
Abstract
Thioredoxin is a small, multifunctional protein with a redox-active disulfide/dithiol in the active site. Thioredoxin plays several important biologic roles both in intracellular and extracellular compartments with its redox-regulating and reactive oxygen intermediates scavenging activities. We assayed the seizure response and the excitotoxic hippocampal injury in thioredoxin transgenic and wild-type C57BL/6 mice. Seizure score after kainic acid treatment was significantly lower in thioredoxin transgenic mice. Seven days after kainic acid administration, the damage in the hippocampal CA1 and CA3 regions was significantly attenuated in thioredoxin transgenic mice. Thioredoxin and redox regulation play an important role in excitotoxic brain damage.
Excessive activation of excitatory amino acid receptors is thought to be involved in several neurodegenerative diseases and ischemic brain injury (Beal, 1992; Choi et al., 1989). Receptors for excitatory amino acids are widely distributed in the brain. They include specific receptors for N-methyl-D-aspartate (NMDA), quisqualate, kainic acid (KA), and α-amino-3-hydroxy-5-methooisoxasole-4-propionate (AMPA) and metabotropic receptors. KA binds to both KA and AMPA receptors (London and Coyle, 1979). A massive release of the excitatory neurotransmitter glutamate acting on the NMDA receptor and other subtype receptors also results in free radical production (Ayata et al., 1997; Lafon-Cazel et al., 1993). Oxidative stress has been thought to be one of the mechanisms of KA-induced neuronal damage (Cheng and Sun, 1994). Stimulation of excitatory amino acid receptors activates membrane voltage-sensitive calcium channels, with subsequent Ca2+ influx into the cytoplasmic compartment leading to the activation of several enzymes that generate free radicals. These include phospholipases, nitric oxide synthesis, and calcium-dependent proteases. Free radicals generated by these enzymes are probably involved in KA-induced excitotoxicity because KA-induced neuronal damage is attenuated by superoxide dismutase (SOD) and other antioxidants (Hirata and Cadet, 1997; Regan and Guo, 1999).
Thioredoxin is a small multifunctional protein with a redox-active disulfide/dithiol in the conserved active site sequence: -Cys-Gly-Pro-Cys- (Holmgren, 1985, Holmgren, 1989). Increasing evidence has indicated that the cellular redox status modulates various aspects of cellular events, including proliferation and apoptosis. Human thioredoxin has been identified as a highly expressed cytokine-like factor in activated T and B cells that up-regulates the interleukin-2 and interleukin-2 receptor α-chain (Tagaya et al., 1988, Tagaya et al., 1989; Yodoi and Uchiyama, 1992). Thioredoxin regulates various intracellular molecules via thiol redox control involving transcription factors such as nuclear factor-κB (NF-κB), activator protein 1 (AP-1), myb, redox factor 1, and mitogen activated protein kinase MAPK). (Abate et al., 1990; Hirota et al., 1997; Meyer et al., 1993; Saitoh et al., 1998Schenk et al., 1994). Further, thioredoxin is a scavenger of reactive oxygen intermediates (ROI) (Mitsui et al., 1992), and recombinant thioredoxin has protective activity against cytotoxicity, in which the generation of ROI seems to be involved in the cytotoxic mechanism (Nakamura et al., 1994). Moreover, recently we have reported that ischemic brain injury was attenuated in thioredoxin transgenic mice (Takagi et al., 1999). These data suggest that thioredoxin plays several important biologic roles both in intracellular and extracellular compartments with its ROI-scavenging activity.
We hypothesized that endogenous thioredoxin functions as a regulator of excitotoxic injury through its ROI-scavenging and redox-regulating activities.
METHODS
Thioredoxin transgenic mice
Human thioredoxin cDNA was inserted between the β-actin promoter and the β-actin terminator and was used to generate transgenic mice. The pronuclei of fertilized eggs from hyperovulated C57BL/6 mice were microinjected with this DNA construct (Takagi et al., 1999). Three lines (β Ac-ADF-2, β Ac-ADF-5, β Ac-ADF-10) of transgenic mice were confirmed by Southern blot analysis. Lysates prepared from various tissues were analyzed by Western blot analysis using antimouse and antihuman thioredoxin antibody. Among the three established lines of thioredoxin transgenic mice, the line that expressed the highest level of the human thioredoxin protein (β Ac-ADF-2) was used for further experiments. The presence of the thioredoxin transgene was also confirmed by reverse transcription polymerase chain reaction analysis at the termination of the ischemic experiments. In some animals, human thioredoxin expression was confirmed by Western blot analysis.
KA treatment
Mice weighing 30 to 40 g each were used for the experiments. KA (Sigma, St. Louis, MO, U.S.A.) was diluted in phosphate-buffered saline at a concentration of 5 mg/mL. All mice were injected intraperitoneally with KA (20 mg/kg). Animals were observed for 1 hour after the injection. The mice were perfused transcardially with phosphate-buffered saline followed by a cold fixative solution (4% paraformaldehyde and 0.1 mol/L phosphate buffer) 7 days after KA administration. The brains were removed and cut into 20-μm-thick coronal sections and stained with cresyl violet to assess the neuronal damage. Nissle-positive undamaged neurons were counted in hippocampal CA1 and CA3 regions (three ×45 fields per section and three sections per brain). Cell counts were performed without knowledge of the genotype or treatment history of the mice.
Seizure score
Ten thioredoxin transgenic and 10 wild-type mice were observed for 1 hour after KA injection. Seizures were scored as previously described (Yang et al., 1997): 1 = arrest of motion; 2 = myoclonic jerks of the head and neck, with brief twitching movement; 3 = unilateral clonic activity; 4 = bilateral forelimb tonic and clonic activity; 5 = generalized tonic-clonic activity with loss of postural tone, including death from continuous convulsions.
Statistical analysis
Results are expressed as mean ± standard deviation. Mean values were compared using one-way analysis of variance. Significance was taken as P < 0.05.
RESULTS
Thioredoxin transgenic mice
The characterization and distribution of human thioredoxin have already been reported (Takagi et al., 1999). Human thioredoxin expression was confirmed by Western blot analysis (Fig. 1). Expression of total thioredoxin was five times greater in the brains of thioredoxin transgenic mice compared with wild-type C57BL/6 mice. Human thioredoxin distributed in pyramidal neurons in the hippocampus and cortex, vascular endothelial cells, and glial cells (Takagi et al., 1999). Cresyl violet staining revealed no major difference in cellular structure between thioredoxin transgenic and wild-type mice.
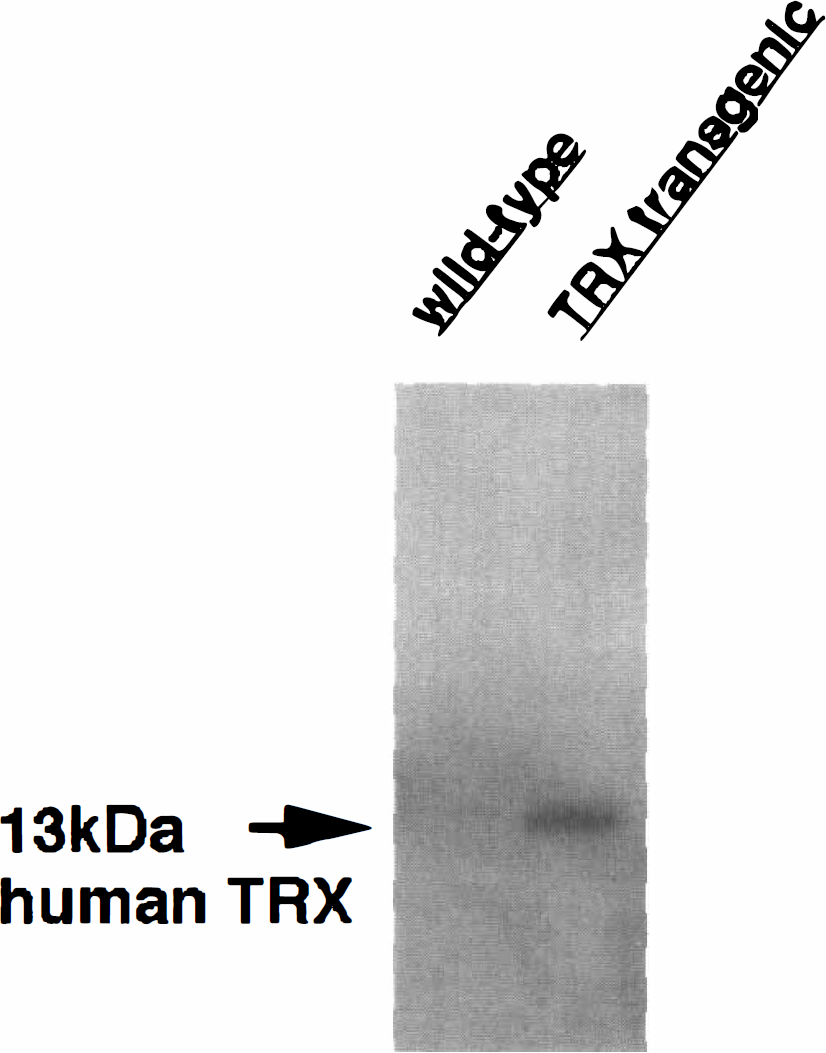
Expression of human thioredoxin in wild-type C57BL/6 and thioredoxin transgenic mice. Western blot analysis showed that human thioredoxin was expressed as a single band of 13 kDa.
Attenuation of KA-induced neuronal damage in thioredoxin transgenic mice
Seven of the 10 thioredoxin transgenic mice and all 10 wild-type C57BL/6 mice exhibited severe seizures. The mean seizure score was significantly lower in thioredoxin transgenic mice than in wild-type mice both 30 and 45 minutes after treatment (Fig. 2). Seven days after KA treatment, hippocampal neuronal damage was assessed by cresyl violet staining (n = 13 per group). Two of the 15 wild-type mice and 1 of the 14 thioredoxin transgenic mice died before the assessment. There were significantly more undamaged neurons in the hippocampal CA1 and CA3 regions in thioredoxin transgenic mice than in wild-type mice (Fig. 3). Hippocampal neuronal damage was attenuated in thioredoxin transgenic mice compared with wild-type mice.
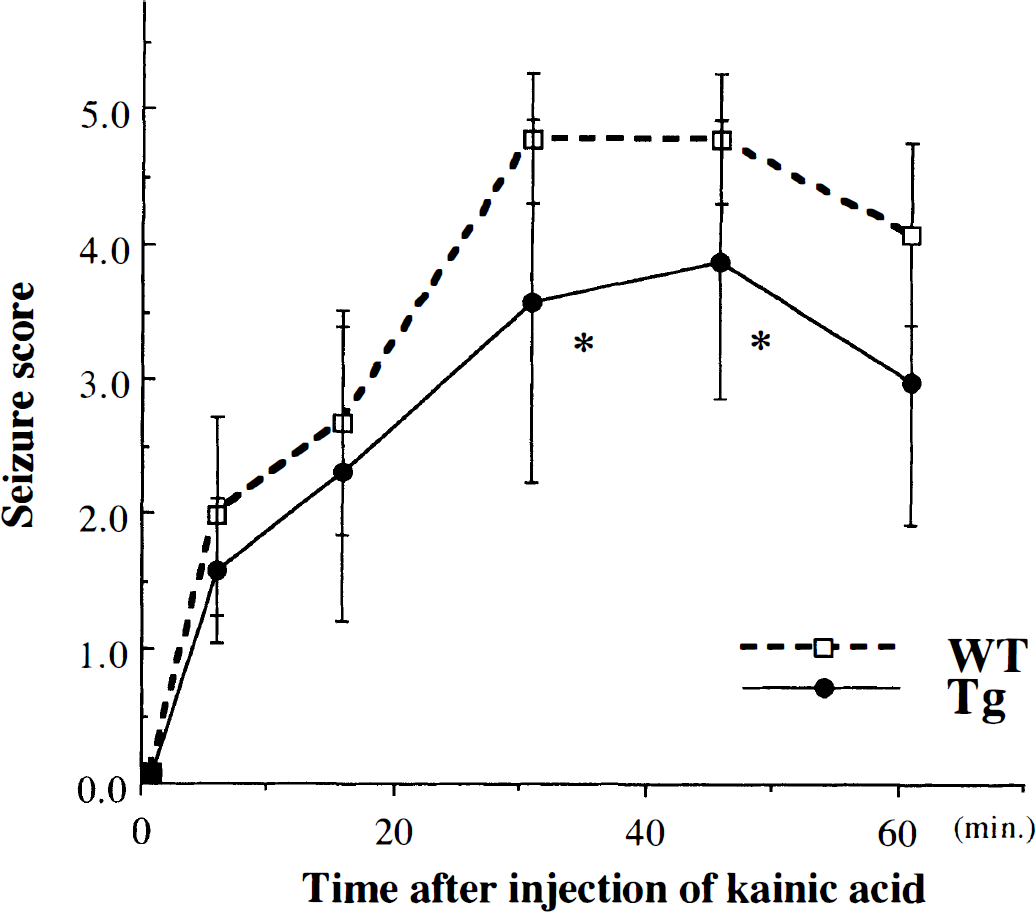
Seizure responses of 10 wild-type C57BL/6 mice and 10 thioredoxin transgenic mice to intraperitoneal injection of kainic acid (20 mg/kg). Seizures were scored as described in text. At 30 and 45 minutes after the treatment, the seizure score was significantly lower in thioredoxin transgenic mice than in wild-type mice (*P < 0.05).
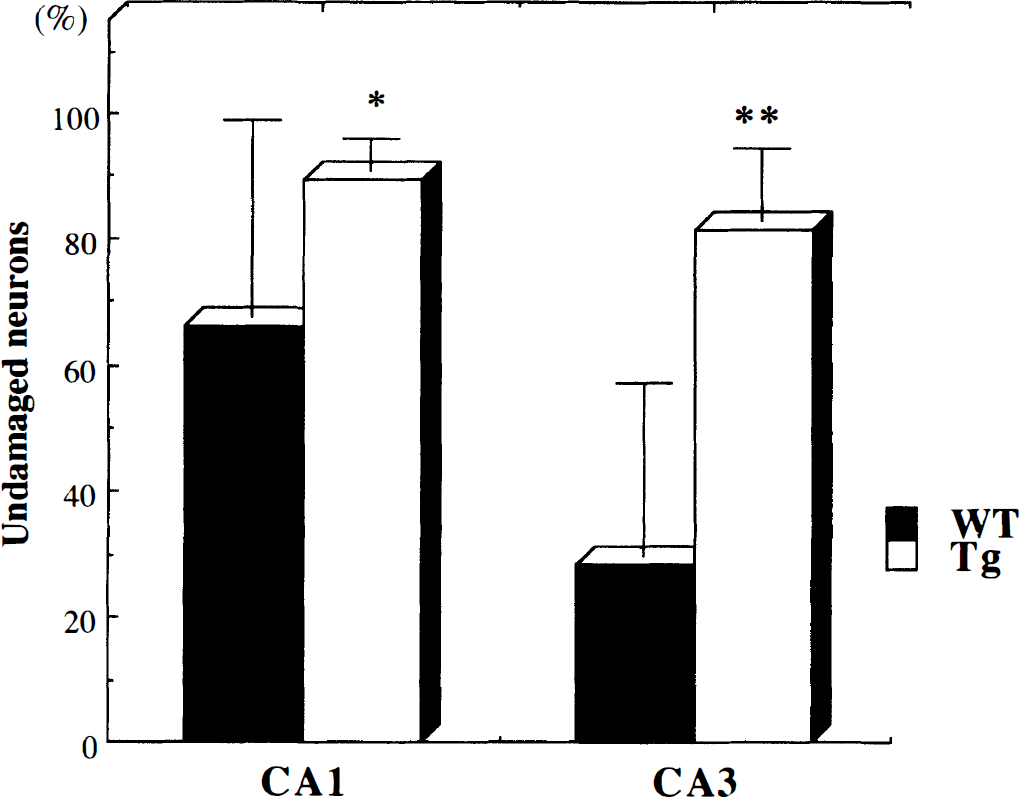
Hippocampal neuronal damage is attenuated in thioredoxin transgenic mice. Kainic acid was administered intraperitoneally to wild-type C57BL/6 mice and to transgenic mice. Seven days later, 13 wild-type and 13 transgenic mice were killed and the damage to CA1 and CA3 neurons was quantified. Values are mean ± SD. (*P < 0.05, **P < 0.01).
DISCUSSION
Redox regulation of intracellular molecules is an interesting and important issue. Thioredoxin is a small, ubiquitous protein with two redox-active half-cystine residues in the active center. In the central nervous system, recombinant thioredoxin has a neuroprotective effect on murine primary cultured neurons obtained from the striatum and cortex (Hori et al., 1994). Thioredoxin functions as a neurotropic factor for central cholinergic neurons (Endoh et al., 1993) and was upregulated in mechanical brain injury (Lippoldt et al., 1995). Previously, we reported that thioredoxin levels are decreased in the ischemic core and increased in the penumbra ischemic region during permanent middle cerebral artery occlusion in rats (Takagi et al., 1998). Moreover, ischemic brain injury was attenuated in thioredoxin transgenic mice (Takagi et al., 1999). Together, these findings indicate that thioredoxin and the redox system modulated by thioredoxin have a function in the cellular defense against oxidative stress in neurons as well as in other cell types and may play a role during brain injury.
This is the first demonstration of attenuated KA-induced hippocampal damage in thioredoxin transgenic mice. Because thioredoxin is a redox regulatory protein, the redox-sensitive signal pathway must be involved in KA-induced cell death. The mechanisms of KA-induced neuronal damage are controversial. Kondo and colleagues (1997) demonstrated that although KA causes hippocampal damage, there was no difference between transgenic mice overexpressing CuZn SOD and wild-type mice. In this report, prolonged expression of c-fos and HSP70 was recognized in CuZn SOD transgenic mice. Lafon-Cazal (1993) and Dugan (1995) and their associates found that superoxide radicals are produced by the activation of NMDA receptor but not kainate receptors. However, several investigators have shown that KA-induced cell damage involves free radical production both in vitro and in vivo (Ayata et al., 1997; Gary et al., 1998; Shultz et al., 1995). Several antioxidants can attenuate KA-induced hippocampal cell damage (Gary et al., 1998; Hirata and Cadet, 1997; Regan and Guo, 1999; Uz et al., 1996). Our results indicated that the mechanism of attenuation of neuronal injury in thioredoxin transgenic mice must be due to modulation of redox-sensitive signals.
The C-Jun N-terminal kinase (JNK)-c-Jun dependent pathway may be responsible for the neuroprotective effect in thioredoxin transgenic mice. Recently, JNK activation was reported to be involved in neuronal apoptosis by KA (Mielke et al., 1999). In the adult rat brain, disruption of the JNK-3 locus protected hippocampal neurons against excitotoxic neuronal cell death (Yang et al., 1997). However, our preliminary immunohistochemical data indicated that JNK, phospho c-Jun, and c-Jun early activation after KA injection did not differ between thioredoxin transgenic and wild-type mice (unpublished data). This observation indicated that a different pathway may participate in neuroprotection during KA-induced hippocampal injury. JNK activation does not inevitably lead to apoptosis because JNKs are expressed in the untreated intact rat brain and are activated after acquisition of novel information (Xu et al., 1997). Previous studies indicated that neuronal apoptosis could occur without JNK-AP-1 activation (Liu et al., 1996; Natoli et al., 1997). One candidate is p38, which belongs to be MAPK family and has been reported to be activated by KA. Another MAPK family member, extracellular signal-regulated kinase, was also activated after KA injection and is another candidate (Mielke et al., 1999). Thioredoxin has been reported to inhibit apoptosis signal-regulating kinase-1 (a MAPK kinase enzyme) directly and to prevent apoptosis (Saitoh et al., 1997). The downstream molecule of these factors has not been clearly defined. The role of ATF-2 or TRAF except for c-Jun and AP-1 has been widely discussed (Goillot et al., 1997; Liu et al., 1996; Natoli et al., 1997). NF-κB plays an important role in neuronal apoptosis. Thioredoxin inhibits NF-κB activation. The effect in thioredoxin transgenic mice may be due to the NF-κB-related pathway. NF-κB has been reported to play a role in neuronal apoptosis without JNK activation (Liu et al., 1996; Natoli et al., 1997). Further investigation of these factors' activation in thioredoxin transgenic and wild-type mice is needed.
In summary, KA-induced hippocampal injury was attenuated in thioredoxin transgenic mice. Thioredoxin and redox-regulating systems must play an important role in neuronal death during neurodegeneration and ischemia.
Footnotes
Acknowledgment
The authors thank Ms. Yoko Kanekiyo for secretarial help.