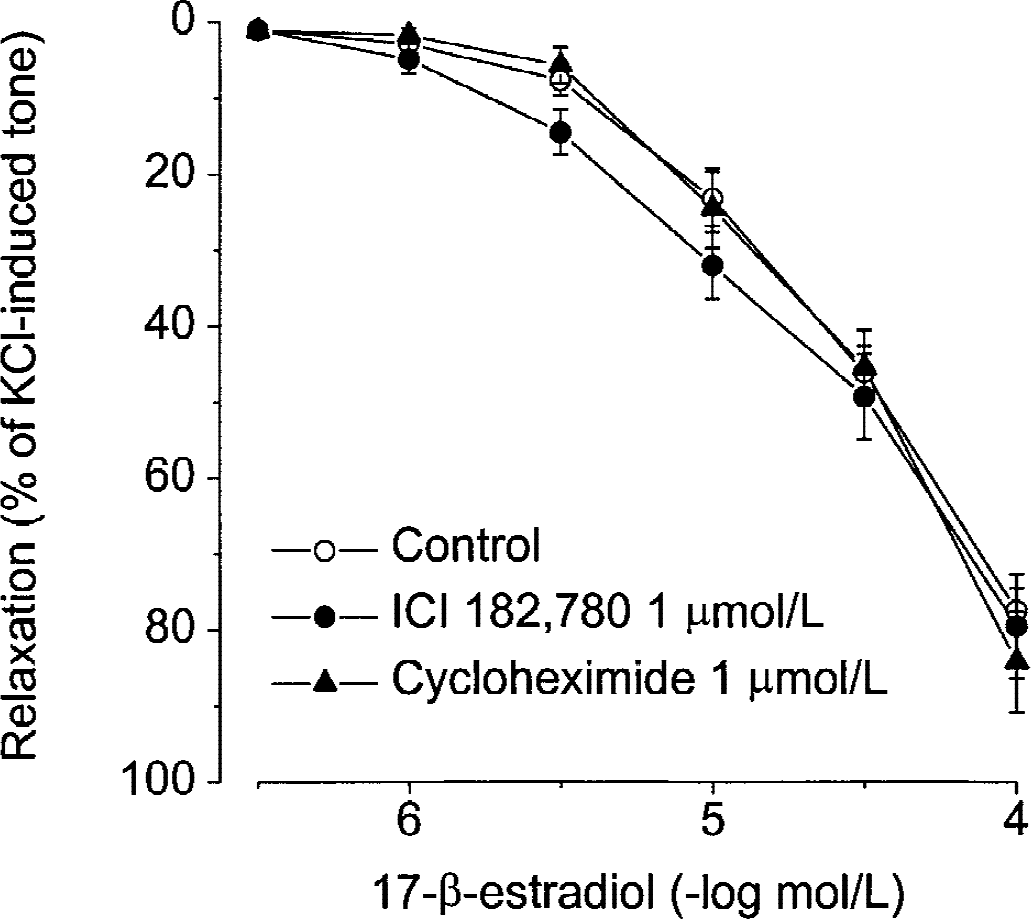
Effects of preincubation of the rabbit basilar artery with the selective estrogen receptor antagonist ICI 182,780 and the protein synthesis inhibitor cycloheximide on concentration-dependent relaxation induced by 17-β-estradiol. Values are means ± SEM (control, n = 74; ICI 182,780, n = 6; cycloheximide, n = 6).
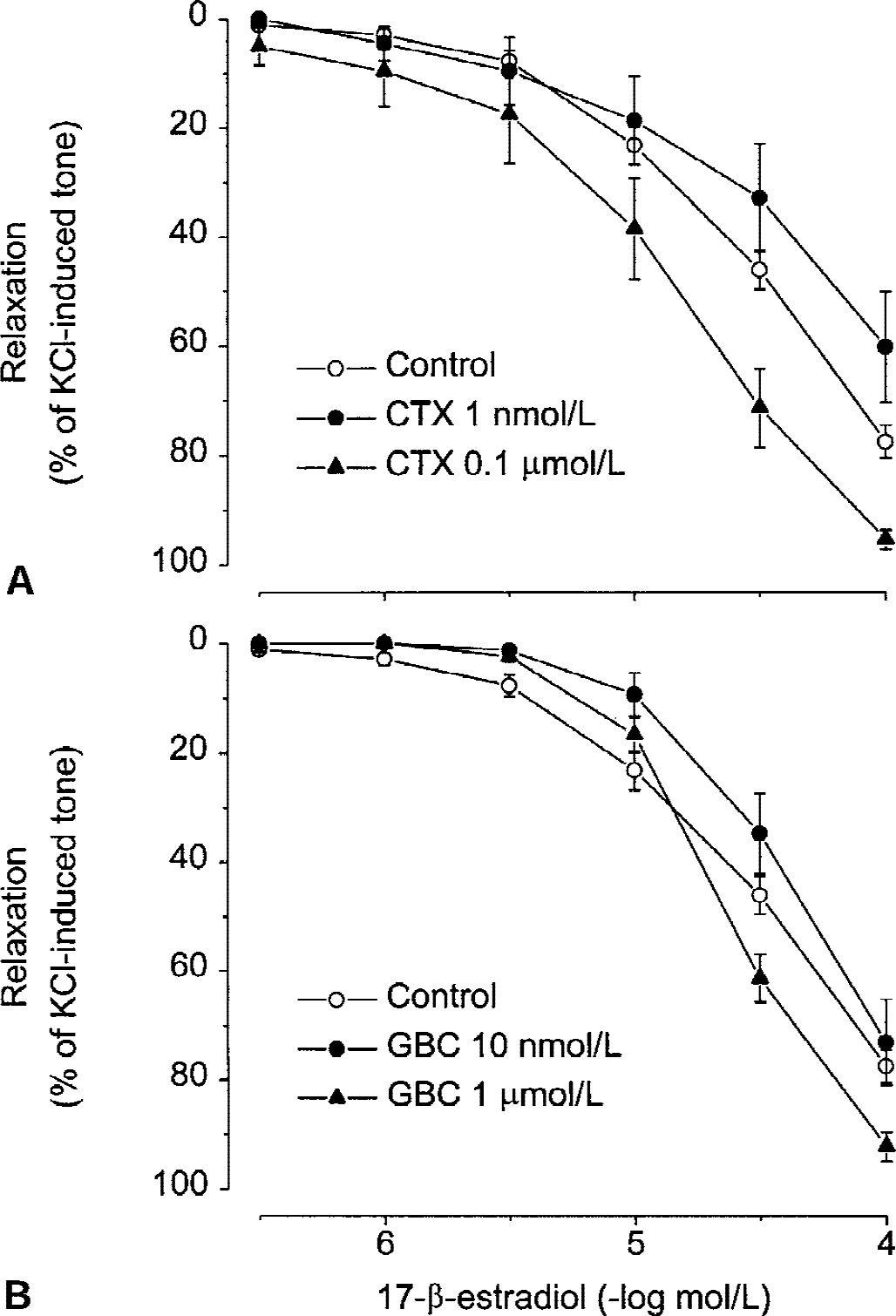
Concentration-dependent relaxations induced by the K+ channel openers NS1619 and pinacidil (0.3 μmol/L to 0.1 mmol/L) in basilar artery are shown in Fig. 4, together with relaxation induced by 17-β-estradiol. Both K+ channel openers relaxed the basilar artery to a different extent than 17-β-estradiol. The Emax of NS1619 was significantly (P < 0.05) greater, whereas the Emax of pinacidil was significantly (P < 0.001) less than the Emax of the estrogen. As shown in Fig. 5, relaxation to 17-β-estradiol was not significantly modified by incubation of basilar arteries from male rabbits with the K+ channel blockers charybdotoxin (1 nmol/L and 0.1 μmol/L) or glibenclamide (10 nmol/L and 1 μmol/L).
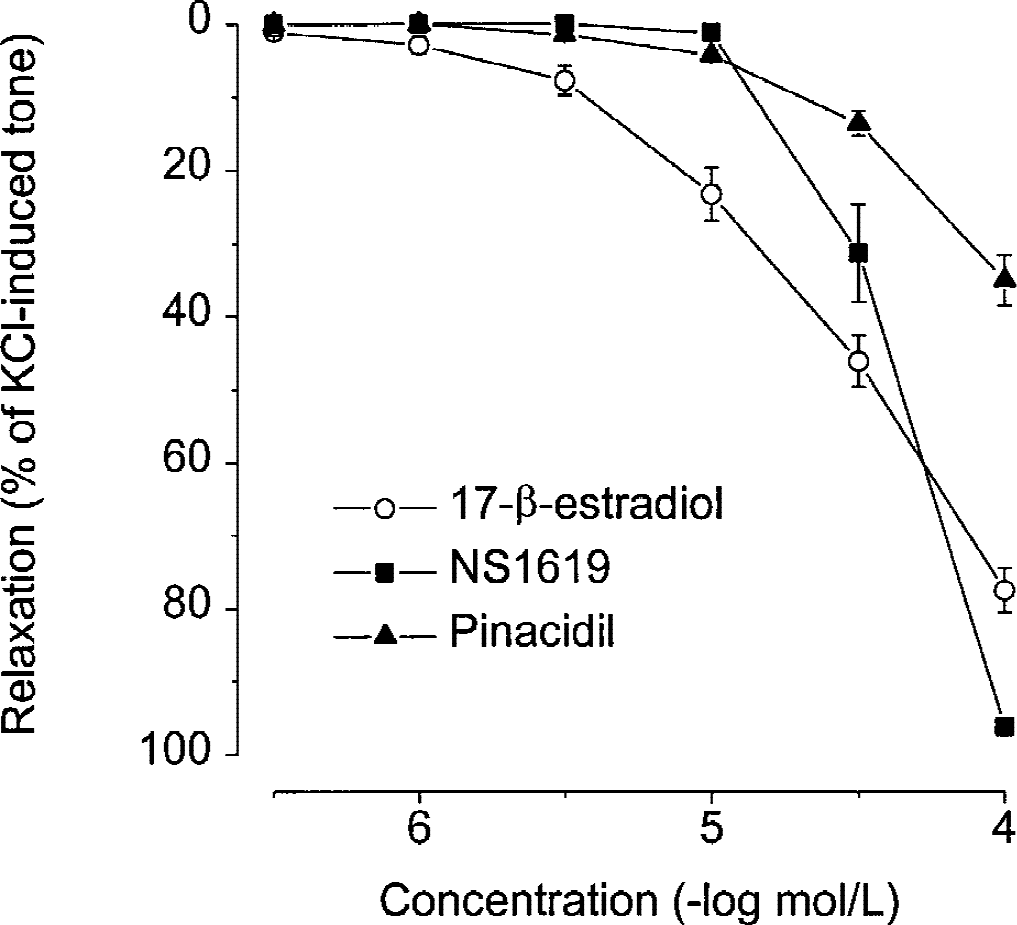
Comparative concentration-dependent relaxant effects of 17-β-estradiol (n = 74), the selective Ca2+-activated K+ channel (KCa) opener NS1619 (n = 8), and the selective ATP-dependent K+ channel (KATP) opener pinacidil (n = 8) in rabbit basilar artery. Values are means ± SEM.
Effects of preincubation of the rabbit basilar artery with
Cumulative doses of CaCl2 (10 μmol/L to 10 mmol/L) induced concentration-dependent contractions in basilar arteries from male rabbits depolarized by 50 mmol/L KCl in Ca2+ free medium. Preincubation with 17-β-estradiol (3 to 100 μmol/L) or the L-type Ca2+ channel blocker nicardipine (0.01 to 10 nmol/L) produced concentration-dependent inhibition of CaCl2-induced contraction (Fig. 6). Table 1 summarizes the inhibitory effects of 17-β-estradiol and nicardipine, at the different concentrations used, on the pEC50 and Emax values of the CaCl2 concentration-response curve.
Pharmacologic parameters for the concentration-effect curve to CaCl2in control conditions and during incubation with different concentrations of 17-β-estradiol or nicardipine
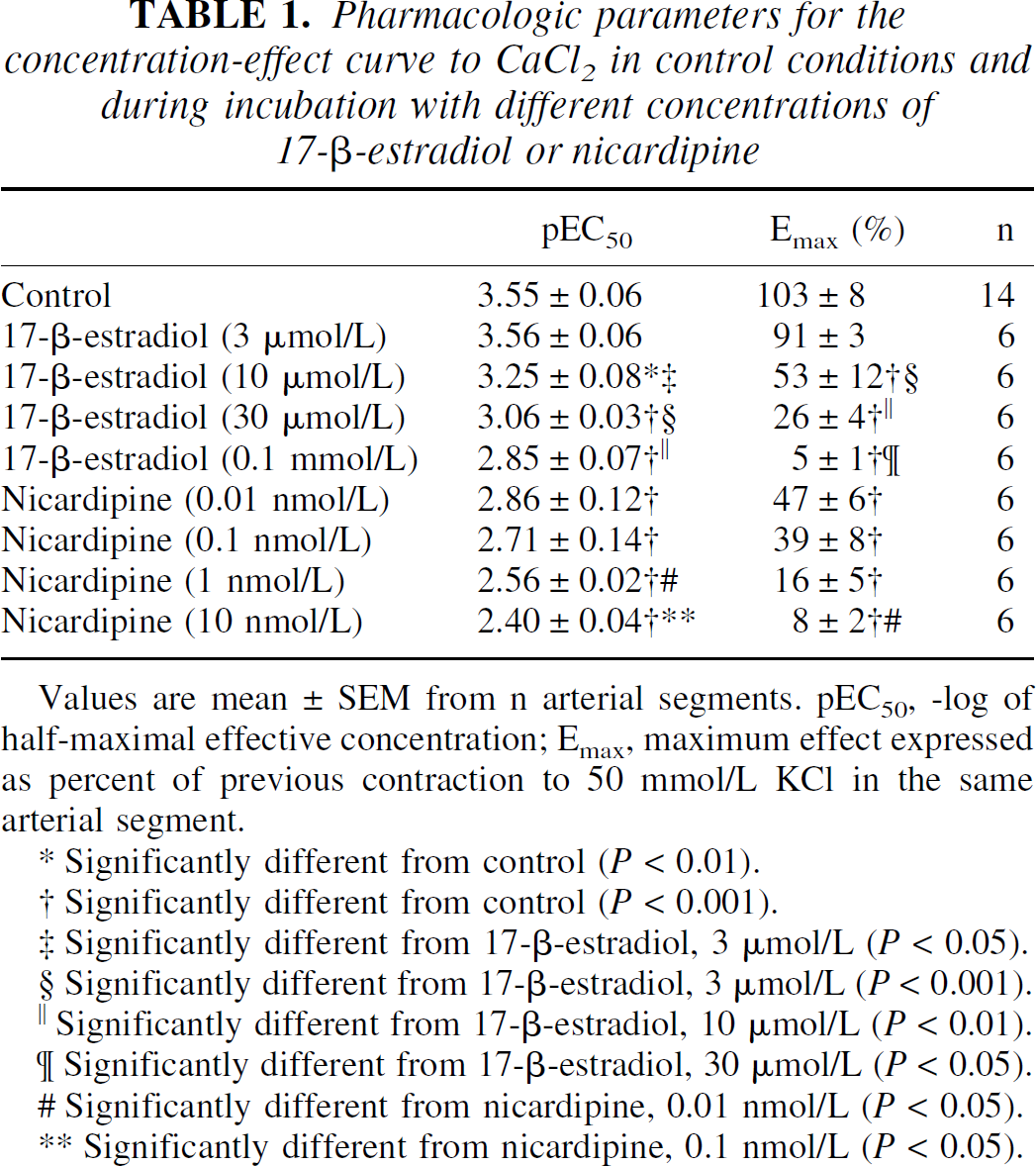
Values are mean ± SEM from n arterial segments. pEC50, -log of half-maximal effective concentration; Emax, maximum effect expressed as percent of previous contraction to 50 mmol/L KCl in the same arterial segment.
Significantly different from control (P < 0.01).
Significantly different from control (P < 0.001).
Significantly different from 17-β-estradiol, 3 μmol/L (P < 0.05).
Significantly different from 17-β-estradiol, 3 μmol/L (P < 0.001).
Significantly different from 17-β-estradiol, 10 μmol/L (P < 0.01).
Significantly different from 17-β-estradiol, 30 μmol/L (P < 0.05).
Significantly different from nicardipine, 0.01 nmol/L (P < 0.05).
Significantly different from nicardipine, 0.1 nmol/L (P < 0.05).
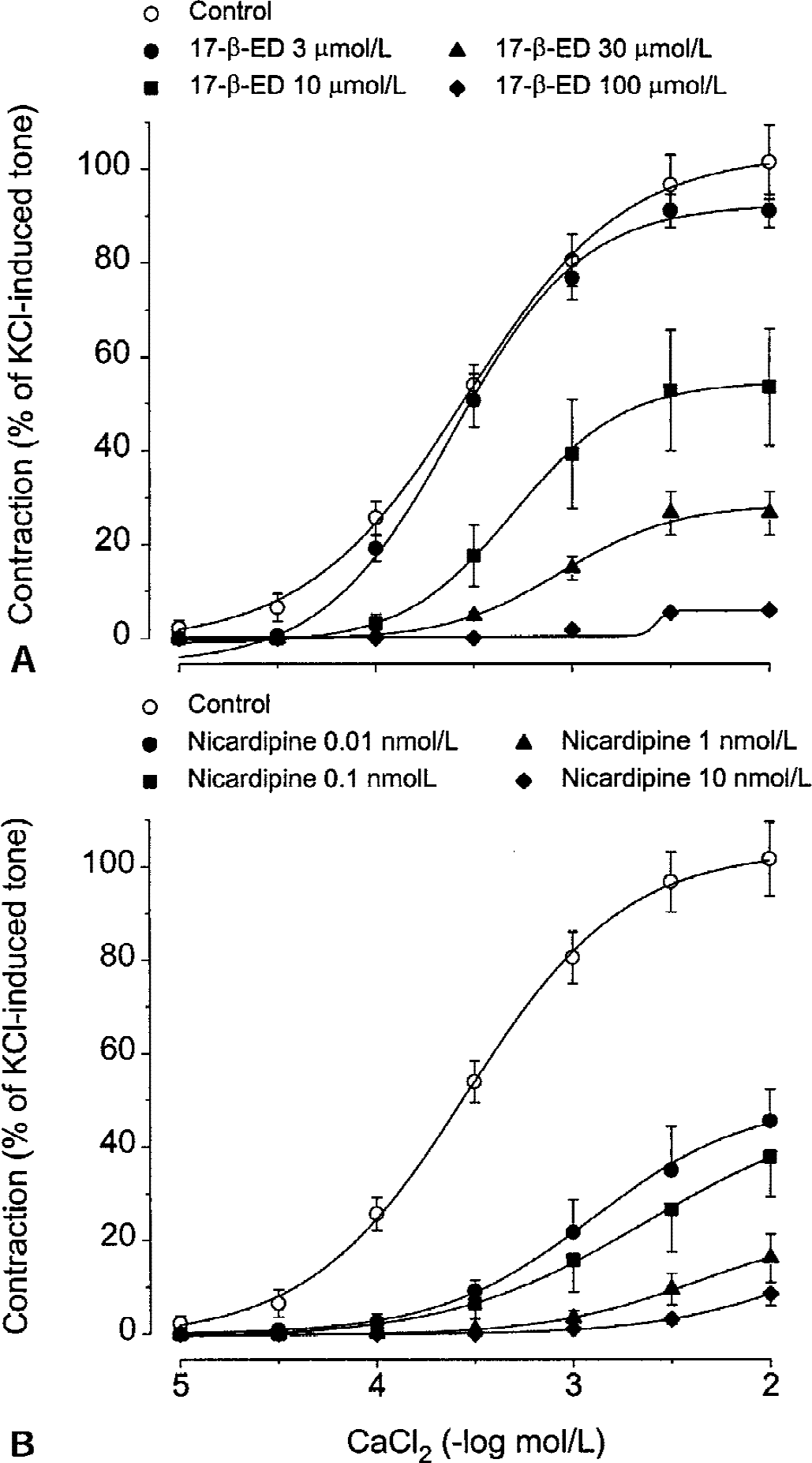
Effects of preincubation of the rabbit basilar artery with different concentrations of
DISCUSSION
The current results show that 17-β-estradiol induces endothelium-independent relaxation in rabbit basilar artery. Slight gender differences were in the relaxant effect of 17-β-estradiol as arteries from female rabbits relaxed less than arteries from male rabbits. In rat femoral artery (Kitazawa et al., 1997) and rabbit coronary artery (Jiang et al., 1991), relaxation by 17-β-estradiol in males and females is similar. This suggests that sex differences in the response to 17-β-estradiol may be specific, but not necessarily exclusive, for cerebral arteries. However, in the current study, the selectively greater relaxant effects of 17-β-estradiol in basilar versus carotid arteries, as well as the differences because of the nature of the active tone, were more evident. Arterial tone reached by depolarization with high K+ was more efficiently relaxed than arterial tone reached by UTP-induced contraction. In contrast, in porcine coronary artery, 17-β-estradiol relaxes prostaglandin F2α- and histamine-precontracted segments to a greater extent than KCl-precontracted segments (White et al., 1995). In rabbit coronary artery, relaxation is similar in KCl- and prostaglandin F2α-precontracted segments (Jiang et al., 1991) and less than in endothelin-1-precontracted segments (Jiang et al., 1992). Therefore, it is quite clear that 17-β-estradiol does not cause arterial relaxation by blocking a specific vascular receptor type responsible for the active tone (for example, purinergic receptors, prostanoid receptors, etc.). The estrogen must be interacting with some mechanism whose contribution to the maintenance of vascular tone varies depending on the agent eliciting the contraction.
The current results show that the presence of a functional endothelium is irrelevant to the relaxant effects of 17-β-estradiol in rabbit basilar artery. Similar relaxation was induced in control intact arteries and in arteries in which endothelium-dependent relaxation (that is, acetylcholine-elicited relaxation) had been almost abolished by mechanical rubbing. Endothelium-independent relaxation to 17-β-estradiol has been also reported in rabbit coronary artery (Jiang et al., 1991, 1992). In porcine coronary artery, relaxation to 17-β-estradiol is induced in the absence of the endothelium, although no intact arteries were assayed (White et al., 1995). This suggests that the relaxant effect of the estrogen is because of a direct action on vascular smooth muscle. Even though the endothelium does not play a role in the acute relaxant effects of 17-β-estradiol, chronic treatment with estrogen increases eNOS protein in rat cerebral microvessels (McNeill et al., 1999) and pial arterioles (Pelligrino et al., 2000), which accounts for restoration of endothelium-dependent vasodilatation lost after ovariectomy (Pelligrino et al., 2000). Moreover, 17-β-estradiol treatment increases endothelial glucose transporter in brain microvessels (Shi et al., 1997). Therefore, the cerebrovascular endothelium shows responses to 17-β-estradiol that could influence cerebral perfusion or glucose uptake.
Human umbilical vein endothelial cells contain membrane binding sites for 17-β-estradiol, which mediate rapid NO release (Russell et al., 2000). According to the current results, this kind of estrogen receptor: (1) does not exist in cerebroarterial endothelium, or (2) is not coupled to NO release able to induce or potentiate vascular relaxation. On the other hand, type β estrogen receptors, which are predominant in human vascular smooth muscle, have been linked to genomic regulation of transcriptional activation involved in, for example, antiproliferative effects (Hodges et al., 2000). In the current study, the pure estrogen receptor antagonist ICI 182,780 did not inhibit the relaxant effects of 17-β-estradiol in rabbit basilar artery, the same as the protein synthesis inhibitor cycloheximide. Therefore, activation of vascular estrogen receptors does not account for the relaxant effects of 17-β-estradiol that do not involve translation of proteins. This is in line with the time course of rapid effects such as vascular relaxation.
Binding of 17-β-estradiol to the regulatory β subunit of the KCa channel induces its activation (Valverde et al., 1999). Opening of membrane K+ channels in vascular smooth muscle increases K+ efflux, which leads to membrane hyperpolarization, closing of voltage-dependent Ca2+ channels, and subsequent relaxation (Nelson and Quayle, 1995). With regard to cerebral arteries, activation of KCa channels contributes to the actions of NO and other nitrovasodilators in rabbit basilar artery (Robertson et al., 1993), and acts as a negative feedback mechanism to regulate the level of resting tone maintained by increased basal Ca2+ influx in dog basilar artery (Asano et al., 1993). However, according to the current results, 17-β-estradiol–induced relaxation of rabbit basilar artery is not because of activation of K+ (KCa or KATP) channels because of the following: (1) concentration-response curves to selective openers of KCa channels (NS1619) and KATP channels (pinacidil) did not resemble the relaxant effects of 17-β-estradiol, and (2) the relaxant effects of the estrogen were unaffected by the selective blockers of KCa channels (charybdotoxin) and KATP channels (glibenclamide). In line with the authors' conclusion, activity of KCa channels does not mediate the reduction in myogenic tone induced by 17-β-estradiol in isolated-pressurized rat cerebral arteries (Geary et al., 1998). There is electrophysiologic evidence that 17-β-estradiol has no effect on the outward K+ currents evoked by membrane depolarization in rabbit basilar artery (Ogata et al., 1996). The current study adds functional evidence that an increase in K+ efflux does not mediate 17-β-estradiol-elicited relaxation in cerebral arteries. In contrast, 17-β-estradiol relaxes porcine coronary artery by opening KCa channels through cGMP-dependent phosphorylation (White et al., 1995).
In the current study, KCl-precontracted arteries were more effectively relaxed than agonist-precontracted arteries, which supports the possibility that 17-β-estradiol affects membrane Ca2+ channel function in rabbit basilar artery. The inhibition of extracellular Ca2+ influx by 17-β-estradiol was confirmed by the current results showing concentration-dependent inhibition of the contraction elicited by CaCl2 in depolarizing conditions. Moreover, there was a close correlation between estrogen concentrations causing relaxation and those producing inhibition of contraction. Electrophysiologic evidence of inhibition of Ca2+ currents by 17-β-estradiol has been reported in rat aorta cultured smooth muscle cells (Zhang et al., 1994), in freshly dispersed smooth muscle cells from rabbit basilar artery (Ogata et al., 1996), and from nonvascular smooth muscle (Kitazawa et al., 1997). In rat aortic rings, 17-β-estradiol inhibits the increase of 45Ca uptake induced by KCl-depolarization (Freay et al., 1997). The current results raise functional evidence for the Ca2+ antagonistic effect of 17-β-estradiol in cerebral arteries and are in line with a similar effect reported in rabbit coronary artery and rat aorta (Jiang et al., 1991). When compared with nicardipine, a classic L-type Ca2+ channel blocker, 17-β-estradiol is a relatively weak Ca2+ channel blocker. Whereas nicardipine showed inhibitory effects at nanomolar concentrations, similar inhibitory effects were obtained with micromolar concentrations of the estrogen. Both 17-β-estradiol and nicardipine showed functional antagonism against CaCl2-induced contraction, producing a parallel decrease in the potency and the efficacy of the contractile agent (Kenakin, 1987).
Contraction and increase in [Ca2+]i because of Ca2+ entry from the extracellular space, but not Ca2+ release from the intracellular stores, are reduced in aortic smooth muscle cells from female rats when compared with male rats (Murphy and Khalil, 2000). Because, as the authors have demonstrated, 17-β-estradiol behaves as a Ca2+ entry blocker, lower dependency on extracellular Ca2+ could explain the relatively reduced relaxant effect of the estrogen in basilar arteries from female rabbits when compared with male rabbits. In line with this suggestion, there is a tendency in rat aorta to decrease relaxant effects to 17-β-estradiol in females when compared with males (Freay et al., 1997).
Micromolar concentrations of 17-β-estradiol producing relaxation of rabbit basilar artery are clearly greater than physiologic serum levels of the estrogen. Therefore, the effects described in the current study are because of a pharmacologic action. In the light of these results, the authors can not conclude if such action is because of binding of 17-β-estradiol to a estrogen receptor coupled to membrane Ca2+ channels or to direct binding to some regulatory subunit in the own Ca2+ channel. The circulating concentration of 17-β-estradiol could approach this high level under abnormal endocrine conditions or during hormone replacement therapy in the clinical setting (Pedersen et al., 1997), and during experimental estrogen administration aiming neuroprotection in animal stroke models (Rusa et al., 1999). Agreeing with the current results in isolated cerebral arteries, exogenous 17-β-estradiol has been reported to increase cerebral blood flow during global cerebral ischemia and ameliorate postischemic hyperemia (Hurn et al., 1995). In contrast, normal endogenous circulating level of 17-β-estradiol is not likely to induce effects on cerebral blood flow through vasodilatory action in situations of compromised brain perfusion (Carswell et al., 2000).
In conclusion, the authors have demonstrated that 17-β-estradiol induces endothelium-independent relaxation of isolated cerebral arteries to a greater extent than larger carotid arteries. Relaxation, which is selectively greater in male than in female arteries, is not mediated by classic estrogen receptor and protein synthesis activation. Functional evidence is provided that the relaxant effect is because of inhibition of extracellular Ca2+ influx to vascular smooth muscle, however, in contrast with coronary arteries, activation of K+ efflux is not involved. Although the relatively high pharmacologic concentrations of estrogen causing relaxation preclude acute effects of physiologic circulating levels on the cerebral circulation, effects during current or future therapies based in exogenous 17-β-estradiol administration should be considered.
Footnotes
Acknowledgments:
The authors thank María C. Tirados and María C. Máñez for their technical assistance.