
Abstract
Striatal large aspiny interneurons were recorded from a slice preparation using a combined electrophysiologic and microfluorometric approach. The role of intracellular Ca2+ stores was analyzed during combined oxygen/glucose deprivation (OGD). Before addressing the role of the stores during energy deprivation, the authors investigated their function under physiologic conditions. Trains of depolarizing current pulses caused bursts of action potentials coupled to transient increases in intracellular calcium concentration ([Ca2+]i). In the presence of cyclopiazonic acid (30 μmol/L), a selective inhibitor of the sarcoendoplasmic reticulum Ca2+ pumps, or when ryanodine receptors were directly blocked with ryanodine (20 μmol/L), the [Ca2+]i transients were progressively smaller in amplitude, suggesting that [Ca2+]i released from intracellular stores helps to maintain a critical level of [Ca2+]i during physiologic firing activity. As the authors have recently reported, brief exposure to combined OGD induced a membrane hyperpolarization coupled to an increase in [Ca2+]i. In the presence of cyclopiazonic acid or ryanodine, the hyperpolarization and the rise in [Ca2+]i induced by OGD were consistently reduced. These data support the hypothesis that Ca2+ release from ryanodine-sensitive Ca2+ pools is involved not only in the potentiation of the Ca2+ signals resulting from cell depolarization, but also in the amplification of the [Ca2+]i rise and of the concurrent membrane hyperpolarization observed in course of OGD in striatal large aspiny interneurons.
On the basis of electrophysiologic, morphologic, and pharmacologic criteria, at least three distinct subtypes of interneurons have been recognized in the mammalian striaturn (Kawaguchi, 1993, 1995; Wilson et al., 1990). Large aspiny (LA) interneurons represent less than 5% of the entire striatal neuronal population (Graybiel, 1990). Despite their rarity, they give rise to the intrinsic cholinergic innervation of the striatum, an area known to be highly enriched with acetylcholine (Bolam et al., 1984; Graybiel, 1990). Functionally, these cholinergic interneurons are essential for motor and cognitive tasks (Apicella et al., 1991; Graybiel et al., 1994). In vivo recordings from the striatum of behaving monkeys have shown that these cholinergic interneurons, named tonically active neurons, respond in a temporally related manner to stimuli serving as instructions, as triggers for learned behavioral reactions, and as signals for reward delivery (Graybiel et al., 1994).
It has long been known that the striatum is extremely susceptible to ischemic damage (Pulsinelli, 1985). Within the striatum, however, neuronal subtypes express a differential vulnerability. Medium-sized spiny cells, representing the large majority of the entire striatal neuronal population, are rapidly lost both in ischemia and during excitotoxic damage (Beal et al., 1986; Francis and Pulsinelli, 1982; Pulsinelli, 1985), whereas cholinergic interneurons are selectively spared under both these pathologic conditions (Ferrante et al., 1985; Francis and Pulsinelli, 1982). Indeed, the cellular factors regulating the selective vulnerability of different striatal neuronal subtypes are not known yet. We have recently shown that LA interneurons respond to combined oxygen/glucose deprivation (OGD) with a membrane hyperpolarization, coupled to an increase in the intracellular calcium concentration [Ca2+]i(Pisani et al., 1999). A pharmacologic analysis of this event provided evidence for a partial involvement of voltage-gated Ca2+ channels as a source for the [Ca2+]i rise during OGD. Although an increase in [Ca2+]i does not necessarily imply a cell injury, the role of [Ca2+]i overload in mediating neuronal death in both ischemic and excitotoxic insults is widely accepted (Choi, 1990; Silver et al., 1997; Tymianski et al., 1994); thus, the ability of endogenous buffers to mitigate the consequences of such a burden might represent the basis for the differential neuronal vulnerability under these pathologic conditions.
Several experimental studies indicate that disturbances of endoplasmic reticulum Ca2+ homeostasis are involved in the pathologic processes underlying neuronal death during ischemia and apoptosis (Paschen and Doutheil, 1999). However, only a few in vitro studies have addressed the role of internal Ca2+ stores during energy deprivation in a slice preparation. Involvement of intracellular Ca2+ pools has been shown in hippocampal CA1 neurons during anoxia (Belousov et al., 1995; Grondahl et al., 1998; Yamamoto et al., 1997) and in hippocampal astrocytes during combined hypoxia and hypoglycemia (Duffy and MacVicar, 1996).
Thus, by a combined approach of simultaneous intracellular recordings and optical measurements of [Ca2+]i, we investigated the possible involvement of intracellular Ca2+ stores in the [Ca2+]i rise observed during OGD in LA interneurons.
METHODS
Preparation and maintenance of slices
These procedures have been described previously (Calabresi et al., 1995, 1997; Pisani et al., 1999). In brief, male Wistar rats (3 to 5 weeks of age) were killed under ether anesthesia by cervical dislocation, the brain was quickly removed, and corticostriatal coronal slices (180 to 200 μm thick) were cut from tissue blocks with a vibratome in an ice-cold (0°C) Krebs solution (see composition below). Slices were maintained at 34°C in an oxygenated solution for approximately 30 minutes. A single slice was then transferred into a recording chamber, mounted on the stage of an upright microscope (Axioskop FS, Zeiss, Oberlochen, Germany) equipped with a ×60, 0.90 n.a. water-immersion objective (LUMPlan FI; Olympus Optical, Hamburg, Germany), and fully submerged in a continuously flowing Krebs solution (33°C, 3 mL/min) gassed with 95% O2 and 5% CO2. The composition of the solution was (in mmol/L): 126 NaCl, 2.5 KCl, 1.3 MgCl2, 1.2 NaH2PO4, 2.4 CaCl2, 10 glucose, and 18 NaHCO3. OGD was induced with a glucose-free solution equilibrated with a 95% N2 and 5% CO2 gas mixture. Glucose was replaced with saccharose to balance osmolarity. Complete replacement of the medium in the chamber took 45 to 60 seconds. As reported previously (Jiang et al., 1991), baseline pO2 is maintained at more than 50 torr throughout brain tissue and rapidly declines during OGD within 30 to 60 seconds.
Combined electrophysiologic and optical recordings
The tip of sharp microelectrodes was filled with 1 mmol/L bis-fura-2 (hexapotassium salt; Molecular Probes, Leiden, The Netherlands) in 1 mol/L KCl. The shank of the electrode was backfilled with a 2 mol/L KCl solution. LA cells were impaled under visual guidance, according to their characteristic shape and size, up to 50 to 70 μm beneath the surface of the slice. After cell impalement, cells were loaded with bis-fura-2 by injecting 0.1 to 0.5 nA negative current through the recording pipet. Electrode resistance dropped from initial values of 120 to 150 mOhm to 35 to 45 mOhm. An Axoclamp 2A amplifier (Axon Instruments, Foster City, U.S.A.) was used for electrophysiology. Electrical samples were displayed on an oscilloscope and digitally stored. Traces were displayed on a high-gain chart recorder (Gould RS 3400. Ilford, U.K.). In the single-electrode voltage-clamp mode, the switching frequency was 3 kHz. The headstage signal was continuously monitored on a separate oscilloscope. Fluorescence of bis-fura-2 was elicited by a 75 W xenon lamp bandpass-filtered at 340 and 380 nm. Emission light was filtered (500 nm) and then detected by a CCD camera (Photonic Science, Millham Mountfield, U.K.). In the OGD experiments, pairs of 340-nm and 380-nm images were acquired at intervals of 12 seconds. During the depolarizing current pulses, the acquisition rate was 1 second. Analysis of the data was performed off-line (IonVision, Coventry, U.K., ImproVision, Northampton, U.K.; Microcal Origin 4.1, Microcal Software, U.S.A., running on PowerMac 8100 and on a PC, respectively). Pairs of 340-nm and 380-nm images were background-subtracted, where backgrounds were regions free of dye fluorescence, and ratio images were obtained. Ion concentration values were calculated according to Grynkiewicz et al. (1985). The calibration parameters Rmin and Rmax were obtained in situ by bathing perforated (ionomycin) cells in Ca2+ free (1 mmol/L EGTA) and in 1 mmol/L Ca2+-containing solution. The value for the apparent Kd was 0.6 μmol/L, Rmin was 0.22, and Rmax was 4. These values gave reliable [Ca2+]i estimates according to our in vitro slice preparation (Garaschuk et al., 1997; Swandulla et al., 1991). Values in the text and in the figures are expressed as mean ± standard deviation. Student's t-test was used for statistical analysis.
Sources and handling of the compounds
Both cyclopiazonic acid (CPA; Calbiochem, LaJolla, CA, U.S.A.) and ryanodine (AlomoncLabs, Jerusalem, Israel) were dissolved in dimethylsulfoxide in a 10-mM stock solution and diluted to the final concentration immediately before use. Bath-application of the compounds was obtained by switching the solution to one containing the desired concentration of the drug. Drug solutions entered the chamber within approximately 20 to 30 seconds after turning a three-way tap on. Caffeine and tetrodotoxin were from Sigma (Milan, Italy).
RESULTS
Identification of recorded cells
Both the dye fluorescence and the typical electrophysiologic features allowed us to identify the recorded cells as LA interneurons. As described earlier (Kawaguchi, 1993; Kawaguchi et al., 1995), these cells had a large soma (approximately 25 to 50 μm), a polygonal shape, and two to four primary dendritic arborizations. Electrophysiologic recordings have shown that these cells are relatively depolarized at rest (−64 ± 6 mV); in addition, they have a low threshold for spike discharge and high input resistance (Calabresi et al., 1997; Kawaguchi, 1993; Kawaguchi et al., 1995; Wilson et al., 1990). A depolarizing current pulse (Fig. 1Aa; 1 nA, 2 seconds; same protocol used in B) evoked an action potential discharge that was limited by a strong spike frequency adaptation and was followed by a pronounced after-hyperpolarization (AHP). During voltage-clamp experiments performed in the presence of 1 μmol/L tetrodotoxin (see Fig. 1Ac), a voltage step in the depolarizing direction (from −65 to −40 mV, 3 seconds) elicited an outward current, followed at the end of the voltage pulse by a slow inward current. This outward current, elicited at depolarized potential, might account for both action potential accommodation and AHP, whereas the slow inward current might explain the slow recovery to the resting membrane potential that follows AHP. Hyperpolarizing current pulses evoked a prominent sag conductance (Ih) (not shown).
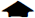
Decremental response of the depolarization-induced intracellular calcium concentration ([Ca2+]i) transients by depletion of internal stores. ( whereas the slow inward current might explain the slow recovery to the resting membrane potential that follows AHP  (
(
On average, in the soma, basal [Ca2+]i was 44.22 ± 2.5 nM; dendritic [Ca2+]i at rest was 52.19 ± 2.2 nM as measured in the regions of interest (n = 98; Fig. 2A). This slight difference in the basal levels of [Ca2+]i may be due to the spontaneous spiking activity observed in these neurons, as previously reported (Pisani et al., 1999). In some cases, to prevent spontaneous firing activity, negative holding current (0.1 to 0.4 nA) was injected into the recorded cell.
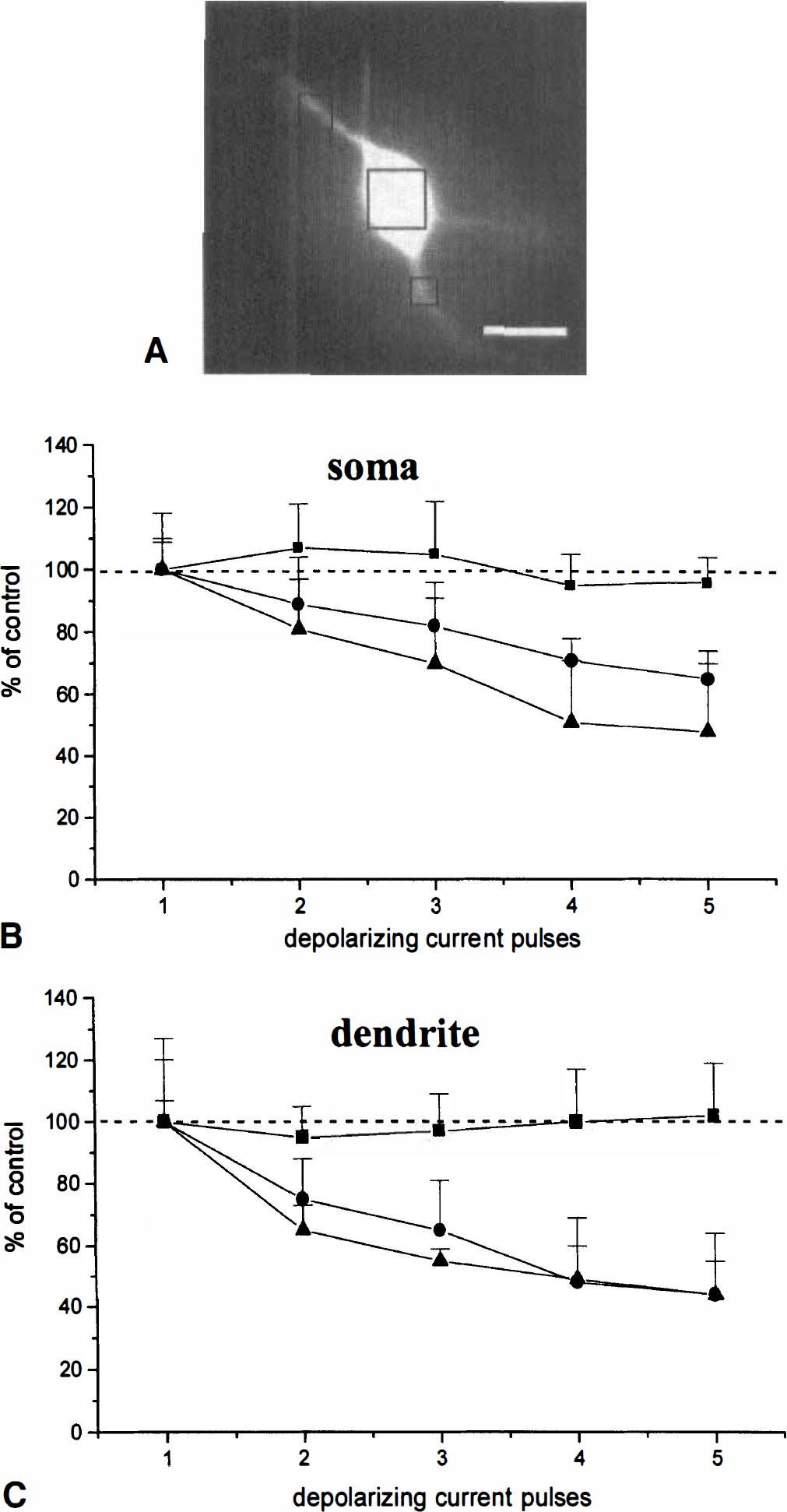
Summary plots of the effect of cyclopiazonic acid and ryanodine on intracellular calcium concentration ([Ca2+]i) transients. (
Effects of CPA and ryanodine on [Ca2+]i transients triggered by membrane depolarization
The mycotoxin CPA is known to be a specific and reversible blocker of the Ca2+ uptake into the endoplasmic reticulum stores, through the inhibition of the sarcoendoplasmic reticulum Ca2+ pumps (Pozzan et al., 1994; Thomas and Hanley, 1994; Tsien and Tsien, 1990). Before addressing the role of internal stores in course of OGD, we first examined the possible involvement of this uptake mechanism under physiologic conditions. We performed a set of experiments analyzing the effects of CPA on [Ca2+]i transients evoked by depolarizing current pulses. Then, in another group of experiments, a similar test protocol was used in the presence of ryanodine (20 μmol/L). At this concentration, ryanodine binds to ryanodine-sensitive channels and locks them in a low conductance state, thereby depleting ryanodine-sensitive stores of their Ca2+ content (Rousseau et al., 1987).
Under control conditions (see Fig. 1B), the bursts of action potentials evoked by five repetitive pulses of depolarizing current were limited by a strong spike adaptation and were followed at the end of each pulse by a pronounced AHP. One of the five depolarizing pulses is shown at a higher sweep speed in a current-clamp recording under control conditions (see Fig. 1Aa) and in ryanodine (see Fig. 1Ab). In voltage-clamp recordings, both the outward and the inward currents observed in controls (see Fig. 1Ac) were significantly reduced in ryanodine (see Fig. 1Ad). Indeed, the number of action potentials induced by the five depolarizing pulses was not significantly different from the first to the fifth pulse. In the presence of both CPA (30 μmol/L, 10 to 20 minutes preincubation) and ryanodine (20 μmol/L, 20 to 30 minutes preincubation), no consistent change in the resting membrane potential and in the input resistance of the cell was detected, as measured at the resting potentials from the amplitude of small (<10 mV) hyperpolarizing electrotonic potentials (not shown). However, both drugs caused a modest but significant increase in the amplitude of the voltage response during the depolarizing current pulses (see Figs. 1Ab, 1C). In addition, a reduced rate of accommodation of the firing activity was observed, resulting in an increased number of action potentials per pulse (see Figs. 1Ab, 1C). Finally, a reduction of the AHP amplitude at the end of each current pulse was recorded both in ryanodine (see Figs. 1Ab, 1C) and CPA (not shown).
As reported for other central neurons (Lev Ram et al., 1992, Midtgaard et al., 1993), dendritic [Ca2+]i reached higher levels compared with the cell body (see Fig. 1B). Under control conditions, the amplitude of the [Ca2+]i transients produced by the depolarizing pulses did not vary significantly among the five pulses (see Fig. 1B, 2; n = 11; on average: first pulse, 91 ± 6.17 nM in the soma, 180 ± 4.96 nM in dendrites; second pulse, 100 ± 8.9 nM and 156 ± 11 nM; third pulse, 105 ± 5.5 and 159 ± 4.6; fourth pulse, 88 ± 4 nM and 181 ± 4.5 nM; fifth pulse, 87 ± 8.8 and 176 ± 7.9 nM).
Bath-application of ryanodine and CPA did not affect the resting [Ca2+]i, either at the dendritic or the somatic level. However, in the presence of ryanodine and CPA, the [Ca2+]i elevations induced by cell depolarization were consistently increased (see Fig. 1C); during the first transient, somatic [Ca2+]i was 218 ± 4.4 nM and dendritic was 328 ± 6.2 nM. This result is likely to be due to the more depolarized membrane potential observed in the presence of these drugs during the depolarizing pulses (see Fig. 1C). Accordingly, by reducing the intensity of the depolarizing current pulses to reach membrane potentials similar to those obtained at the peak of the control pulses, the amplitude of the [Ca2+]i transients did not differ from those measured under control conditions (data not shown). Moreover, both these drugs caused a progressive decrement in the amplitude of the [Ca2+]i transients evoked by cell depolarization (see Figs. 1C, 2C). This decrement of the amplitude of [Ca2+]i transients was observed even at lower stimulus intensities, producing [Ca2+]i transients similar to those measured under control conditions.
Expressed as a percentage of control, assumed to be the [Ca2+]i value reached by the first transient, in the presence of ryanodine, the second transient was 75% ± 13% of control in the dendritic region and 89% ± 15% in the soma, the third was 65% ± 16% and 82% ± 14%, the fourth was 48% ± 21% and 71% ± 7%, and the fifth was 44% ± 16% and 65% ± 9% (see Figs. 1, 2; n = 11; P < 0.005). Similarly, in CPA, the [Ca2+]i transients evoked by the current pulses were progressively smaller in amplitude (see Fig. 2, n = 12; P < 0.005): during the second pulse, the [Ca2+]i was 65% ± 8% of control in the dendrites and 81% ± 16% in the soma; the respective figures were 55% ± 4% and 70% ± 21% for the third pulse, 49% ± 11% and 51% ± 20% for the fourth pulse, and 44% ± 11% and 48% ± 20% for the fifth pulse.
A pharmacologic approach was also tested to evoke [Ca2+]i release from ryanodine-sensitive internal pools. However, bath-applied caffeine (10 to 20 mmol/L) failed to affect either resting membrane potential or basal [Ca2+]i (n = 5; not shown). The lack of effect observed with caffeine may be explained, as recently suggested (Garaschuk et al., 1997), by the slow bath-application technique used in the present work.
Effect of CPA and ryanodine on OGD-induced responses in LA cells
Recently we have characterized the time course of the electrical and [Ca2+]i changes occurring during OGD in LA interneurons (Pisani et al., 1999). Approximately 35% of the [Ca2+]i rise during OGD in LA cells was ascribed to an influx of Ca2+ from the extracellular space through high-voltage activated Ca2+ channels. The observed membrane hyperpolarization was Ca2+-dependent, because it was almost completely blocked by intra-cellular injection of BAPTA and by bath-applied charybdotoxin, a blocker of Ca2+-dependent K+ channels. LA interneurons responded to combined OGD with a membrane hyperpolarization whose magnitude was directly related to the time of energy deprivation. We used an exposure time to OGD ranging from 2 to 6 minutes, which was shown to be sufficient to generate significant changes both in membrane potential and [Ca2+]i levels under our experimental conditions (Pisani et al., 1999). Compared with the control responses to OGD (see Fig. 3A, B), in CPA (30 μmol/L, 10 to 20 minutes preincubation), both the OGD-induced membrane hyperpolarization and the [Ca2+]i elevation were significantly reduced: the membrane hyperpolarization was 56% ± 8% of control; the concomitant [Ca2+]i increase was 51% ± 9% and 60.8% ± 5.7% of control in the cell body and in the dendrites, respectively (see Figs. 3C, 3D, 4; n = 14; P < 0.005). The inhibitory effect exerted by CPA was reversible: as shown, after 30 to 60 minutes of washout of the drug, the OGD-induced changes were comparable to the control ones (see Figs. 3E, 3F). Likewise, in the presence of ryanodine (20 μmol/L, 20 to 30 min), the membrane hyperpolarization and the [Ca2+]i rise caused by OGD were largely decreased (see Fig. 4; 64% ± 4% of control membrane hyperpolarization; 48.2% ± 4% somatic [Ca2+]i; 60% ± 5.3 dendritic [Ca2+]i; n = 11; P < 0.005). In this case, the inhibition produced by ryanodine did not reverse, even after 60 to 90 minutes of washout.
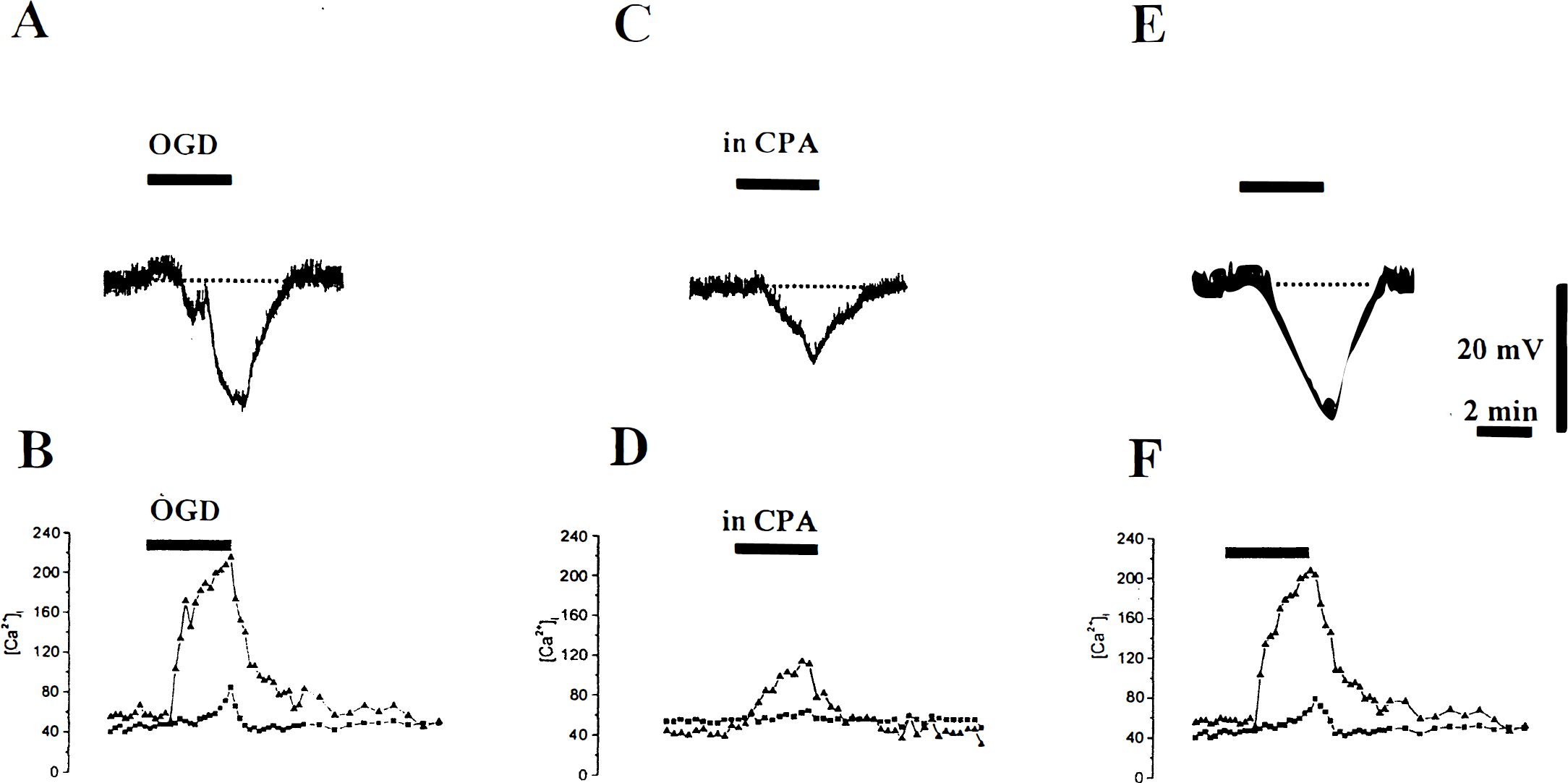
Cyclopiazonic acid (CPA) reversibly reduces the membrane hyperpolarization and intracellular calcium concentration ([Ca2+]i) increase induced by oxygen/glucose deprivation. Membrane hyperpolarization and the [Ca2+]i increase both in the soma ▪ and in the dendrites ▴ induced by 3 minutes of oxygen/glucose deprivation (
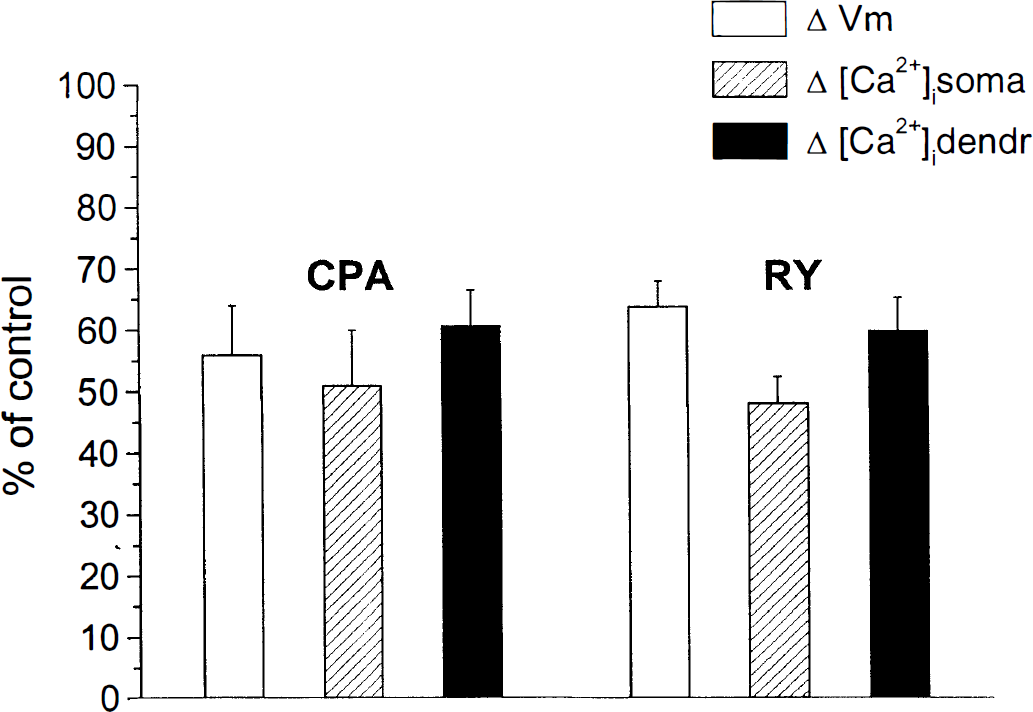
Bar histograms summarizing the inhibitory action of cyclopiazonic acid (CPA) and ryanodine on the membrane hyperpolarization and intracellular calcium concentration ([Ca2+]i) increase induced by oxygen/glucose deprivation (OGD). The vertical bars show the pharmacologic effects of CPA and ryanodine on membrane hyperpolarization (ΔVm) and somatic and dendritic changes in [Ca2+]i caused by OGD. Values are expressed as percentage of change (mean ± SD, at least eight independent observations for each drug) of OGD-induced alterations recorded under control conditions.
DISCUSSION
In this work we extended our previous observations on the effects of combined OGD in LA striatal interneurons. In particular, we tried to verify the hypothesis of involvement of internal Ca2+ stores as a source of [Ca2+]i during OGD. We demonstrated that [Ca2+]i released from ryanodine-sensitive intracellular stores may either amplify the [Ca2+]i signals evoked in physiologic conditions, such as during membrane depolarization, or it may boost the [Ca2+]i rise occurring in pathophysiologic circumstances, such as OGD.
[Ca2+]i release from internal stores in LA interneurons
The key role of Ca2+ signaling in neuronal functioning is well established (Berridge, 1998; Miller, 1991). Intra-cellular Ca2+ stores contribute to the dynamics of Ca2+ signaling, regulating [Ca2+]i homeostasis and thereby exerting a profound influence on the excitability of neurons (Berridge, 1998; Garaschuk et al., 1997). In different neuronal subtypes, Ca2+ entry through voltage-gated Ca2+ channels was shown to induce release of Ca2+ from ryanodine-sensitive pools (Friel and Tsien, 1992; Hua et al., 1993; Llano et al., 1994; Usachev and Thayer, 1997). In the present work we have shown that depolarization-evoked [Ca2+]i transients are affected by depleting ryanodine-sensitive stores. The observation that under control conditions the [Ca2+]i transients induced by depolarizing current steps were homogeneous in amplitude suggests that even at rest LA cells have a releasable pool of Ca2+, which is rapidly restored after a sustained membrane depolarization. Thus, the inhibitory effect on the amplitude of [Ca2+]i signals observed in the presence of ryanodine indicates that release from ryanodine-sensitive Ca2+ stores in LA interneurons occurs during cell depolarization and firing activity, contributing to the maintenance of steady levels of [Ca2+]i in the cytosol, which may be critical for information processing, as shown for other neuronal subtypes (Llano et al., 1994; Usachev and Thayer, 1997). Another explanation for the decremental response observed in the presence of ryanodine may lie in its complex mode of action, which is both dose-dependent and use-dependent. The latter property would allow exogenous ryanodine to bind more ryanodine receptors once Ca2+ release from internal stores is triggered by Ca2+ influx from the extracellular space. In addition, the analogous result obtained with CPA suggests that this releasable pool of Ca2+ is maintained by a continuous uptake of Ca2+ into ryanodine-sensitive stores operated by the sarcoendoplasmic reticulum Ca2+ pump. Variations of [Ca2+]i levels are crucial for regulating neuronal excitability.
A distinctive feature of LA cells is a prominent, long-lasting AHP after spiking activity that prevents the cell from firing continuously (Kawaguchi, 1993; Wilson et al., 1990) and thus regulates the firing rate of these cells. AHPs have been shown to arise from the opening of Ca2+-activated K+ conductances. In particular, the slow, long-lasting component of AHP seems to be generated by a K+ conductance activated by [Ca2+]i released from ryanodine-sensitive pools (Berridge, 1998). Indeed, the evidence that both ryanodine and CPA were able to reduce not only the AHP after spike discharge but also the accommodation rate during depolarizing current pulses indicates that ryanodine-sensitive Ca2+-activated K+ conductances, active at depolarized potentials, promote firing accommodation, hence limiting cell excitability.
The finding that the inhibitory effect exerted on [Ca2+]i transients by ryanodine and CPA was more evident at the dendritic recording sites is in accordance with recent immunohistochemical findings showing the presence of intense labeling for the cardiac form of ryanodine receptors in striatal LA cholinergic interneurons; indeed, at the subcellular level, extensive labeling was detected in dendrites (Martone et al., 1997). Conversely, these cells exhibit virtually no labeling for inositol-3-trisphosphate (IP3) receptors (Martone et al., 1997).
Role of ryanodine-sensitive [Ca2+]i stores during OGD
Under conditions of energy limitations, ion homeostasis in neurons is rapidly lost. It is accepted that even brief periods of disruption of energy supply can lead to permanent damage (Erecinska and Silver, 1994). A large body of experimental evidence supports the hypothesis that disturbances of the functioning of the endoplasmic reticulum play a key role in determining cell damage under a variety of pathophysiologic conditions (Paschen and Doutheil, 1999). Impairment of endoplasmic reticulum Ca2+ homeostasis has been established in experimental models of cerebral and myocardial ischemia (Fukumoto et al., 1991; Higashi et al., 1994; Paschen et al., 1998), apoptosis (Paschen and Doutheil, 1999; Wei et al., 1998), excitotoxicity (Mody and MacDonald, 1995), and degenerative disorders (Beal, 1995; Mattson, 1994). In particular, it has been proposed that dysfunction of the endoplasmic reticulum Ca2+ homeostasis would result in a common cascade of events involving suppression of global protein synthesis and activation of stress gene expression, leading ultimately to cell damage (Paschen and Doutheil, 1999). We have recently reported that during OGD these cells hyperpolarize, and that this event is directly related to an increase in [Ca2+]i levels, which is partially due to an influx through high-voltage activated Ca2+ channels (Pisani et al., 1999). The findings that the reversal potential for the membrane hyperpolarization was close to the K+ equilibrium potential, and that blockade of the hyperpolarization was observed in the presence of both BAPTA or charybdotoxin, a BK K+ channel blocker, suggest that during OGD a Ca2+-dependent K+ conductance is activated by Ca2+ influx. The data obtained in this work support the involvement of ryanodine-sensitive [Ca2+]i stores in the response of LA cells to energy deprivation. Indeed, the inhibitory effect on both membrane hyperpolarization and [Ca2+]i increase observed in CPA or ryanodine indicates that during OGD the initial Ca2+ entry through high-voltage activated channels is amplified by Ca2+ release from ryanodinesensitive pools. Therefore, in contrast to the view that ryanodine-sensitive stores act essentially as buffers, contributing to the clearance of [Ca2+]i, is the present evidence that, at least in these neuronal subtype, these stores amplify the incoming Ca2+ signals independently on the situation triggering them.
Functional implications
The striatum is particularly vulnerable to ischemic and excitotoxic damage. Interestingly, striatal neuronal subtypes express a differential susceptibility: LA interneurons have been shown to be exceptionally resistant to such pathophysiologic conditions (Ferrante et al., 1985; Francis and Pulsinelli, 1982). Currently, however, the cellular mechanisms underlying this peculiar feature have not been identified. The efficacy in buffering excessive [Ca2+]i accumulation might be one crucial factor in this regard. Surprisingly, it has been demonstrated that striatal LA interneurons are devoid of any known Ca2+-binding protein (Bennett and Bolam, 1993; Kita et al., 1990), ruling out a possible contribution of any of these proteins as endogenous Ca2+ buffers in these cells in response to energy deprivation. Moreover, the present results support an involvement of ryanodine-sensitive stores during OGD as amplifiers rather than buffers of the Ca2+ signals. However, the hyperpolarizing response under conditions of energy limitation may be interpreted as a defense mechanism, allowing the cell to keep its membrane potential distant from the firing threshold, thereby limiting ion overload. In conclusion, further work is required to address the role of other potential endogenous buffers to clarify the peculiar behavior of these striatal neurons.
Footnotes
Acknowledgments
The authors thank Drs. P. Gubellini and E. Scarnati for helpful comments and Mr. M. Tolu for his excellent technical support.