
Abstract
Several groups have provided evidence that positron emission tomography (PET) and single-photon emission computed tomography (SPECT) neuroreceptor imaging techniques might be applied to measure acute fluctuations in dopamine (DA) synaptic concentration in the living human brain. Competition between DA and radioligands for binding to D2 receptor is the principle underlying this approach. This new application of neuroreceptor imaging provides a dynamic measurement of neurotransmission that is likely to be informative to our understanding of neuropsychiatric conditions. This article reviews and discusses the body of data supporting the feasibility and potential of this imaging paradigm. Endogenous competition studies performed in rodents, nonhuman primates, and humans are first summarized. After this overview, the validity of the model underlying the interpretation of these imaging data is critically assessed. The current reference model is defined as the occupancy model, since changes in radiotracer binding potential (BP) are assumed to be directly caused by changes in occupancy of D2 receptors by DA. Experimental data supporting this model are presented. The evidence that manipulation of DA synaptic levels induces change in the BP of several D2 radiotracers (catecholamines and benzamides) is unequivocal. The fact that these changes in BP are mediated by changes in DA synaptic concentration is well documented. The relationship between the magnitude of BP changes measured with PET or SPECT and the magnitude of changes in DA concentration measured by microdialysis supports the use of these noninvasive techniques to measure changes in neurotransmission. On the other hand, several observations remain unexplained. First, the amphetamine-induced changes in the BP of D2 receptor antagonists [123I]IBZM and [11C]raclopride last longer than amphetamine-induced changes in DA extracellular concentration. Second, nonbenzamide D2 receptor antagonists, such as spiperone and pimozide, are not affected by changes in DA release, or are affected in a direction opposite to that predicted by the occupancy model. Similar observations are reported with D1 radiotracers. These results suggest that the changes in BP following changes in DA concentration might not be fully accounted by a simple occupancy model. Specifically, the data are reviewed supporting that agonist-mediated receptor internalization might play an important role in characterizing receptor-ligand interactions. Finally, it is proposed that a better understanding of the mechanism underlying the effects observed with benzamides is essential to develop this imaging technique to other receptor systems.
Keywords
Over the last decade, numerous studies have provided data suggesting that, under specific conditions, in vivo neuroreceptor imaging with positron emission tomography (PET) and single-photon emission computed tomography (SPECT) could be used to measure acute fluctuations in synaptic concentration of neurotransmitters. Competition between endogenous transmitters and radioligands for binding to neuroreceptors is the principle underlying this technique: changes in transmitter synaptic concentration translate in changes in transmitter receptor occupancy that can be detected as changes in the binding potential (BP) of the radioligand. This promising new application of neuroreceptor imaging has the potential to enable direct measurement of synaptic transmission in the living brain and correlations of these dynamic measurements with behaviors and symptoms. This idea has been applied mostly to the measurements of changes in dopamine (DA) concentration in the milieu surrounding DA D2 receptors.
The idea to apply competition techniques and PET to the assessment of DA synaptic level was proposed as early as 1984 (Friedman et al., 1984). Nonetheless, endogenous competition studies became a major focus of PET and SPECT research only during the last decade. A possible reason for this delay is that during the late 1970s and most of the 1980s, the butyrophenone [3H] spiperone or its methylated analog [11C]N-methyl-spiperone ([11C]NMSP) were the most widely used radioligands to label D2 receptors in vivo (Leysen et al., 1978; Wagner et al., 1983; Lyon et al., 1986). Studies performed with these ligands did not support the feasibility of this application. The development of substituted benzamides such as [11C]raclopride and [123I]IBZM as imaging agents (Ehrin et al., 1985; Kohler et al., 1985; Kung et al., 1988a, b ) was essential in transforming this theoretical idea into a valuable research tool.
The development of the endogenous competition concept in the brain imaging field also is linked to the controversies and debates that followed publications of discrepant results regarding the density of D2 receptors in schizophrenia. Using [11C]NMSP, Wong et al. (1986b) reported a large increase in the density of D2 receptors in patients with schizophrenia; while using [11C]raclopride, Farde et al. (1990) reported no change. In two noted communications, Seeman et al. (1988, 1989) argue that this discrepancy might arise from a different “sensitivity” of [11C]raclopride and [11C]NMSP binding to endogenous competition by DA. This hypothesis played an important role in bringing the endogenous competition concept to the attention of the imaging field.
This interest translated in numerous investigations that characterized in detail the phenomenon of endogenous competition between DA and various D2 receptor radiotracers. The use of this technique to measure DA transmission has been extensively validated and was successfully applied to characterize alterations of DA transmission in clinical conditions. On the other hand, application of the same principle to other transmission systems has not substantially emerged. The difficulties encountered in extending this idea to other receptor systems call for a critical examination of the data obtained with D2 receptors.
This line of research has emerged under a theoretical framework referred to here as the occupancy model (Fig. 1). This model predicts that challenges that increase DA synaptic concentration will result in increased occupancy of D2 receptors by DA, and reduced availability of D2 receptors for binding of the radiotracer. On the other hand, manipulations that decrease DA synaptic concentration will reduce D2 receptor occupancy by DA and increase D2 receptor availability to radiotracer binding. This article first reviews studies in rodents, nonhuman primates, and humans that evaluated in vivo competition between DA and radiolabeled D2 and D1 receptor ligands. After presenting these data, this report addresses several issues pertinent to the validity of the theoretical framework (i.e., the occupancy model) supporting the interpretation of these results. Results incongruent with the occupancy model receive special attention. This article concludes by examining the limitations of the occupancy model and speculating about other mechanisms that might be involved in altering radioligand binding after acute fluctuations of synaptic neurotransmitter levels.
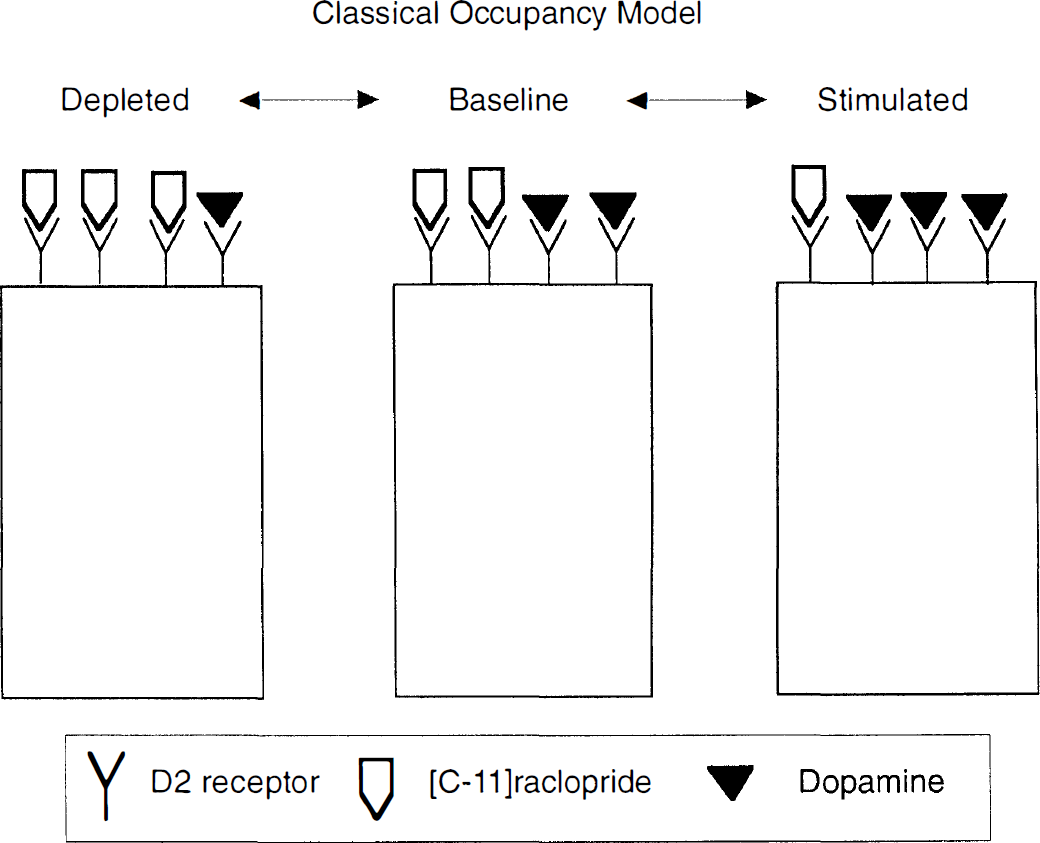
Schematic representation of the classic occupancy model adopted to explain the increase and decrease of [11C]raclopride binding following depletion or stimulation, respectively, of dopamine concentration in the vicinity of D2 receptors. This model postulates that changes in [11C]raclopride binding potential are directly related to the changes in occupancy of D2 receptors by dopamine. The figure is misleading inasmuch as [11C]raclopride, at tracer dose, occupies only a fraction of the available receptors. The effective binding of [11C]raclopride is related to the binding potential, which in turn is affected by the presence of dopamine in a complex manner, since dopamine affects both the number and affinity of available sites.
LITERATURE REVIEW
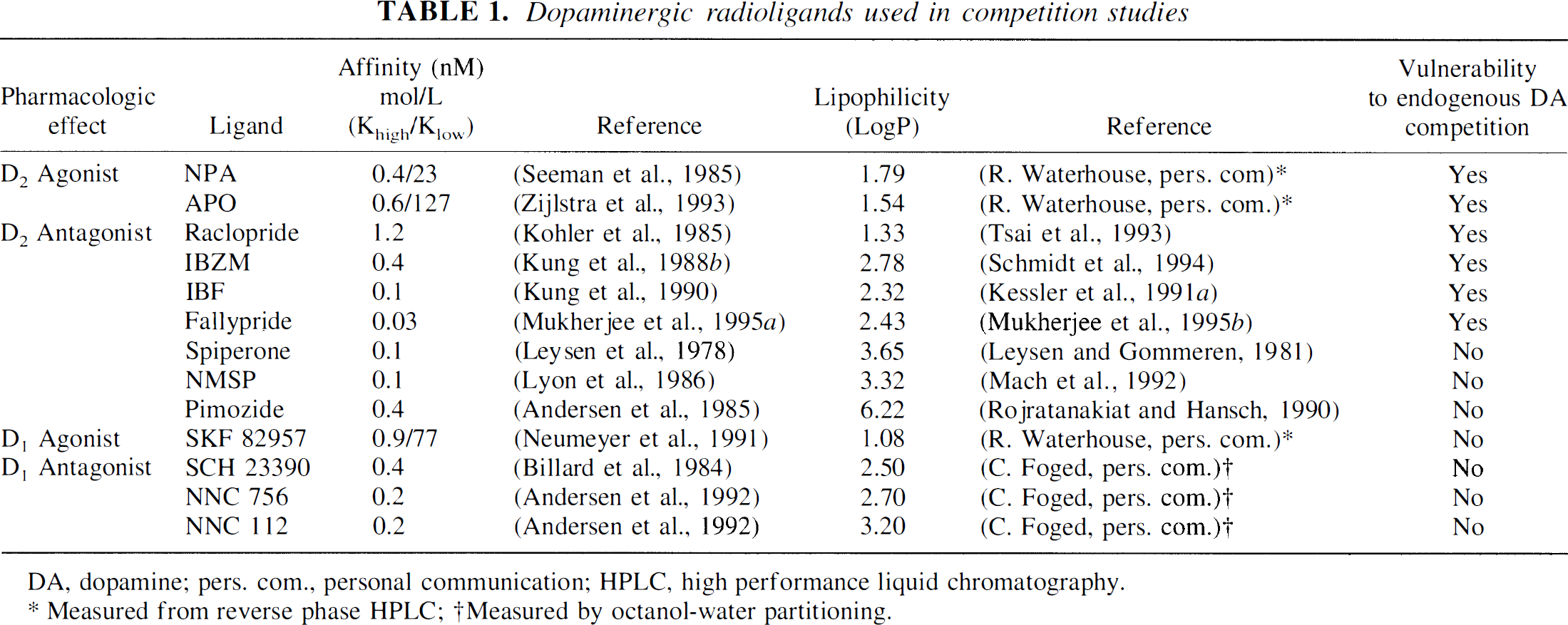
A variety of D2 and D1 receptor radioligands have been used in experiments evaluating the impact of manipulation of synaptic DA on radioligand binding in vivo. D2 receptor radioligands include the agonists propylnorapomorphine (NPA) and apomorphine (APO), and a variety of antagonists including raclopride, IBZM, IBF, fallypride, spiperone, N-methyl-spiperone (NMSP), and pimozide. D1 receptor radioligands include the agonist SKF 82957 and the antagonists SCH 23390, NNC 756, and NNC 112. Table 1 lists the affinity and lipophilicity of these agents. A representative value of Kd or logP was chosen when several values were available in the literature. For logP, values determined by the octanol-water partitioning method (shake-flask method) were selected when available.
Dopaminergic radioligands used in competition studies
DA, dopamine; pers. com., personal communication; HPLC, high performance liquid chromatography.
Measured from reverse phase HPLC
Measured by octanol-water partitioning.
Table 1 also lists the vulnerability of these tracers to endogenous DA competition, as derived from the literature review detailed later. Inspection of Table 1 rapidly reveals that neither pharmacologic effect (agonist versus antagonist), affinity, or lipophilicity are appropriate predictors of vulnerability to endogenous DA competition. Rather, this property appears to be associated to the chemical class. The in vivo binding of catecholamines (NPA and APO) and benzamides (raclopride, IBZM, IBF, fallypride) is affected by endogenous DA in a manner consistent with the occupancy model, whereas other radiotracers (butyrophenones, benzazepines) show either no change or change in the direction opposite to predictions of the occupancy model.
Studies in rodents
Table 2 lists in vivo studies in rodents (rats and mice) that measured changes in the accumulation of a variety of DA receptor radioligands after pharmacologic challenges or other manipulations expected to decrease or increase synaptic DA concentration. Studies are sorted by receptor systems, radioligands, and type of challenge (expected effect of the challenge on DA synaptic concentration, i.e., inhibition or augmentation of synaptic DA). For each study, Table 2 lists the observed effect of the challenge on radioligand binding (increase, decrease, no change), the consistency of the results with the simple occupancy model (yes, no, or paradoxical), and the reference. Challenges aimed at depleting or inhibiting DA synaptic concentration include pharmacologic administration of the vesicle-depleting agent reserpine, the neuronal impulse flow inhibitor γ-butyrolactone, and the tyrosine hydroxylase inhibitor alpha-methyl-para-tyrosine (AMPT), as well as chemical destruction of DA neurons with 6-OH-dopamine. Inhibition of DA release also was indirectly achieved by increasing GABAergic transmission (flunitrazepam, NNC 711). Direct augmentation of DA synaptic concentration was achieved with the DA releaser amphetamine, inhibitors of dopamine transporters (DAT) including methylphenidate, cocaine, buproprion, amfonelic acid, nomifensine, GBR 12909, and the DA precursor
In vivo competition studies between DA and radiotracers: Ex vivo studies in rodents
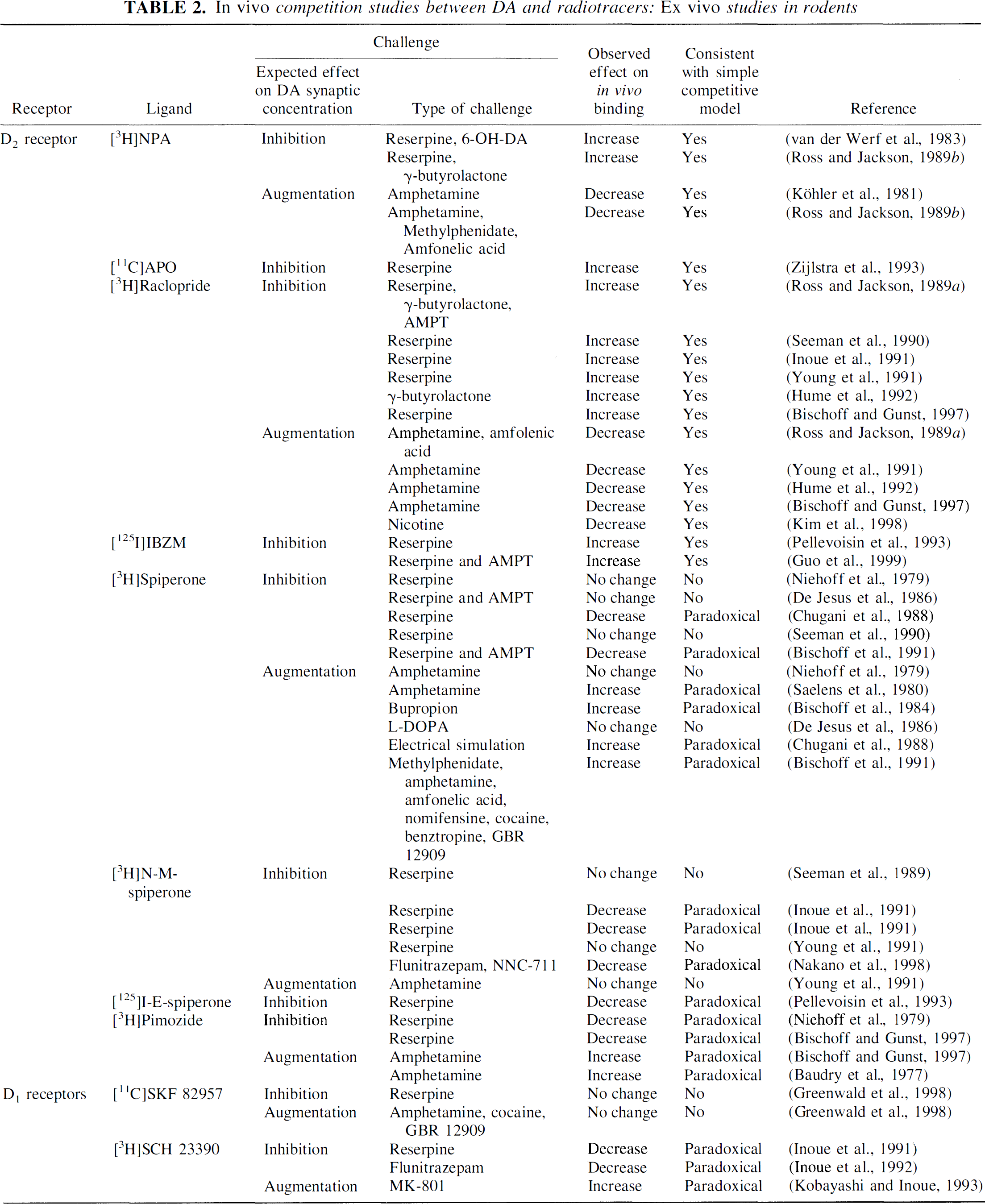
The model referred to here as the occupancy model (increase in DA synaptic levels results in decrease in tracer accumulation, and vice versa) is fully supported by data obtained with [3H]NPA, [11C]APO, [3H]raclopride, or [123I]IBZM. In contrast, data obtained with [3H]spiperone and [3H]pimozide do not support this model. In fact, most of the results obtained in rodents with these two radioligands indicate regulations in the direction opposite to predictions of the occupancy model: inhibition of DA release resulted in paradoxical decrease in radioligand-specific binding, whereas stimulation of DA release resulted in paradoxical increase in radioligand-specific binding.
Most rodent studies published during the 1970s and 1980s were performed with these “paradoxical” radiotracers (spiperone and pimozide). In 1988, the only data available to support the occupancy model were the studies of van der Werf et al. (1983) and Köhler et al. (1981) with the D2 receptor agonist [3H]NPA. That year, Chugani et al. (1988) published a seminal report proposing that the paradoxical behavior of [3H]spiperone uptake agonist challenge resulted from agonist-promoted receptor internalization, a phenomenon resulting in an effective “trapping” of [3H]spiperone in the cells.
The following year was a turning point with the publication of the elegant and influential studies by Ross and Jackson (1989a, 1989b). By performing experiments at various levels of [3H]NPA-specific activities, they established that amphetamine, amfolenic acid, and methylphenidate decreased the affinity of [3H]NPA for D2 receptors with no changes in Bmax. Reserpine and γ-butyrolactone pretreatment results in an increased affinity of [3H]NPA with no changes in Bmax. These data support the occupancy model and the competitive nature of the interaction between DA and [3H]NPA. In a companion report, the same authors presented the results of an equally elegant set of studies performed with [3H]raclopride. In contrast to results obtained with [3H]NPA, reserpine and γ-butyrolactone affected both the KD and the Bmax of [3H]raclopride. This result suggests the presence of both competitive and noncompetitive components in the interaction between [3H]raclopride and DA in vivo.
The studies of Inoue et al. (1991), Young et al. (1991), and Bischoff et al. (1997) compared modulations of [3H]raclopride and [3H]NMSP in rodents by pharmacologic challenges under comparable and carefully controlled conditions, and definitively established the differences in the behavior of these radiotracers vis-à-vis DA synaptic concentration. Opposite effects also were observed after indirect challenges. Gamma-aminobutyric acid agonists that are expected to inhibit DA release were shown to decrease [3H]spiperone binding (Nakano et al., 1998), but to increase [11C]raclopride binding (Dewey et al., 1992). The NMDA antagonists (ketamine and MK-801) increased [11C]NMSP binding (Onoe et al., 1994) but decreased [11C]raclopride binding (Breier et al., 1998; Ginovart et al., 1998; Smith et al., 1998).
Interestingly, D1 receptor radiotracers, whether agonists or antagonists, behave more like spiperone than raclopride. The uptake of the D1 receptor agonist [11C]SKF 82957 was not affected by reserpine or amphetamine (Greenwald et al., 1998). For the antagonist [3H]SCH 23390, inhibition of DA release with reserpine (Inoue et al., 1991) or flunitrazepam (Inoue et al., 1992) induced decreased radiotracer accumulation, whereas stimulation of DA release with MK-801 induced increased accumulation (Kobayashi and Inoue, 1993). Thus, like spiperone and its analog, D1 receptor radiotracers appear to show either no change or paradoxical response to acute DA manipulations.
Studies in nonhuman primates
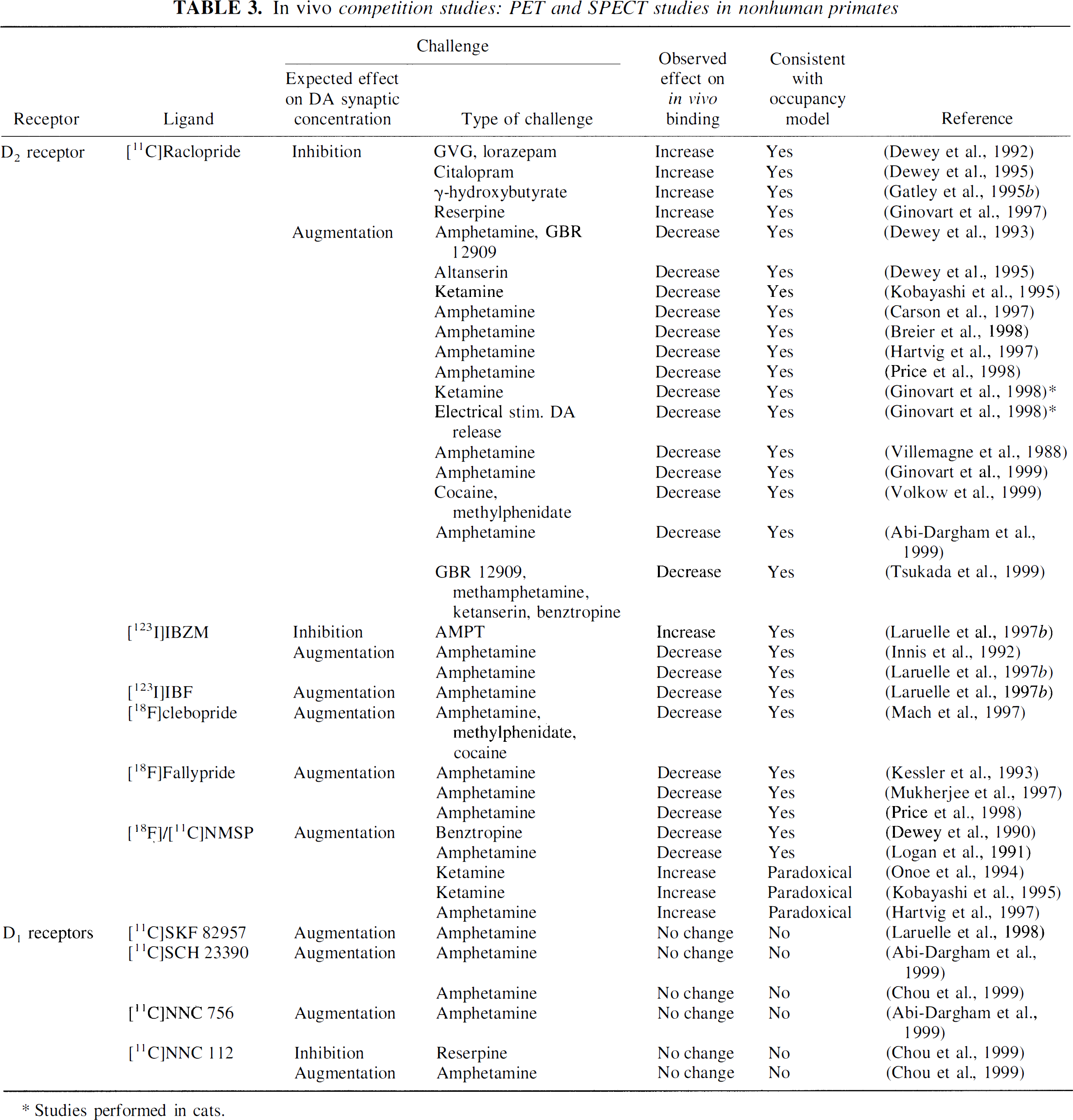
Studies performed with PET and SPECT in nonhuman primates are listed in Table 3. The first two articles reporting DA—ligand interactions with PET were performed with [18F]NMSP (Dewey et al., 1990; Logan et al., 1991) and report results consistent with the occupancy model (but inconsistent with the rodent studies reviewed earlier). In 1990, Dewey et al. (1990) reported that the anticholinergic and DA uptake inhibitor drug benztropine reduced the striatal uptake of [18F]NMSP in the baboon brain. The following year, the same group reported that amphetamine also reduced [18F]NMSP striatal uptake in the baboon brain (Dewey et al., 1991). This report was followed by a more comprehensive kinetic analysis of the same data by Logan et al. (1991).
In vivo competition studies: PET and SPECT studies in nonhuman primates
Studies performed in cats.
Starting in 1992, several of studies in primates have reported the effect of direct DA enhancing challenges on [11C]raclopride in baboon: [11C]raclopride BP was consistently shown to be reduced by amphetamine, GBR 12909, cocaine, methylphenidate, and electrical stimulation of dopaminergic neurons (Villemagne et al., 1988; Dewey et al., 1993; Carson et al., 1997; Hartvig et al., 1997; Ginovart et al., 1998; Price et al., 1998; Abi-Dargham et al., 1999; Ginovart et al., 1999; Volkow et al., 1999). Direct depletion of endogenous DA by reserpine or gamma-hydroxybutyrate increased [11C]raclopride BP (Gatley et al., 1995b; Ginovart et al., 1997). [11C]Raclopride BP was sensitive to indirect manipulation of DA transmission, and these interactions were consistent in their directionality with the occupancy model. Challenges expected to decrease DA release (by increasing gamma-aminobutyric acid and 5-hydroxytryptamine [5-HT] transmission) resulted in increased [11C]raclopride binding (Dewey et al., 1992; Dewey et al., 1995), whereas the NMDA antagonist ketamine, a drug expected to increase DA transmission, decreased [11C]raclopride binding (Ginovart et al., 1998).
Studies in primates with the raclopride analog [123I]IBZM report similar interactions. Augmentation and inhibition of DA release were reported to decrease and increase, respectively, the specific binding of [123I]IBZM in baboons (Innis et al., 1992; Laruelle et al., 1997b). Several other benzamides, such as [123I]IBF, [18F]FCP, and [18F]fallypride, also were shown to be affected by endogenous DA augmentation (Kessler et al., 1993; Laruelle et al., 1997b; Mach et al., 1997; Mukherjee et al., 1997; Price et al., 1998).
Although the first studies published in baboons were performed with [18F]NMSP and reported positive results, a recent study compared, in the same laboratory, the effects of amphetamine on [11C]NMSP and [11C]raclopride binding in primates (Hartvig et al., 1997). This article reported the usual effect of amphetamine on [11C]raclopride binding but failed to detect an amphetamine effect on [11C]NMSP binding. In the same context, the study that reported a paradoxical increase in [11C]NMSP accumulation after ketamine is noticeable (Onoe et al., 1994), especially in light of the opposite effect of ketamine on [11C]raclopride (Ginovart et al., 1998). Thus, the lack of effect, or the presence of a paradoxical effect of DA release reported in the rodent literature, is supported by some (Onoe et al., 1994; Hartvig et al., 1997) but not all (Dewey et al., 1990; Logan et al., 1991) PET studies in baboons.
Finally, studies performed in baboons with several D1 radiotracers failed to detect an effect of amphetamine on [11C]SKF 82957, [11C]SCH 23390, and [11C]NNC 756 (Laruelle et al., 1998; Abi-Dargham et al., 1999; Chou et al., 1999).
Studies in healthy humans
Since primates studies clearly established a robust effect of endogenous DA manipulation on [11C]raclopride and [123I]IBZM BP, it is not surprising that these radiotracers were selected to evaluate the existence of such interactions in humans (Table 4).
In vivo competition studies: PET and SPECT studies in healthy humans
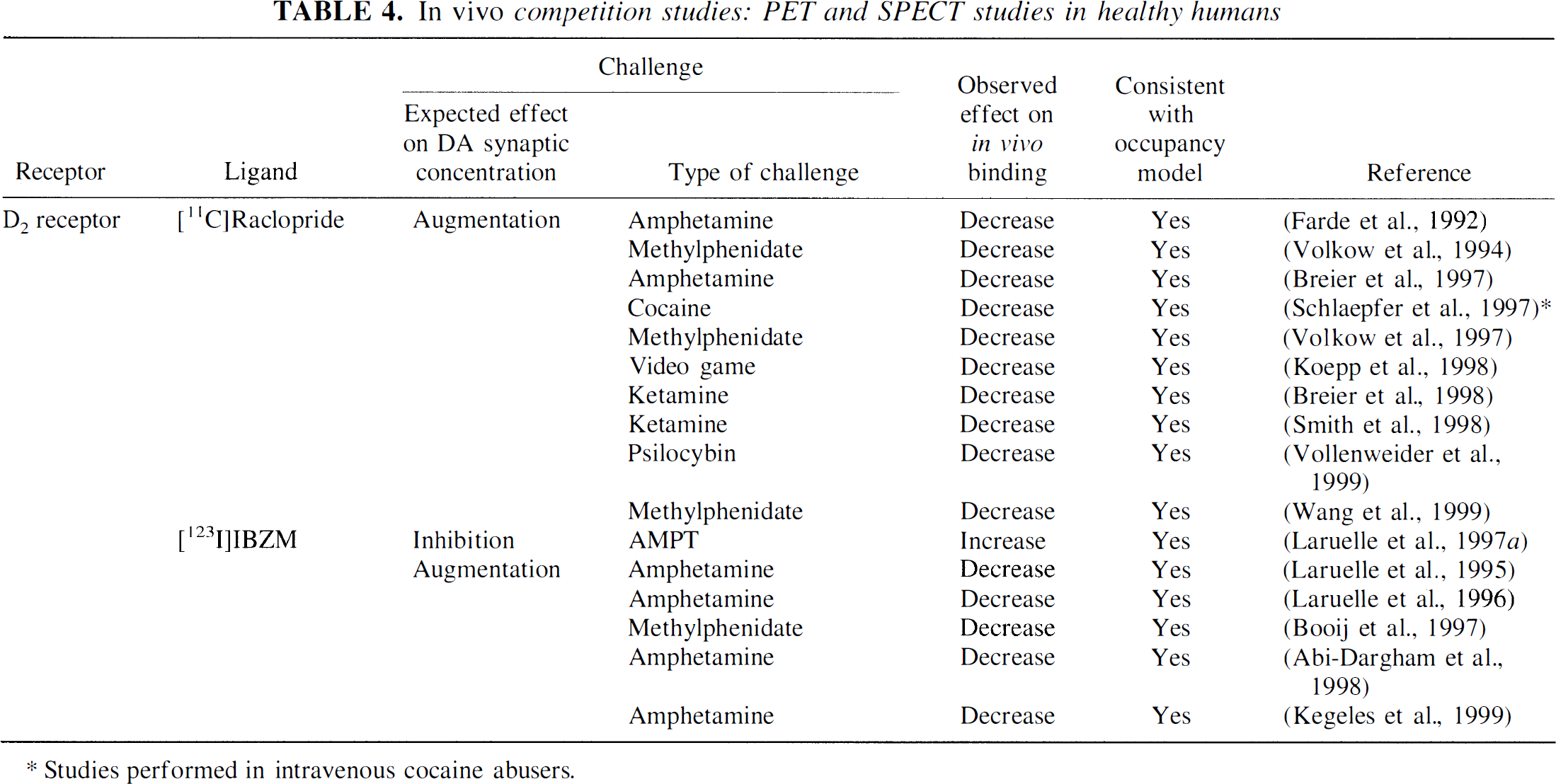
Studies performed in intravenous cocaine abusers.
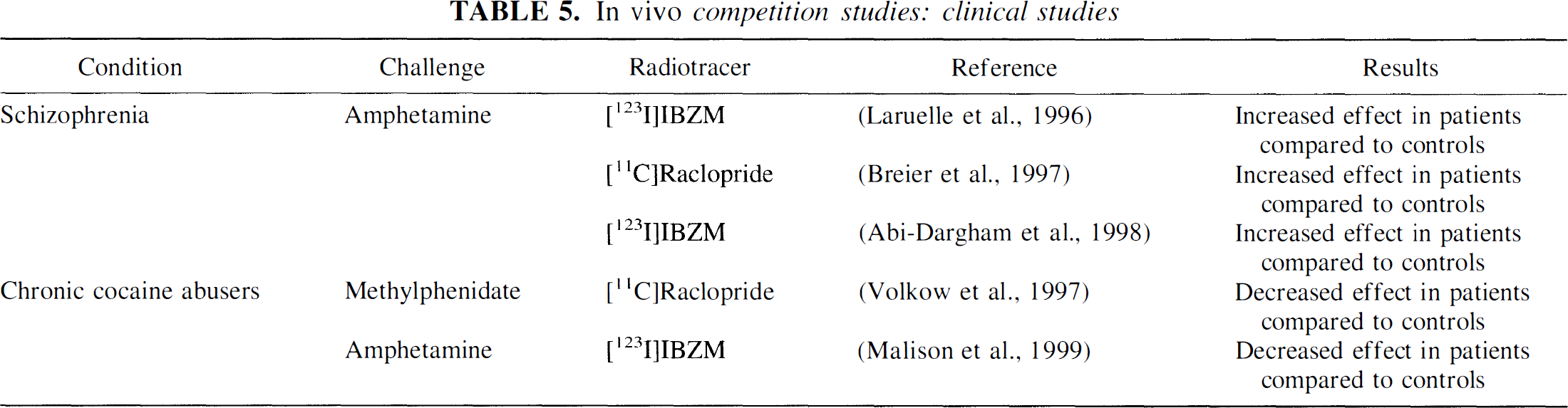
In vivo competition studies: clinical studies
The first literature report of an endogenous competition study in humans is buried in the discussion of an article examining occupancy of D2 receptors with antipsychotic drugs (Farde et al., 1992). An oral dose of 30 mg of amphetamine was reported to decrease [11C]raclopride BP by an average of 10% in three healthy volunteers. The first comprehensive report of this effect in humans was published by Volkow et al. (1994). Methylphenidate given intravenously (0.5 mg/kg) reduced [11C]raclopride BP by an average of 23%. This article was followed by a report of the effect of amphetamine (0.5 mg/kg) on [123I]IBZM BP in humans (Laruelle et al., 1995). Amphetamine (0.3 mg/kg intravenously) decreased [123I]IBZM BP by 15%. Both studies revealed relationships between the magnitude of DA release as assessed by the radiotracer displacement and the subjective states reported by the subjects, either before (Volkow et al., 1994) or after (Laruelle et al., 1995) the psychostimulant challenge.
The vulnerabilities of [11C]raclopride and [123I]IBZM to psychostimulant challenges in humans were independently confirmed with amphetamine, which induced a 16% decrease in [11C]raclopride BP (Breier et al., 1997), and methylphenidate, which induced a 8% decrease of [123I]IBZM (Booij et al., 1997).
Test-retest studies demonstrate that the amphetamine-induced decrease in [123I]IBZM BP could be measured with high reliability in humans (Kegeles et al., 1999). The methylphenidate-induced decrease in [11C]raclopride BP was less reproducible (intraclass correlation coefficient of 0.58, Wang et al., 1999) than the amphetamine-induced decrease in [123I]IBZM BP (intraclass correlation coefficient of 0.89, Kegeles et al., 1999), maybe because the effect of methylphenidate on DA release is more affected by the subjective environment and experience compared with the amphetamine effect.
Cocaine (48 mg) also was reported to affect [11C]raclopride uptake (11% decrease) in chronic cocaine abusers (Schlaepfer et al., 1997). The NMDA antagonist ketamine decreased [11C]raclopride BP by 13% (Smith et al., 1998) and 10% (Breier et al., 1998). The hallucinogen psilocybin, a potent 5HT2A agonist, decreased [11C]raclopride BP by 20% (Vollenweider et al., 1999). Video game playing was showed to be associated with a decrease in [11C]raclopride BP (13%), similar in magnitude to the decreases BP induced by amphetamine (0.5 mg/kg intravenously) and cocaine (48 mg intravenously) (Koepp et al., 1998).
Finally, DA depletion studies in humans showed that acute AMPT challenge resulted in 29% increase in [123I]IBZM BP (Laruelle et al., 1997a).
Clinical studies
Five clinical studies compared the effects of stimulation of DA release on in vivo binding of [11C]raclopride and [123I]IBZM in controls and patients with psychiatric conditions (Table 5). Using [123I]IBZM, Laruelle et al. (1996) and Abi-Dargham et al. (1998) report increased amphetamine-induced (0.3 mg/kg) DA release in patients with schizophrenia compared with matched healthy controls. In patients with schizophrenia, the magnitude of amphetamine-induced DA release estimated by the displacement of [123I]IBZM was related to a transient increase in positive symptoms. A similar result was published by Breier et al. (1997) using [11C]raclopride and a lower dose of amphetamine (0.2 mg/kg). This result also was observed in patients experiencing a first episode of illness who were never previously exposed to antipsychotic medications (Laruelle et al., 1999). Volkow et al. (1997) report a blunting of methylphenidate-induced DA release measured with [11C]raclopride in chronic cocaine abusers compared with controls, a finding replicated by Malison et al. (1999) with [123I]IBZM and amphetamine. These clinical results illustrate the potential of this technique to study alterations of DA transmission in neuropsychiatric conditions.
Outcome measures for reversible ligands (tracer doses)

Bmax, density of available binding sites; KD, affinity of the radiotracer for available binding sites; f1, radiotracer plasma free fraction; V2, distribution volume of nondisplaceable (free and nonspecifically bound) compartment; K1 and k2, rate constants for transit of the radiotracer between plasma and nondisplaceable compartment; k3 and k4, rate constants for transit of the radiotracer between nondisplaceable and specific binding compartment; DV, total tissue distribution volume relative to total radiotracer concentration.
THE OCCUPANCY MODEL
As reviewed earlier, numerous independent laboratories have established that [11C]raclopride and other benzamides behave as predicted by the occupancy model: higher synaptic DA levels are associated with lower ligand binding and vice versa. If the occupancy model is correct, these changes in binding reflect changes in endogenous DA. The next section reviews several arguments that either support or question the effectiveness of these imaging methods to measure changes in synaptic DA concentration, and the adequacy of the occupancy model on which the interpretation of these data is based.
First to be discussed are several issues and results that, in our opinion, support the use of this imaging technique to measure DA release. Appropriate model-based quantitative approaches have unequivocally documented that the decrease in benzamide-specific uptake measured following these manipulations results from a reduction in BP and not from challenge-induced changes in regional CBF (rCBF), peripheral clearance, or nonspecific binding. This article also reviews the experimental data confirming that these effects are mediated by DA release, and that the magnitude of these effects are correlated with the magnitude of changes in DA level as measured with microdialysis. Also reviewed is the evidence supporting that these effects are mainly caused by synaptic changes in endogenous DA as opposed to extrasynaptic changes. Also discussed are the data supporting the use of these techniques to estimate DA levels in the “baseline” or “unchallenged” state, measurements that might be important to the study of pathophysiologic conditions.
Next, this article focuses on data that raise concerns about the validity of the simple occupancy model. First, this report demonstrates that the temporal discrepancies between PET or SPECT and microdialysis measurements significantly question this model. Next, I address the issues raised by the results obtained with spiperone and D1 radiotracers. A discussion of the role of radiotracer affinity in competition studies is needed to fully appreciate the significance of the spiperone and D1 radiotracer data. This discussion leads to the proposition that the simple occupancy model might be limited in its ability to account for all of the experimental data, and that other factors might play a significant role in the alterations of BP observed following DA manipulations.
Data supporting the occupancy model
The importance of model-based methods to measure changes in binding potential. The first question is whether the changes in radiotracer binding measured after these challenges truly reflect changes in receptor availability (in this report, the term receptor availability is used to denote the BP of the receptors in the presence of a competitor such as DA). Pharmacologic interventions that induce significant changes in DA synaptic concentration frequently affect multiple physiologic parameters such as body temperature, rCBF, peripheral rate of clearance of the radiotracer, blood pH, and radiotracer plasma protein binding. For example, in anesthetized baboons, a single amphetamine injection (0.4 mg/kg) decreases rCBF by as much as 50% (Hartvig et al., 1997). These physiologic factors are known to affect radiotracer uptake. Therefore, a receptor parameter quantification method resistant to variations in these physiologic parameters must be implemented for appropriate evaluation of the effect of these challenges on receptor availability. Several quantification methods have been developed to address these concerns, referred to here under the generic term of model-based methods. I consider here the relatively simple case of reversible radiotracers such as [11C]raclopride or [123I]IBZM.
Regarding blocking experiments, a variety of model-based methods have been used to analyze the challenge effects on BP. For [11C]raclopride, these methods include kinetic three-compartment modeling using the arterial metabolite-corrected tracer concentration as input function (Carson et al., 1997; Price et al., 1998; Abi-Dargham et al., 1999), the Logan et al. (1990) graphical method for reversible tracers (Dewey et al., 1992; Dewey et al., 1993; Volkow et al., 1994; Dewey et al., 1995; Volkow et al., 1997; Smith et al., 1998; Volkow et al., 1999), the two-compartment model using the cerebellum as input function (Kobayashi et al., 1995; Koepp et al., 1998), or the peak equilibrium method (Farde et al., 1992; Ginovart et al., 1997; Hartvig et al., 1997; Ginovart et al., 1998). These methods generally enable measurements of receptor parameters that are independent from rCBF or peripheral clearance. For example, derivation of [11C]raclopride k3/k4 ratio by the three-compartment kinetic modeling and the graphical method of Logan et al. (1990) are unaffected by changes in rCBF (Logan et al., 1994).
However, these methods assume the rCBF to be constant during the time frame of the scan. These methods effectively correct for changes in rCBF between a baseline and a challenge scan if these changes are sustained during the scanning period, but they are vulnerable to variations in rCBF that occur during the scan. An initial decrease followed by an increase in rCBF might cause a time-activity curve kinetically indistinguishable from a curve resulting from a decrease in BP availability. The potential of these rCBF effects to induce artifactual results might not have received enough attention. These phenomena might contribute to some of the surprising results found in the literature, suggesting, for example, that video game playing elicit a greater DA release than intravenous cocaine.
Results from displacement experiments performed during the regional washout phase of the radiotracer are even more difficult to interpret. In several studies, the effect of the challenge was assessed by comparing the radiotracer washout rate following the challenge to this rate measured under control conditions. For example, amphetamine increased the striatal washout rate of [123I]IBZM (Innis et al., 1992), [18F]fallypride (Kessler et al., 1993; Mukherjee et al., 1997), and [18F]FCP (Mach et al., 1997). Yet, increase in the peripheral clearance of the tracer or increase in rCBF also increases the radiotracer washout rate. This issue was formally addressed by Dagher et al. (1998), who showed that both an increase in k2 (from increased rCBF) or a decrease in k3 (from decreased BP) induced during the washout phase could result in similar curves. Thus, this design is not appropriate to establish an effect of the challenge on receptor availability. For [123I]IBZM and [18F]fallypride, the amphetamine-induced reductions in BP initially suggested by the wash-out rate method (Innis et al., 1992; Kessler et al., 1993; Mukherjee et al., 1997) were confirmed with more robust methods (Laruelle et al., 1997b; Price et al., 1998).
A better method for displacement experiments is to administer the radiotracer as a bolus followed by constant infusion, wait for the establishment of a sustained equilibrium state, and inject the challenge drug during the equilibrium period (Fig. 2). Under those conditions, an effect on the peripheral clearance will result in a change in the plasma steady-state level of the radiotracer, and the incorporation of this information in the outcome measure will correct for this effect. Variations in rCBF will not affect the equilibrium because, during sustained equilibrium state, there is no net radiotracer transfer across the blood-brain barrier (BBB). Thus, the robust amphetamine-induced decreases in [123I]IBZM and [11C]raclopride-specific binding demonstrated during constant infusion experiments (Laruelle et al., 1995; Carson et al., 1997; Laruelle et al., 1997b) represent an unequivocal decrease in BP.
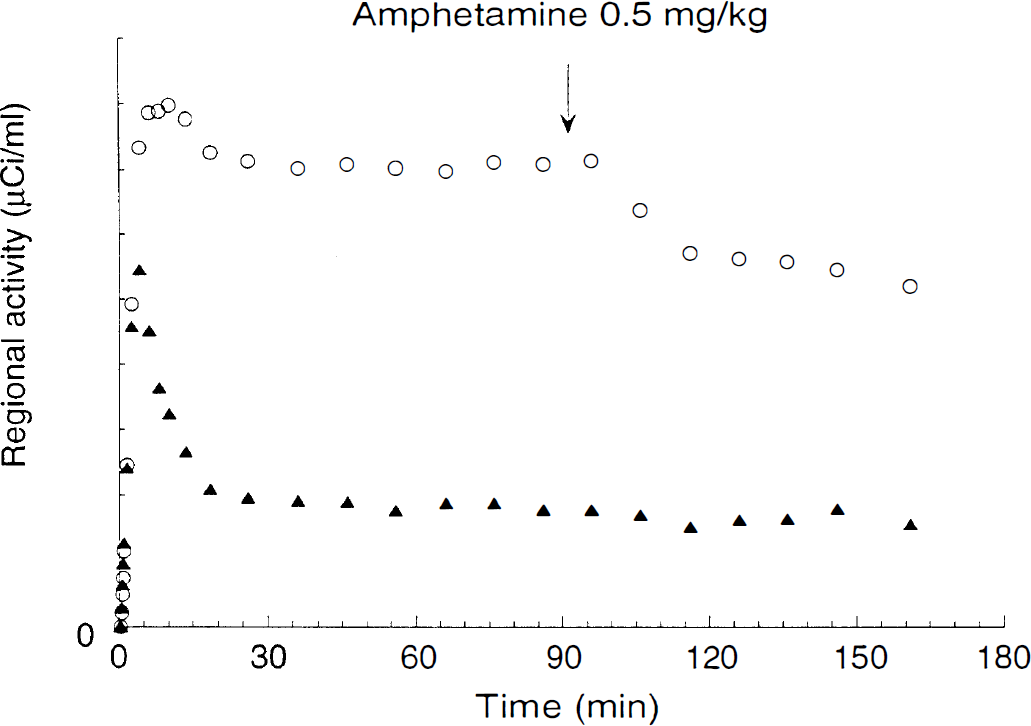
Regional time—activity curves during [11C]raclopride bolus plus a continuous infusion experiment in a baboon and injection of amphetamine (0.5 mg/kg) at 90 minutes (arrow). Regions represented are cerebellum (triangles) and striatum (open circles). Equilibrium was achieved by 30 minutes. Amphetamine induced a prolonged decrease in striatal activity and no change in cerebellar activity.
In vivo competition studies with psychostimulants and [11C]raclopride or [123I]IBZM: effect range
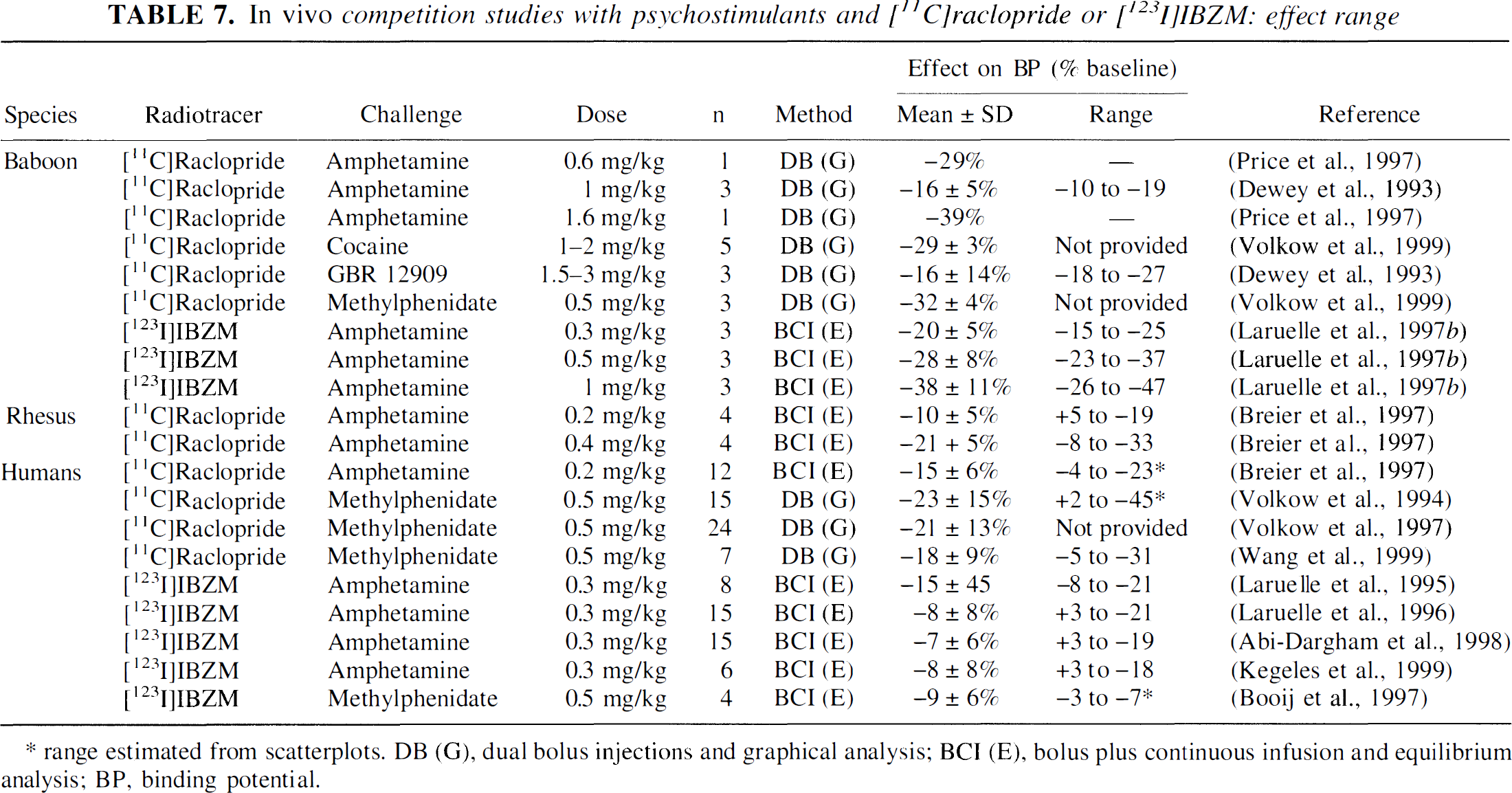
* range estimated from scatterplots. DB (G), dual bolus injections and graphical analysis; BCI (E), bolus plus continuous infusion and equilibrium analysis; BP, binding potential.
Ignoring the presence of the competitor DA, the radioligand specifically bound concentration (B) is, at equilibrium, related to the radioligand free concentration (F), the maximal number of D2 receptors (Bmax), and the equilibrium dissociation rate constant of radioligand for D2 receptors (KD) by the Michaelis-Menten equation equilibrium, related to the radioligand free concentration (F), the maximal number of D2 receptors (Bmax), and the equilibrium dissociation rate constant of radioligand for D2 receptors (KD) by the Michaelis-Menten equation
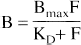
If the radioligand is administered at tracer dose, F is negligible relative to KD and this equation simplifies to
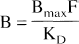
Eq. 2 can be rearranged to demonstrate the equivalence between the BP and the B/F ratio.
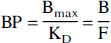
Eq. 3 indicates that BP is equal to the ratio of the specifically bound (B) concentration to the free ligand concentration in the vicinity of the receptors (F), when these terms are at equilibrium and when the tracer doses of the radioligand are used.
The radiotracer usually is assumed to distribute into three compartments: the plasma compartment (C1), the brain nondisplaceable compartment (C2, which includes both free and nonspecifically bound radiotracer), and the receptor compartment (C3). Defining that Vi is the equilibrium distribution volume of a brain compartment relative to the free radiotracer concentration in the brain, V2 and V3 are the distribution volumes of the nondisplaceable and receptor compartments, respectively, and VT is the total distribution volume (VT = V2 + V3). Eq. 3 shows that BP is equal to V3, that is, the bound over free ratio (B/F) at equilibrium.
The radioligand specifically bound concentration, B, is directly measurable as the specific binding (e.g., as the difference between striatum and cerebellum activities at equilibrium), but F, the free concentration of the radiotracer in the brain, is not directly measurable. Because the radiotracer crosses the BBB by passive diffusion, it can be assumed that at equilibrium (when there is no net transfer of radiotracer across the BBB), the free concentration is equal on both sides of the BBB, and the free plasma concentration can be substituted to the free brain concentration in Eq. 3 to calculate BP. This assumption has been validated by two observations: (1) at equilibrium, the tracer concentration in the CSF is equal to the free concentration in the plasma (Kawai et al., 1991; Laruelle et al., 1994a); and (2) the KD measured in vivo equals the in vitro KD when the free plasma concentration is used as the concentration parameter (Laruelle et al., 1994a, b ). The radiotracer free fraction in the plasma and in the nondisplaceable compartment usually are noted as f1 and f2, respectively. At equilibrium, C1f1 = C2f2. It follows that the nondisplaceable distribution volume, V2, is equal to 1/f2. Thus, calculation of BP as V3 represents the “ideal” measurement of BP because the denominator F does not include any nonspecific component. The use of this outcome measure requires the measurement of the free fraction in the plasma (f1).
The plasma free fraction (f1) is difficult to measure with accuracy, especially for radiotracer with high binding to plasma proteins. Therefore, BP sometimes is expressed relative to the total (bound or unbound) plasma concentration, and the term V3′ is used to designate this ratio. More often, BP is expressed relative to the total (free plus nonspecifically bound) brain concentration in a region of reference devoid of specific binding, and the term V3″ is used to designate this ratio. In the terminology of Carson et al. (1997), V3, V3′ and V3″ are designated S′, S, and R, respectively.
Using the kinetic notation of a three-compartment model (plasma, nondisplaceable and specific compartments), V3 is given by K1k3/k2k4f1, V3′ is given by K1k3/k2k4, and V3″ is given by k3/k4. The equivalence between this terminology and the terminology derived from the graphical analysis of Logan et al. (1990) also is given in Table 6. Each of these outcome measures have distinct advantages and limitations (see discussion in Abi-Dargham et al., 1999).
The importance of the choice of outcome measure in the estimation of changes in receptor parameters induced by a challenge can be appreciated in Table 2 of Carson et al. (1997). Using graphical analysis, amphetamine (0.4 mg/kg) induced a 42% ± 17% reduction in V3 (S′), a 29% ± 21% reduction in V3′ (S), and a 36% ± 15% reduction in V3′ (R).
A major advantage of V3″ is the highest test-retest reproducibility of this outcome measure compared with V3′ and V3 (Logan et al., 1994; Abi-Dargham et al., 1995; Carson et al., 1997). Because the magnitude of the challenge effect generally is small, it is reasonable to select the outcome measure attached with the lowest tes-t—retest variability, and V3″ has been the outcome measure of choice for clinical studies (Laruelle et al., 1996; Breier et al., 1997; Volkow et al., 1997; Abi-Dargham et al., 1998). This choice is based on the assumption that the challenge will not affect the nondisplaceable distribution volume in the brain (V2), that is, the nonspecific binding.
Yet, this assumption generally is not valid. For example, changes in radiotracer cerebellum distribution volume (V2' = V2f1) often are measured during challenge tests (see, for example, Table 1 in Dewey et al., 1993). If f1 is not measured (a situation where V3 is not available), it is impossible to decide if this change in cerebellum distribution volume results from a change in nonspecific binding in the plasma (1/f1) or the cerebellum (1/f2). Using V3″ as outcome measure effectively controls for changes in f1, since f1 cancels out in the V3/V2 ratio, but leads to artifactual results if the change is in f2. Using V3′ as outcome measure effectively controls for changes in f2, since V2 is not included in the outcome measure, but the result will be biased if the change in V2' is caused by change in f1. Since the amphetamine effect on [11C]raclopride-specific binding has been demonstrated with each of the three outcome measures, there is no doubt that, at least with this tracer, amphetamine induces a reduction of the Bmax/KD ratio. However, in the absence of accurate measurement of f1, it is not possible to control for the potential impact of the challenge on plasma and brain nonspecific binding.
DA release is required for the effect of amphetamine on D2 receptor binding potential. In the previous section, this article reviews the evidence supporting that changes in [11C]raclopride- or [123I]IBZM-specific uptake after DA manipulations effectively result from changes in BP, and not from artifacts caused by alterations in rCBF, peripheral metabolism, or nonspecific binding. The next question is whether these changes in BP are mediated by changes in synaptic DA concentration. Several experiments directly confirm that changes in benzamide's BP following amphetamine are dependent on changes in DA release.
Amphetamine induces DA release by reverse transport of DA from the cytoplasmic pool to the synapse through the DAT (Fischer and Cho, 1979; Sulzer et al., 1995). Thus, blocking DAT with DA uptake inhibitors such as nomifensine results in a blunting of amphetamine-induced DA release (Fischer and Cho, 1979; Raiteri et al., 1979; Connor and Kuczenski, 1986; Parker and Cubeddu, 1986; Butcher et al., 1988; Simon et al., 1991). Because amphetamine releases DA from the cytoplasmic pool, amphetamine-induced DA release is dependent on new DA synthesis and on the functional activity of tyrosine hydroxylase. Thus, the inhibitor of tyrosine hydroxylase, AMPT, significantly reduces amphetamine-induced DA release (Butcher et al., 1988; Nash and Yamamoto, 1993; Cadoni et al., 1995; Laruelle et al., 1997b). Finally, at high doses, amphetamine redistributes DA from the vesicular to the cytoplasmic pool (Sulzer et al., 1993; Sulzer et al., 1995). Thus, reserpine pretreatment affects amphetamine-induced DA release, at least after high doses of amphetamine (Dluzen and Liu, 1994; Cadoni et al., 1995; Florin et al., 1995; Pifl et al., 1995). It is therefore expected that the DA uptake blockers AMPT and reserpine should diminish the effect of amphetamine on [11C]raclopride or [123I]IBZM BP.
The first demonstration that this was the case was provided by Innis et al. (1992) in baboons. Reserpine pretreatment resulted in significant blunting of the amphetamine-induced increase in [123I]IBZM washout rate. Next, it was established that AMPT pretreatment blocked the effect of amphetamine on [123I]IBZM BP, as measured during radiotracer constant infusion paradigm (Laruelle et al., 1997b). Recently, Villemagne et al. (1988) showed that pretreatment with the DAT blocker GBR 12909 blocked the effect of amphetamine on [11C]raclopride BP, as measured during radiotracer constant infusion paradigm. Together, these data provide firm evidence that the effect of amphetamine on benzamides BP requires enhanced DA release.
Quantitative relationship between DA release and change in D2 receptor binding potential. Because this imaging technique was proposed as a measure of DA transmission, it was important to characterize experimentally the relationship between extracellular DA and radioligand displacement.
The first step was to demonstrate the existence of a dose-effect relationship between challenge drug and radiotracer displacement. Laruelle et al. (1997b) showed a dose-dependent effect of three doses of amphetamine on [123I]IBZM BP in baboons: [123I]IBZM BP was reduced by 20% ± 5%, 28% ± 7%, and 38% ± 10% after 0.3-, 0.5-, and 1.0-mg/kg amphetamine doses (n = 3), respectively. Breier et al. (1997) report the effect of two doses of amphetamine on [11C]raclopride BP in rhesus monkeys: [11C]raclopride BP was reduced by 10% ± 5% and 21% ± 5% after 0.2- and 0.4-mg/kg amphetamine doses, respectively (n = 4). Hartvig et al. (1997) administered amphetamine using constant infusion to achieve steady-state plasma amphetamine ranging from 0.2 to 25 ng/mL in rhesus monkeys and showed an increased reduction in [11C]raclopride BP following an increasing amphetamine plasma level.
The next step was to characterize the magnitude of DA release elicited by these various doses of amphetamine in anesthetized primates. Laruelle et al. (1997b) report microdialysis measurements of DA release in one vervet monkey after five doses of amphetamine, ranging from 0.03 to 1.5 mg/kg. Breier et al. (1997) performed simultaneous microdialysis and PET experiments (four rhesus monkeys and two amphetamine doses). In both studies, the relationship between combined microdialysis measurements and combined radiotracer displacements at each amphetamine dose was characterized. These analyses yielded consistent results. Linear correlation between microdialysis and SPECT measurements revealed that each percent decrement in [123I]IBZM BP corresponded to a 44% increase of peak DA concentration following DA (Fig. 3). Likewise, each percent decrement in [11C]raclopride BP corresponded to a 44% DA increase for the 0.2-mg/kg dose, and 64% DA increase for the 0.4-mg/kg dose. Both studies indicate that a large increase in extracellular DA release (range, 400% to 1500%) is associated with a relatively small effect on radiotracer BP (decrease range, 10% to 38%), but that these effects were correlated, supporting the usefulness of the imaging paradigm in providing noninvasive measurement of DA release.
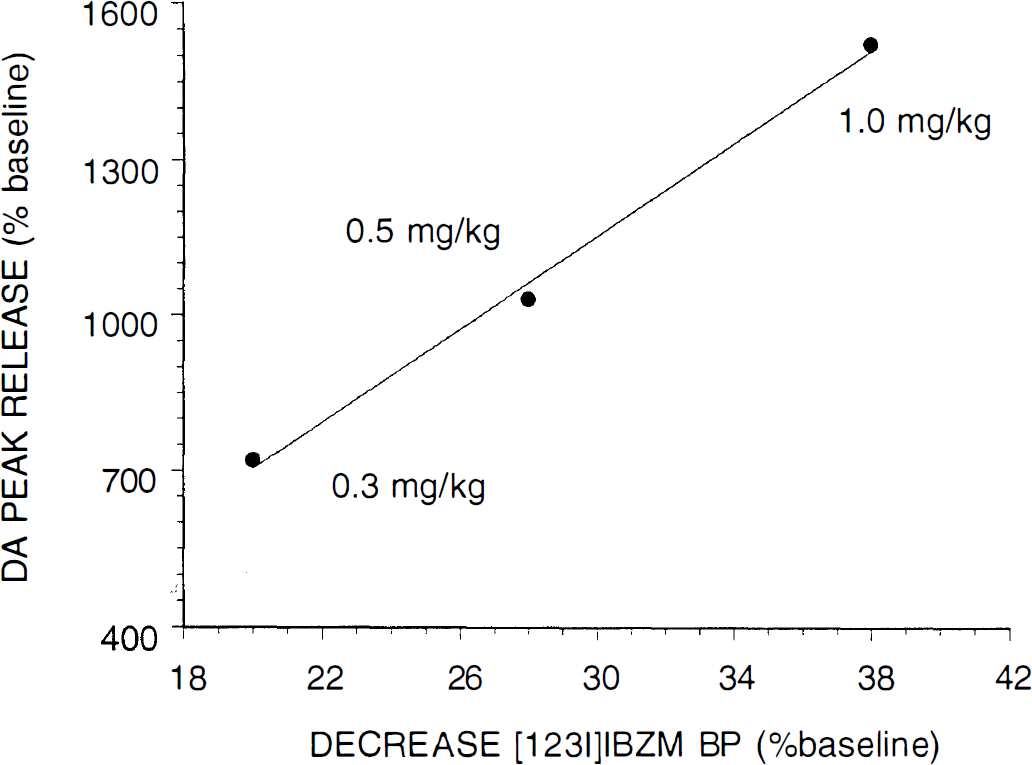
Correlation between amphetamine-induced peak dopamine (DA) release, measured with microdialysis (y axis) and the decrease in [123I]IBZM D2 binding potential (BP), measured with single-photon emission computed tomography (each point is the mean of three experiments). This relationship was linear (y = −190 + 44x, r = 0.99). Constraining the intercept to zero, the equation was y = 39x. Thus, each percent decrement in [123I]IBZM BP corresponded to about a 40% increase over baseline of DA concentration.
However, closer examination of the data reveals some limitations to these conclusions. In the study by Breier et al. (1997), the microdialysis and PET measurements were obtained simultaneously. Therefore, these data provided the opportunity to examine the relationship between microdialysis and PET measurements within each experiment. Examination of the data from Tables 1 and 2 of the report by Breier et al. (1997) reveals a lack of relationship between microdialysis and PET measurements collected within the same experiment. For example, rhesus 2 showed the larger increase in extracellular DA after the 0.2-mg dose (+1000%), but [11C]raclopride BP was not affected. Rhesus 3 showed the larger effect of the 0.2-mg/kg amphetamine dose on [11C]raclopride BP (decrease by 19.2%) but showed only a modest 116% increase in extracellular DA. These discrepancies suggest that a large noise is associated with both measurements and that the data at each dose must be averaged yield a meaningful relationship. Alternatively, these results might indicate that the extracellular DA level is not the only factor playing a role in determining the magnitude of the change in [11C]raclopride BP.
Discussion of the ceiling effect. Decreases in [11C]raclopride and [123I]IBZM BP have been measured after several challenges, and the literature is consistent in the range of radiotracer displacement (less than 50%). Table 7 lists the mean ± SD and the range of [11C]raclopride and [123I]IBZM BP reduction after a single administration of psychostimulants measured with PET or SPECT in nonhuman primates and humans. The sorting order in the Table 7 is species, radiotracer, challenge drug, and dose. Inspection of Table 7 clearly indicates that only about half of the radiotracer-specific binding is affected by psychostimulant challenges.
This ceiling effect is not inconsistent with the occupancy model. The D2 receptors are configured in states of high or low affinity for agonists, with approximately 50% of the receptors contributing to each state in vitro (Zahniser and Molinoff, 1978; Sibley et al., 1982; George et al., 1985a; Seeman and Grigoriadis, 1987; Richfield et al., 1989). The antagonists [123I]IBZM and [11C]raclopride bind with equal affinity to both states. The agonist DA is not expected to compete efficiently with [123I]IBZM or [11C]raclopride binding to D2 receptors in the agonist low-affinity configuration. This factor alone would leave only about 50% of the antagonist binding susceptible to endogenous competition. In addition, not all receptors configured in the high-affinity state are available to the binding of the radiotracer at baseline, since a proportion of these high-affinity state receptors is expected to be occupied by baseline levels of endogenous DA. Assuming that 50% of the sites are configured in the agonist high-affinity state and that 20% are already occupied by DA at baseline (i.e., before the challenge), only about 30% of the total receptor population (i.e., 37.5% of receptors contributing to baseline [11C]raclopride or [123I]IBZM BP) will be susceptible to an additional occupancy by DA following the challenge (Fig. 4).
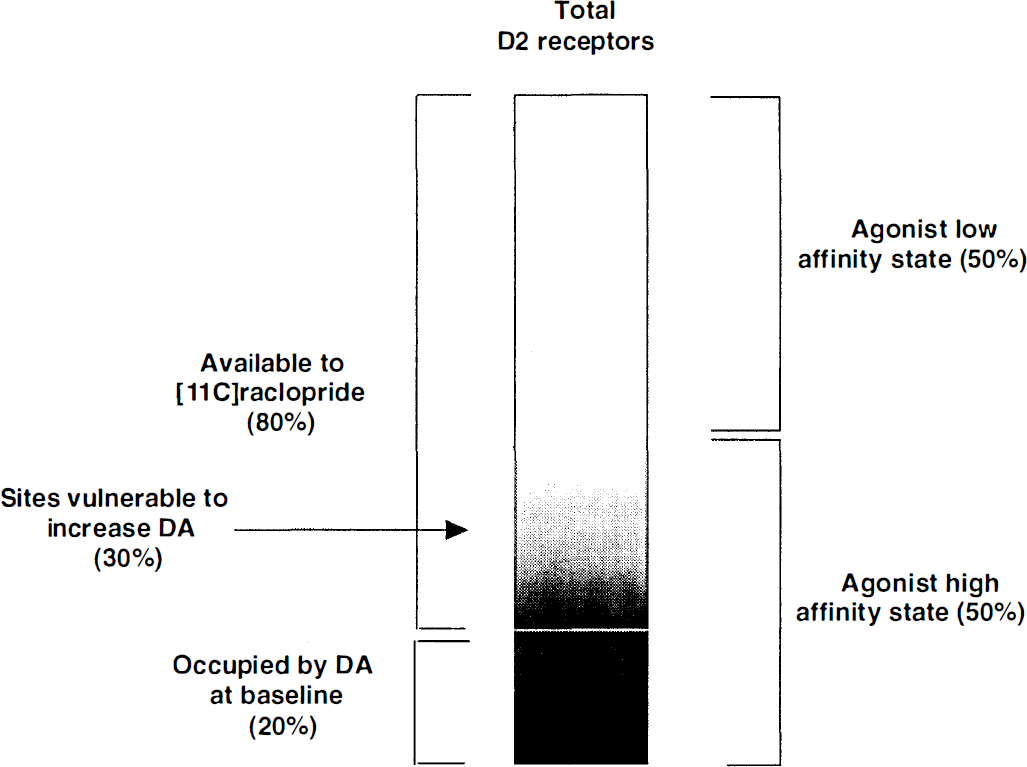
Schematic representation of D2 receptor subgroups accounting for the existence of a ceiling effect in the magnitude of [11C]raclopride BP reduction following psychostimulant challenges (Table 7). It is assumed that (1) 50% of receptors are in high and low affinity states for DA, respectively; (2) 20% of receptors are already occupied by DA at baseline, that is, before the challenge, and are not available for [11C]raclopride binding; and (3) DA does not bind effectively to D2 receptors that are in the low-affinity configuration. It follows that only 30% of total D2 receptors (i.e., 37.5% of receptors available to [11C]raclopride) will be vulnerable to an increase above baseline of DA concentration.
Displacement studies with agonists in mice confirm that only 10% to 30% of the D2 sites satisfy both conditions of being in the high agonist affinity states and not being occupied by DA under resting conditions (Ross and Jackson, 1989b). Moreover, many D2 receptors are not located in the synaptic cleft, and extrasynaptic receptors might be less exposed to changes in DA release compared with receptors located at the synaptic level (Levey et al., 1993; Smiley et al., 1994; Hersch et al., 1995; Yung et al., 1995; Caille et al., 1996). Given these factors, it is not surprising that DA would displace only up to 50% of the in vivo binding of [11C]raclopride and [123I]IBZM, and the existence of this plateau effect is not inconsistent with the occupancy model.
Synaptic versus extrasynaptic location of the effect. As eluded to in the previous section, not all D2 receptors are located within the synaptic clef. In striatal dendritic spines, as much as 50% of D1 and D2 receptors might not be associated with synapses (Yung et al., 1995). In addition, receptors associated with synapses were more often associated with asymmetrical synapses that usually receive input from glutamateric corticostriatal afferents than with symmetrical synapses, usually associated with terminals of nigrostriatal projections (Levey et al., 1993; Hersch et al., 1995; Yung et al., 1995). Thus, whereas the proportion of synaptic versus nonsynaptic D1 and D2 receptors is not precisely known, it is clear that a non-negligible number of these receptors are located at sites distant from dopaminergic synapses.
Microdialysis measurements are performed in the extrasynaptic extracellular compartment, where the baseline concentration of DA is estimated to be in the 20- to 40-nmol/L range (Church et al., 1987). The DA concentration probably is higher within the synapse. Using fast-scan voltametry measurement of DA release at the synaptic interface, Kawagoe et al. (1992) estimated that the synaptic concentration of DA rapidly varies from as high as 200 nmol/L to as low as 6 nmol/L, with a temporal average of about 100 nmol/L (May 1988). In normal conditions, rapid uptake of synaptic DA by DAT is thought to prevent DA released in the synapse from diffusing to the extrasynaptic space (Grace, 1993). Supporting this view is the absence of changes in extracellular DA elicited by electrical stimulation of DA neurons at frequencies lower than 10 Hz (Kuhr et al., 1984; Kuhr and Wightman, 1986; May, 1988). Intrasynaptic DA is increased at these frequencies, since DA-mediated behavioral effects are induced. The role of DAT in preventing intrasynaptic DA to diffuse to extrasynaptic space is confirmed by the increase in extrasynaptic DA elicited at these low frequencies when a DAT blocker is coadministered (see discussion in Grace, 1993). Grace (1991, 1993) proposed the terms phasic release to characterize the intrasynaptic release elicited by cell firing and tonic release to characterize the low level of extrasynaptic release that is independent of cell firing.
If the effects of endogenous DA competing with radioligand were located mostly at the nonsynaptic D2 receptors, it would be expected that microdialysis measurements and decrease in [11C]raclopride BP would be related across several pharmacologic challenges. Evidence suggests that this is not the case. Nicotine, a drug that stimulates DA cell firing without blocking DAT, induces only a modest increase in extrasynaptic DA concentration. In rodent, a high dose of nicotine (5 mg/kg) elicits only a 29% increase in extrasynaptic DA measured with microdialysis (Kim et al., 1998). Yet, this dose of nicotine was associated with a 21% decrease in [3H]raclopride in vivo binding (Kim et al., 1998). In contrast, the dose of amphetamine (0.4 mg/kg) associated with the same 21% decrease in [11C]raclopride in rhesus monkeys elicits a 1365% increase over baseline in extrasynaptic DA (Breier et al., 1997).
This apparent discrepancy is explainable by the differences in the DAT blocking properties of these drugs. Extrasynaptic DA reflects intrasynaptic DA following DAT blockers such as amphetamine, but not following drugs like nicotine, which stimulate DA release without blocking DAT. Because a similar reduction in raclopride BP (21%) is observed following nicotine (5 mg/kg) and amphetamine (0.4 mg/kg) challenges associated with 29% and 1365% increase in extracellular DA, respectively, the challenge must affect mainly the BP of D2 receptors located within the synaptic cleft.
Similar results have been reported (Tsukada et al., 1999). The DA extracellular concentrations and [11C]raclopride displacements were measured in unanesthetized monkeys after the administration of “direct” DA enhancers (DAT blockers GBR 12909 and methamphetamine), and “indirect” enhancers, that is, the muscarinic antagonist benztropine (at a dose not expected to block DAT) and the 5HT2A antagonist ketanserin. Again, at similar level of [11C]raclopride BP decrease, the magnitude of DA extracellular increase following direct enhancers was much larger than following indirect enhancers, which is expected because only direct enhancers blocked DAT. These results also are consistent with a predominant intrasynaptic location of the effect measured with PET.
Thus, the discrepancies across pharmacologic agents in their ability to affect DA microdialysis and benzamide BP measurements is not inconsistent with the occupancy model, if it is accepted that the effect on benzamide BP primarily occurs within the synaptic space.
Occupancy of D2 receptors by baseline levels of dopamine. Endogenous competition studies thus might be the only tool available to estimate intrasynaptic D2 receptor occupancy by DA and intrasynaptic DA levels. Across studies, results are consistent in indicating that a substantial proportion of D2 receptors are occupied by DA under physiologic conditions. Inhibition and depletion studies presented in Tables 2 to 4 indicate that 20% to 30% of D2 receptors are “unmasked” by removal of endogenous DA. These estimates are valid to the extent that the DA depletion is sufficiently acute to allow unmasked D2 receptors to be measured before significant receptor upregulation occurs (Laruelle et al., 1997a).
Knowledge of the baseline occupancy permits an estimation of baseline DA concentration in the vicinity of D2 receptors (Ross and Jackson, 1989b; Laruelle et al., 1997a). These estimates are based on Eq. 4 describing the equilibrium binding of a radioligand in the presence of a competitor, in this case, DA (Bylund and Yamamura, 1990)
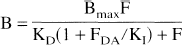
where FDA is the temporal average concentration of free DA in the vicinity of receptors, and KI is the inhibition constant of DA for radioligand binding to D2 receptors. Assuming that the KI of DA to inhibit [3H]NPA binding in vivo was similar to the value measured in vitro at 37°C (101 nmol/L), and assuming that reserpine (5 mg/kg subcutaneously 3 days before experiment) pretreatment resulted in complete DA depletion, Ross and Jackson (1989b) estimated the DA synaptic concentration at 36 nmol/L. Using a similar approach, a KI value of DA to inhibit [123I]IBZM binding of 160 nmol/L (Brücke et al., 1988), and data from AMPT depletion challenge in humans, Laruelle et al. (1997a) estimated the average synaptic DA concentration in humans within the 45 to 72 nmol/L range. These estimates are not too far from the temporal average synaptic DA concentration of 100 nmol/L derived from voltametry studies (Kawagoe et al., 1992). Nonetheless, given the number of unknowns such as the exact KI of DA for D2 receptors in vivo, the proportion of high and low affinity state and their respective Khigh and Klow, the proportion of synaptic versus extrasynaptic and internalized versus noninternalized receptors, these quantitative estimates should be viewed with caution. The important point is that the increase in benzamide BP induced by acute DA depletion might be used to quantify the proportion of D2 receptors unmasked by the removal of endogenous DA (Laruelle et al., 1997a). This method might provide an indirect measure of synaptic DA concentration under baseline conditions, which is driven by the phasic activity of DA neurons.
Equation 4 assumes that the interaction between DA and D2 receptor antagonists is purely competitive, that is, that the presence of the competitor DA affects the apparent KD of the radiotracer but not the Bmax. The question of the competitive or noncompetitive nature of the interaction between DA and D2 receptor antagonists is important. If the interaction was purely competitive, DA levels would affect KD and not Bmax, and the KD of the radiotracer measured in vivo might provide an indirect information about levels of endogenous DA. If the interaction was purely noncompetitive, DA levels would affect Bmax but not KD. If the interaction was mixed, DA levels would affect both KD and Bmax.
The nature of the interaction between dopamine and antagonist at D2 receptors. In vitro data are consistent in showing that interactions between DA and D2 receptor antagonist follow a mixed model: this interaction appears to be competitive at the receptors configured in low agonist affinity state, and noncompetitive at the receptors configured in high agonist affinity state. Saturation experiments performed with [3H]spiperone in the absence and presence of the agonist NPA (50 nmol/L) reveal that NPA reduced the Bmax of [3H]spiperone, with no apparent change in the KD (Sibley and Creese, 1980). However, when the experiment was performed in the presence of nonhydrolyzable G-protein analogs to convert the high agonist affinity sites into low agonist affinity sites, the interaction was strictly competitive, that is, NPA affected the KD but not the Bmax of [3H]spiperone (Sibley et al., 1982). Interactions between [3H]spiperone and DA and other agonists also was shown to be mixed, with both competitive and noncompetitive components (Leysen and Gommeren, 1981; Chatterjee et al., 1988). Guanine nucleotides increase the Bmax of spiperone with no effect on the KD, whereas this effect is abolished by extensive washing of the membranes (which removes endogenous DA) and is restored by adding DA to the incubation medium (George et al., 1985b). This mixed interaction, originally described for [3H]spiperone, also was noted with [3H]raclopride (Seeman et al., 1989; Hall et al., 1990; Seeman et al., 1990).
In vivo, the situation is less clear. Although the interaction between [3H]raclopride appears to be mixed in mice (acute DA depletion reduces KD and increases Bmax) (Ross and Jackson, 1989a), recent data in anesthetized primates obtained with [11C]raclopride are consistent with a pure competitive interaction (Ginovart et al., 1997). Thus, whereas in vitro studies are consistent in detecting both competitive and noncompetitive interactions between DA and [11C]NMSP or [11C]raclopride, the existence of both components has not been consistently documented in vivo, and more data are needed to resolve this issue.
Whether the presence of endogenous DA affects the antagonist Bmax, KD, or both is an important question, but this issue is not crucial to the interpretation of changes in radioligand BP following DA manipulations when measured at tracer doses of the radioligand. Increased DA level might reduce the number of sites available to the radiotracer or might reduce the affinity of these sites for the radiotracer. Both of these effects will translate in a reduction of BP (Bmax/KD). Thus, the existence of these complex interactions does not invalidate per se the occupancy model
Data questioning the occupancy model
After examining the results that support or are consistent with the general occupancy model as a theoretical construct for the interpretation of these experiments, this report focuses on results that are not easily accounted for by this model.
Long-lasting effect of amphetamine on binding potential. A first observation difficult to reconcile with the occupancy model is the long-lasting effect of the short-lived amphetamine-induced DA surge on D2 receptor availability. Studies performed with amphetamine and [123I]IBZM reveal a significant temporal discrepancy between microdialysis and SPECT measurements. Following amphetamine, the rapid increase and decrease of extracellular DA in the extracellular space did not match the slow and prolonged decrease in [123I]IBZM BP induced by amphetamine (Laruelle et al., 1997b). Microdialysis data show that extracellular DA peaked within 10 to 20 minutes after amphetamine and that this peak was followed by a rapid decrease in extracellular DA. Microdialysis measurements were obtained up to 120 minutes following amphetamine. In the 100- to 120-minute postamphetamine collection (endpoint of data collection), extracellular DA concentration was still elevated compared with baseline but at levels much lower than peak levels (34% ± 5.3% of peak value). Thus, the DA surge in extracellular milieu is relatively short-lived.
In contrast, the specific binding of [123I]IBZM decreased for 60 to 90 minutes following amphetamine and stabilized thereafter at levels lower than baseline. [123I]IBZM-specific binding was measured for up to 240 to 300 minutes after amphetamine challenge in numerous experiments (Laruelle et al., 1997b). Without exception, the [123I]IBZMBP still was decreased at its nadir level at these later time points. Such information was not available until recently for [11C]raclopride. Experiments performed with multiple injections of [11C]raclopride confirm the original observation made with [123I]IBZM (R. Carson, personal communication, 1999). Up to 5 hours following amphetamine, [11C]raclopride BP still is decreased at its lower level.
The delay between the peak of DA release and the time to reach a new and lower “equilibrium” level (40 to 60 minutes with [11C]raclopride, 60 to 120 minutes for [123I]IBZM) is easily explainable by the relatively slow dissociation of the radiotracers from the receptors in vivo (Perry et al., 1980; Frost and Wagner, 1984; Frey et al., 1985; Laruelle et al., 1994a; Endres and Carson, 1998; Gifford et al., 1998). In contrast, the sustained and stable nature of the decreased BP is more difficult to explain. At the end of the displacement phase, BP appeared to stabilize at a level lower than baseline, as if a new sustained equilibrium state had been established. Yet, the microdialysis data showed a constant decrease in DA concentration during this period of stable reduced BP.
Consider the case of a radiotracer constant infusion (constant level of free radiotracer in the brain). After the challenge, the radioligand should first predominantly dissociate for as long as the actual bound is higher than the bound predicted by the receptor availability and the Michaelis-Menten equilibrium equation. However, once equilibrium is achieved, this equilibrium should be short-lived. After this nadir, the radiotracer should predominantly associate, since the actual bound would be lower than the bound predicted by the receptor availability and the Michaelis-Menten equilibrium equation. Thus, the lack of reassociation of the radiotracer during that period is not consistent with the hypothesis that changes in radiotracer BP are primarily driven by changes in DA occupancy of the D2 receptor.
Endres et al. (1997) developed a mathematical model to assess the effect of a DA pulse on [11C]raclopride-specific binding achieved during radiotracer constant infusion. Using simulations, these authors demonstrated that, for an average DA pulse, [11C]raclopride-specific binding should return to half of its baseline value by the 110- to 120-minute postamphetamine injection. Since this is not the case, these simulations establish more formally that the simple occupancy model based on microdialysis data fails to account for the prolonged decrease in [11C]raclopride BP observed following amphetamine. Thus, these kinetic analyses are important to clarify the lack of agreement between the simple occupancy model and the experimental data.
The only way to reconcile this observation with the occupancy theory is to postulate that, within the synapse, elevated and stable levels of DA are maintained during this long period. Microdialysis measurements do not rule out such a phenomenon, since extrasynaptic DA concentrations are not necessarily a good measure of intrasynaptic DA concentration (see earlier). In addition to its DAT blocking effect, amphetamine inhibits monoamine oxidase activity (Green and El Haut, 1978) and stimulates DA synthesis (Uretsky and Snodgrass, 1977). It is plausible that these DA-enhancing properties are more important at this later phase than the DAT blocking-reverse transport mechanism, which could explain a time-dependent dissociation between extrasynaptic and intrasynaptic DA concentration.
Also, in a recent report, Volkow et al. (1999) showed a smaller decline in [11C]raclopride following cocaine (1 mg/kg) when cocaine was administered 30 minutes before [11C]raclopride (−13% decrease, n = 1) and 5 minutes before [11C]raclopride (−29 + 3%, n = 3). These data suggest that, at least after cocaine, the decrease in [11C]raclopride BP is transient, which is consistent with the short duration of the effect of cocaine on DA transmission. However, additional experiments are warranted to further document this important question.
Another explanation for the long duration of decreased BP following amphetamine is that the DA surge would induce a long-lasting change in D2 receptor that would not be dependent on sustained increased occupancy of these receptors by DA. Rapid agonist-mediated receptor downregulation, internalization, or phosphorylation might account for this long-lasting decrease in BP. This point is revisited later, after discussion of the spiperone and D1 receptor data. For now, it is proposed that the long-lasting reduction in [11C]raclopride and [123I]IBZM BP following amphetamine challenge raises doubt about the validity of the simple occupancy model, without refuting it definitively.
Radiotracer characteristics and vulnerability of endogenous DA competition. The discrepancies between the occupancy model predictions and the data collected with spiperone and D1 radiotracers constitute a second line of evidence against this model. However, the relevance of the spiperone and D1 radiotracers data can be appreciated only after clarification of the role of radiotracer affinity in vulnerability to endogenous DA competition within the framework of the occupancy model. The role of radioligand affinity in vulnerability to endogenous DA is a complex topic. The initial proposition of Seeman (1989) that low-affinity radiotracers such as raclopride bind more “loosely” to D2 receptors, and, therefore, are more vulnerable to DA competition has gained wide acceptance, possibly because of the intuitive nature of this proposition. However, this proposition violates well established rules of receptor-ligand competition. To fully appreciate this violation, two situations must be considered, that is, when levels of DA are constant, and when they are rapidly changing.
First examine the case when both the ligand and the competitor (DA) are at equilibrium. Thus, the first question is whether the KD of a radiotracer influences the ability of a competitor to affect its binding at equilibrium. The answer is negative if the radiotracer is present at tracer doses, or positive if the radiotracer is present at significant concentration. This answer is derived from the Cheng and Prusoff equation (Cheng and Prusoff, 1973), which relates the intrinsic affinity of a competitor (K1) to its observed potency (IC50) to displace a radioligand with an affinity of KD, when the radioligand is present at concentration L:
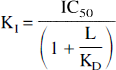
When L is close to 0 (tracer dose), the denominator tends to 1, and the IC50 tends to K1 and becomes independent from KD. The same conclusion can be drawn from Eq. 4. Table 8 shows results derived with Eq. 4 for two radioligands, [3H]spiperone (KD = 0.1 nmol/L) and [3H]raclopride (KD = 1 nmol/L), assuming DA affinity for D2 receptors of 50 nmol/L and a DA concentration of 25 nmol/L. At tracer dose (L = 0.01 nmol/L), both tracers are similarly affected by DA (the presence of DA reduces the radioligand-specific binding by 33% in both cases). As L increases, the effect of DA is reduced for both radioligands. For example, when L is equal to KD (i.e., 50% occupancy of D2 receptors by the radioligand), DA will reduce the specific binding by only 20%. Since the KD of [3H]spiperone is smaller than the KD of [3H]raclopride, the reduction of the DA effect resulting from nontracer doses of the radioligand will be noted at lower concentration of [3H]spiperone than of [3H]raclopride. Results from Table 8 are consistent with the results derived for [11C]NMSP by Logan et al. (1991), who evaluated with another equation the impact of DA on [11C]NMSP binding under equilibrium conditions, using a [11C]NMSP concentration of 1.9 pmol/mL (Logan et al., 1991, Table VIB). At this radioligand concentration, DA has a lower impact on [11C]NMSP than on [11C]raclopride. The authors state that their finding is in contradiction with an earlier statement by Farde et al. (1989). This apparent discrepancy results from the fact that Logan et al. (1991) performed their analysis at non-negligible radioligand concentration, whereas Farde et al. (1989) considered the case of a tracer dose.
Effect of DA on specific bound of [11C]spiperone and [11C]raclopride under equilibrium: effect of radiotracer concentration
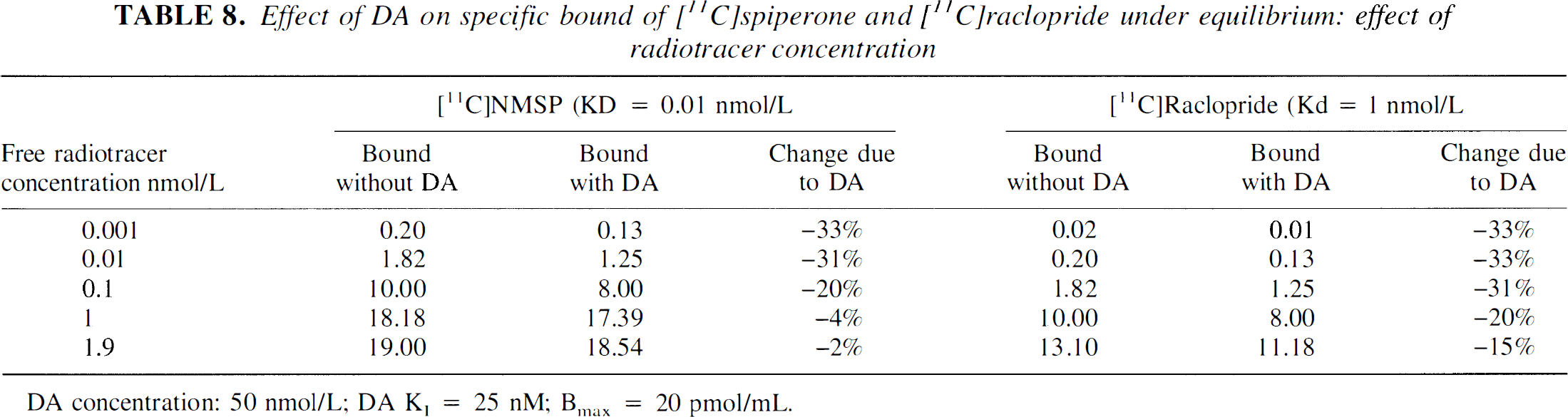
DA concentration: 50 nmol/L; DA K1 = 25 nM; Bmax = 20 pmol/mL.
Once it has been established that, under equilibrium conditions and tracer doses, the affinity of the radioligand does not affect its vulnerability to endogenous DA competition, the case of a pulse of DA must be considered, that is, the effect of a rapidly changing DA concentration. Here, the system is not at equilibrium, and the occupancy model predicts that the affinity of the radioligand (or more exactly, its dissociation rate, koff) will be an important factor. Endres and Carson (1998) provide a comprehensive analysis of the impact of radiotracer affinity to vulnerability of the binding to a DA pulse. They examine these interactions for both single-bolus and constant-infusion administration of radiotracer. In both situations, the decrease in BP measured following a DA pulse will be larger when the rate of dissociation from the receptors (koff) is faster (i.e., lower radioligand affinity since KD = koff/kon), and when the rate of clearance from the brain (k2) is faster (a result previously derived by Logan et al., 1991). According to these analyses, tracers with low affinities are more suitable to endogenous competition studies involving transient changes in DA. As noted by Endres and Carson (1998), there is a limit to this logic, since tracers with low affinity provide such a low signal-to-noise ratio that the effect of the endogenous DA pulse might not be accurately measurable.
In conclusion, within the limits of the simple occupancy model, the affinity of the radiotracer does not impact on its vulnerability to endogenous competition by DA if DA levels are constant during the time frame of the experiment and if the radioligand is given at tracer dose. In contrast, the affinity of a radiotracer will affect its vulnerability to a DA pulse. These conclusions are supported by studies performed with the high-affinity radiotracer [123I]epidipride (KD = 0.024 nmol/L) (Kessler et al., 1991a, b ). Whereas amphetamine-induced DA surge did not affect the washout rate of this radiotracer (Al-Tikriti et al., 1994), sustained decrease in endogenous DA achieved by AMPT administration resulted in increased [123I]epidepride BP in humans (M. Fujita and R. Innis, personal communication, 1999).
The important conclusion is that the resistance of [3H]spiperone to competition by endogenous DA should not be attributed to its higher affinity for D2 receptors. Whereas this factor could explain resistance of [3H]spiperone binding to a DA pulse, the lack of effect of DA depletion is not explainable by the affinity factor (Table 2). Thus, other factors must be involved in the different sensitivity of raclopride and spiperone binding to DA depletion. These other factors are discussed in the next section.
The spiperone question. As reviewed earlier, results obtained with spiperone and analogs are inconsistent with the occupancy model. The first question is whether these paradoxical effects on spiperone BP are real or are artifacts of the difficulties to model appropriately spiperone uptake in the living brain.
First considered is the analysis of [11C]NMSP (or [18F]NMSP) uptake with PET. The modeling approach for [11C]NMSP is different than for [11C]raclopride and other reversible radiotracers. [11C]NMSP does not achieve equilibrium during the time frame of a scan. As a result, neither the dissociation term k4 nor BP can be measured with accuracy, and the outcome measure related to receptor density is the association rate k3 (equal to the product of the association constant kon and receptor density Bmax). A commonly used method to derive k3 is to linearize [11C]NMSP uptake relative to the input function using the Patlak transformation (Patlak et al., 1983; Wong et al., 1986a). Linear regression provides the constant Kin as the slope of the regression line, Kin, which is related to k3 by Kin = K1k3/(k2+k3). K1 and k2 can be derived from the initial portion of the curve and the cerebellum distribution volume, respectively, making it possible to solve for k3. The use of the parameter Kin as an outcome measure has serious limitations: for [11C]NMSP, the normalized derivatives of Kin relative to k3 and K1 were reported as 0.51 and 0.45, respectively (Logan et al., 1991). Thus, a change of 10% in k3 will result in a change of only 5% in Kin, and Kin is similarly affected by rCBF and k3. By extension, the use of striatum to cerebellum ratios at a single time point or other measures based directly on Kin, such as the incorporation quotient (Dewey et al., 1990), will be vulnerable to flow effects.
Logan et al. (1991) report a significant decrease in [18F]NMSP k3 following injection of amphetamine, 1 mg/kg. However, only three experiments are reported, with changes of k3 (i.e, kon Bmax, noted λk3 in this report) of −13%, −29%, and −94%, corresponding to an average of −45% ± 43%. The SD of the effect is much larger than the already large SD of the effect of 1 mg/kg amphetamine on [11C]raclopride BP in baboons (e.g., 15% ± 9% in Dewey et al., 1993). In addition, the experiment resulting in a 94% decrease in k3 appears as a clear outlier in the literature; an amphetamine effect of this magnitude on D2 receptor BP has not been reported elsewhere. Using a larger range of amphetamine injections and a larger number of experiments (n = 8), Hartvig et al. (1997) failed to detect a significant effect of amphetamine on [11C]NMSP k3. If anything, these authors detected a trend toward an increase in [11C]NMSP k3 with larger doses of amphetamine. Thus, a fair conclusion would be that no convincing effect of amphetamine on [11C]NMSP-specific binding has been demonstrated with PET, and that more experiments are warranted to elucidate this issue. This lack of effect is generally consistent with the rodent literature.
Since early [3H]spiperone studies in rodents typically used striatal-to-cerebellar ratios at one time point as outcome measures, results of these studies also are potentially affected by rCBF effects, although the kinetics of spiperone uptake is faster in rodents than in primates (Friedman et al., 1984). Could the lack of change or the paradoxical change in BP reported in rodents with [3H]spiperone constitute artifactual results from rCBF effects? Close examination of the data reveals that appropriate modeling approaches were used in at least some rodent studies (Inoue et al., 1991; Young et al., 1991). In addition, electrical stimulation of substantia nigra was shown to lead to increased accumulation of [3H]spiperone, with no change in flow measured with [14C]iodoantipyrine uptake. The large injected mass in some experiments (e.g., 10 μg/kg in Chugani et al., 1988) raises concerns about significant occupancy of D2 receptors by spiperone, a situation that would dramatically reduce the effect of the competitors. However, other experiments were performed at tracer dose (e.g., 0.01 μg/kg in Inoue et al., 1991).
In conclusion, it seems unjustified to disregard the results of these studies as mere experimental artifacts, and it must be concluded that most of the data indicate that manipulation of DA system does not impact on spiperone-specific binding in a manner consistent with the occupancy model. It has already been established that the relatively higher affinity of [11C]spiperone for D2 receptors is not an adequate explanation for the behavior of this radioligand vis-à-vis DA, despite being frequently quoted. Although koff and k2 might play a role in the vulnerability to a DA surge, as discussed earlier, these factors are not expected to play a role in depletion studies.
If spiperone, at tracer dose, is not affected by sustained change in endogenous concentration, it is important to consider the hypothesis that the binding sites of spiperone and benzamides to D2 receptors might be different, and that DA would display higher affinity for benzamides than spiperone. This hypothesis is well supported by in vitro data from laboratories that measured DA inhibition of [3H]raclopride and [3H]NMSP binding in the same conditions. In vitro, 100 nmol/L DA displaced 19% of [3H]NMSP but 50% of [3H]raclopride binding (Seeman et al., 1989). Hall et al. (1990) report DA Khigh and Klow values of 17.8 and 1100 nmol/L for [3H]raclopride, and 714 and 65, 500 for [3H]NMSP, respectively. The best explanation for this difference is that the raclopride binding site on D2 receptor is more closely related to the DA binding site than the spiperone binding site.
Other interesting differences between spiperone and raclopride binding to D2 receptors have been reported. In vitro, the K1 of most D2 antagonists measured against [3H]raclopride is lower than measured against [3H]spiperone binding (Seeman et al., 1997; Seeman and Tallerico, 1998). The same phenomenon is observed in vivo: the plasma haloperidol concentration corresponding to 50% occupancy of [11C]NMSP and [11C]raclopride binding sites to D2 receptor are 3 to 5 ng/mL and 0.4 to 0.6 ng/mL, respectively (Wolkin et al., 1989; Nyberg et al., 1995; Kapur et al., 1997). Despite this apparent higher potency of neuroleptic drugs to displace [11C]raclopride compared with [11C]NMSP binding to D2 receptors, chronic neuroleptic treatment upregulates spiperone binding sites more than raclopride binding sites (Tarazi et al., 1997). The in vitro Bmax of [3H]raclopride has been reported by some to be 1.3 to 1.8 times higher than the Bmax of spiperone in striatum homogenate preparations and in cells expressing the cloned D2 receptor (Niznik et al., 1985; Terai et al., 1989; Seeman et al., 1992; Ng et al., 1994; Jerning et al., 1995; but see Vile et al., 1995; Malmberg et al., 1996). In vivo, [11C]raclopride Bmax values measured with PET in healthy controls are in the 30- to 40-nmol/L range (Farde et al., 1990; Hietala et al., 1994), whereas [11C]NMSP values range from 15 to 25 nmol/L (Wong et al., 1986b; Tune et al., 1993; Nordström et al., 1995). To explain this apparent difference in the number of binding sites, Seeman et al. (1992) postulate that D2 receptors may exist in either monomeric or dimeric configurations. Benzamides may bind to both configurations, whereas spiperone would bind only to the monomeric configuration. Therefore, depending on the dimer-monomer equilibrium, the number of benzamide binding sites may exceed the number of spiperone binding sites. The monomer-dimer theory received recent support from the identification of two species of immunoreactive D2 receptors, of approximately 44 and 93 kDa, respectively (Ng et al., 1994), and from the direct observation that benzamides label both dimers and monomers, whereas butyrophenones label only monomers (Ng et al., 1996a, b ). This preferential binding of spiperone-like compounds to the monomer configuration also was confirmed with photolabeling experiments (Zawarynski et al., 1998). However, the potential relationship between D2 receptor oligomeric states and vulnerability to endogenous DA competition has not been documented.
The documentation of a lower affinity of DA for spiperone—butyrophenone binding sites than for raclopride—benzamides binding sites would be consistent with little or no effect of DA manipulation on [3H]spiperone or [11C]NMSP. But this factor alone does not explain the paradoxical effects observed in many studies (Saelens et al., 1980; Chugani et al., 1988; Bischoff et al., 1991; Inoue et al., 1991; Pellevoisin et al., 1993; Hartvig et al., 1997). A hypothesis to explain this effect has been advanced by Chugani et al. (1988). These authors propose that agonist-mediated internalization of D2 receptors result in a trapping of [3H]spiperone inside the cells, a mechanism that would be responsible for an apparent increased affinity (i.e., slower koff) and increased BP. Before discussing this view in greater detail, this article examines results from endogenous competition studies performed with D1 receptor radioligands.
The D1 receptor question. In vivo, the DA pulse induced by amphetamine failed to affect several D1 radiotracers, including [11C]NNC 756 (Abi-Dargham et al., 1999), [11C]SCH 23390 (Abi-Dargham et al., 1999; Chou et al., 1999), and [11C] NNC 112 (Chou et al., 1999), as well as the binding of the D1 agonist [11C]SKF 82957 (Greenwald et al., 1998; Laruelle et al., 1998). The DA depletion by reserpine decreased the binding of [3H]SCH 23390 in rodents (Inoue et al., 1991) and did not affect the binding of [11C]NNC 112 in primates. Again, the “affinity” factor is unlikely to explain these findings, since the affinities of these radiotracers are in the same range as the affinities of several D2 receptors ligands that are vulnerable to endogenous DA (Table 1). The lack of in vivo effect of amphetamine on [11C]SCH 23390 was not expected, since Gifford et al. (1996), using a superfused brain slice preparation, detected a significant displacement of [3H]SCH 23390 binding following electrically stimulated DA release. This discrepancy shows the limits of in vitro preparations to mimic the in vivo situation.
The lack of effect of amphetamine on D1 radioligand binding might be explainable by factors specifically related to D1 receptors. Yet, factors potentially accounting for differences in vulnerability to endogenous competition between D1 and D2 antagonists binding are uncertain. Affinity of DA for D1 receptors measured in vitro appears to be in the same range as its affinity for D2 receptors (see affinity tables in Seeman, 1993). For example, in primate striatum, Madras et al. (1988) measured comparable affinity of DA for D1 receptors labeled with [3H]SCH 23390 (Ki(H) = 4.61 nmol/L, Ki(L) = 558 nmol/L) and D2 receptors labeled with [3H]spiperone (Ki(H) = 13.8 nmol/L, Ki(L) = 1850 nmol/L). In contrast, in human striatum, Hall et al. (1988) report lower affinities of DA for [3H]SCH 23390 binding (Ki(H) = 897 nmol/L, Ki(L) = 7580 nmol/L) than for [3H]raclopride binding ((Ki(H) = 64 nmol/L, Ki(L) = 3540 nmol/L). The difference in the affinities of DA for D1 receptors, as reported by both laboratories, illustrates how sensitive is the determination of these values to assay conditions and analysis methods. Another unknown is the proportion of D1 receptors in high- versus low-affinity states for DA in vivo. Again, in vitro studies do not document a systematic difference between D1 and D2 receptors in terms of their respective proportion of receptors in the high- versus low-affinity state (Hall et al., 1988; Madras et al., 1988). Moreover, D1 receptors that are in high-affinity state for agonists are expected to be preferentially labeled with the agonist [11C]SKF 82957. Preliminary results indicate that [11C]SKF 82957 BP is not affected either by reserpine or amphetamine (Greenwald et al., 1998; Laruelle et al., 1998).
The location of receptors relative to the DA release site is another factor that must be taken into consideration. A study from Ann et al. (1998) documents that a lower proportion of D1 receptors are located in the synaptic cleft compared with D2 receptors. This factor might explain why D1 radiotracer binding is less affected by changes in endogenous DA compared with D2 radiotracers. However, D1 receptors are rapidly internalized in vivo following amphetamine (Dumartin et al., 1998), suggesting that, despite their predominant extrasynaptic location, D1 receptors are significantly affected by amphetamine-induced DA release.
In addition, the paradoxical increase in [3H]SCH 23390 reported in one study following MK-801 is intriguing (Kobayashi and Inoue, 1993). The increased DA release elicited by NMDA blockade has been consistently shown to decrease raclopride binding (Kobayashi et al., 1995; Ginovart et al., 1998; Breier et al., 1998; Smith et al., 1998) and to increase spiperone binding (Onoe et al., 1994; Kobayashi et al., 1995). Thus, after NMDA blockade, [3H]SCH 23390 behaves more in a spiperone-like manner than in a raclopride-like manner. Further exploration of this interesting response is warranted.
In conclusion, the lack of effect or the paradoxical effect of DA manipulations on D1 radiotracer binding are not easily accounted for within the framework of the occupancy theory. The paradoxical response detected in several experimental conditions forces the potential importance of other mechanisms, such as agonist-mediated endocytosis, to be considered.
Agonist-mediated receptor internalization. Endocytosis-mediated internalization of G-protein-coupled receptors in response to agonist stimulation is one of the numerous mechanisms by which cellular responses to agonists are rapidly attenuated (Feger et al., 1994; Grady et al., 1997; Koenig and Edwardson, 1997). Rapid agonist-mediated internalization to endosomal compartment has been documented for numerous G-protein-coupled receptors (Maloteaux and Hermans, 1994; Faure et al., 1995; Fonseca et al., 1995; Mantyh et al., 1995; Roettger et al., 1995; Sternini et al., 1996; Barak et al., 1997; Roth et al., 1998). Following endosome formation, acidification of the endosomes permits dissociation of the agonist-receptor complex. The receptor can be recycled to the membrane, whereas the agonist is degraded (see references in Grady et al., 1993). Rapid internalization of D2 receptors in face of an agonist challenge has been demonstrated in several transfected cells (Barton et al., 1991; Itokawa et al., 1996; Barbier et al., 1997; Ng et al., 1997; Ito et al., 1999; Iwata et al., 1999; Vickery and von Zastrow, 1999). For D1 receptors, this mechanism has been documented and visualized both in transfected cells (Trogadis et al., 1995; Ariano et al., 1997; Barbier et al., 1997) and in vivo in rat striatum (Dumartin et al., 1998). Intrastriatal injection of the D1 agonist SKF 82958, or intraperitoneal injection of amphetamine, changes the pattern of D1 immunoreactivity in rat striatum from a homogenous staining of plasma membranes to heterogeneous and intense cytoplasmic concentrations (Dumartin et al., 1998). This effect is detectable as early as 4 minutes after injection.
Agonist-mediated internalization has received little attention in the “endogenous competition” literature, with the exception of the report by Chugani et al. (1988). These authors propose that the paradoxical response of spiperone to DA synaptic levels results from trapping of spiperone within the endocytic vesicles. They propose that DA promotes spiperone-receptor complex internalization. The acidic environment of the vesicle results in protonization of spiperone molecules, which in turn limits its diffusion across membranes, ensuing in an apparently slower koff and increased BP. In support of their hypothesis, Chugani et al. (1988) showed that the rate of [3H]spiperone dissociation from microsomal membranes prepared from rat striatum following in vivo binding was fivefold slower than its dissociation following in vitro binding, and that this rate of dissociation was accelerated by mild detergent. In addition, pretreatment of rats with chloroquine, a drug that prevents receptor recycling but not internalization, significantly increased spiperone binding in the microsomal compartment. The trapping of [3H]spiperone and other radioligands such as [3H]ketanserin or [3H]dexetimide in the endocytic compartment of intact cells has been well documented (Maloteaux et al., 1983; Gossuin et al., 1984).
If such a mechanism modulates spiperone binding following changes in synaptic DA, how does it impact on benzamide binding? Agonist-induced internalization of D2 receptor differentially affects radioligand binding, depending on the ability of the ligand to diffuse in the cells. In Y-79 human retinoblastoma cells induced to express D2 receptors, preincubation of the cells with DA promotes a long-lasting and time-dependent increase in [3H]NMSP binding and a decrease in the binding of the benzamide [125I]iodosulpride (Barton et al., 1991). Since the effects of DA exposure on ligand binding are observed after washing DA out of the medium, these effects cannot be attributed to changes in occupancy of D2 receptors by DA. Barton et al. (1991) propose that [3H]NMSP binds to both surface-exposed and internalized receptors, whereas [125I]iodosulpride would bind only to surface-exposed receptors. They attribute this difference to the different lipophilicity of the compounds. [3H]NMSP, being more lipophilic than sulpride, would access internalized receptors, whereas [125I]iodosulpride would not. The authors also note that the Bmax of [3H]NMSP in these cells is larger (60 fmol/mg protein) than the Bmax of [125I]iodosulpride (40 fmol/mg protein). Since DA promotes internalization, the balance between internalized and exposed D2 receptors is shifted toward the internalized compartment, thereby reducing the Bmax of [125I]iodosulpride but not of [3H]NMSP. In addition, [3H]NMSP displayed higher affinity for internalized receptors, an observation consistent the increased in [3H]NMSP binding following agonist-mediated internalization.
This observation was partially replicated in Chinese hamster ovary cells expressing the short (D2S) and long (D2L) isoforms of D2 receptors (Itokawa et al., 1996). The DA exposure caused a 44% ± 3% decrease in [3H]sulpride binding measured by incubation of the treated cells with [3H]sulpride at 4°C for 4 hours after the DA was washed out. The half-life of the decrease in binding was estimated to be 18 ± 2 minutes, and the concentration of DA giving a half-maximal effect was estimated to be 180 ± 90 nmol/L. The binding of sulpride was restored after incubation of the cells at 37°C, indicating that the DA-induced loss of [3H]sulpride binding sites was reversible. In contrast, the binding of [3H]spiperone was not affected by DA exposure under the same experimental conditions. Itokawa et al. (1996) also propose that the different behaviors of the ligands results from the increased lipophilicity of spiperone relative to sulpride. In other terms, agonist exposure results in a reversible sequestration of D2 receptors in a compartment not accessible to sulpride but accessible to spiperone.
Similarly, in transfected COS-7 cells, exposure followed by removal of the D2 receptor agonist pergolide does not affect [3H]spiperone binding but dramatically reduces [3H]sulpride binding, a result interpreted as evidence of pergolide-induced internalization (Barbier et al., 1997).
These results indicate rapid changes in cellular localization of D2 receptors on agonist exposure, and that these changes might have a different impact on antagonist binding, dependent on the properties of the antagonist. Chugani (1988) proposes that the spiperone-D2 receptor complex itself is internalized, and that this internalization is followed by a trapping of spiperone within the acidified endosomes. However, internalization of the antagonist-receptor complex has not been demonstrated for D2 receptors. In fact, it could be predicted that because internalization is promoted by agonist binding, the D2 receptors occupied by spiperone would be protected from internalization. However, a receptor—ligand equilibrium binding state is characterized by constant dissociation and association of the receptor—ligand complex. The formation of the agonist-receptor complex promotes internalization, followed by dissociation of this complex, resulting in a redistribution of free receptors from a membrane compartment to a endosomal compartment. It follows that the cellular redistribution of the receptors might result in apparent changes in BP of antagonists (Fig. 5).
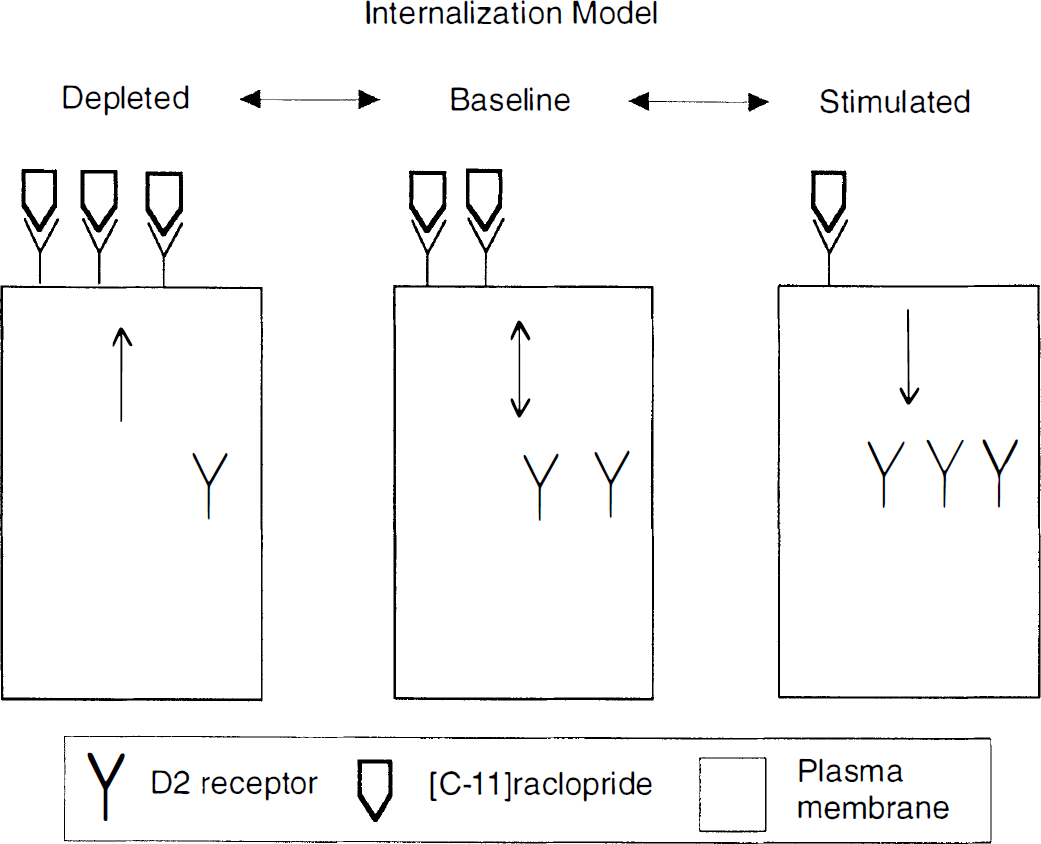
Schematic representation of the internalization model. The model stresses the fact that D2 receptors are distributed between a pool of receptors externalized on the plasma membrane and a pool of receptors internalized in the endosomal compartment. The distribution of receptors between these two compartments is dependent on agonist stimulation. Stimulation of dopamine release shifts the distribution toward the internalized compartment, whereas dopamine depletion shifts the distribution toward externalized compartment. [11C]Raclopride binds preferentially to the externalized receptor because its relatively low lipophilicity prevents its diffusion inside the cell and because the low sodium, high proton environment of the internalized receptors decreases its affinity. In contract, a more lipophilic ligand such as spiperone would access both externalized and internalized receptors. In addition, spiperone binding is less sensitive than raclopride binding to sodium and proton concentration, and trapping mechanisms might provide the appearance of increased affinity of spiperone for internalized receptors, resulting in increased spiperone accumulation after DA stimulation.
Thus, agonist-mediated internalization and subsequent endosomal trapping might account for both the increase in [3H]spiperone binding and the decrease in [3H]raclopride binding following DA pulse if it is accepted that, like sulpride, raclopride's low lipophilicity prevents its diffusion across the plasma membranes. This proposition is directly supported by the data of Rayport and Sulzer (1995), who measured the potency of various antipsychotic drugs to affect the endosomal pH in cultured ventral midbrain DA neurons. Because these drugs are weak bases, their accumulation within the acidic vesicles results in a dissipation of the pH gradient between vesicles and cytoplasm. This pH gradient can be directly visualized with vital dye acridine orange (AO) under fluorescein epifluorescence. Under normal conditions, the acidic milieu of the endosomes is visualized as punctate AO staining. The accumulation of the ligands in the endosomal compartment results in dissipation of the punctate AO staining. Table 9 lists the threshold concentration of various D2 receptor antagonists at which the punctate AO staining dissipates (Rayport and Sulzer, 1995). For example, spiperone dissipates punctate AO staining at 1 μmol/L, whereas 1000 μmol/L of raclopride is required to achieve the same effect. The AO threshold was well correlated with logP (Rayport and Sulzer, 1995). Thus, whereas both spiperone and raclopride rapidly cross the BBB, they exhibit significant differences in their ability to cross the plasma membrane. Spiperone accesses both externalized and internalized receptor pools, whereas raclopride might not significantly access the internalized receptors.
Diffusion of D2 receptor antagonists within the endosomial compartment

Values reproduced from Rayport and Sulzer (1995).
The ability of [123I]IBZM and other benzamides to diffuse within the cell has not been evaluated. Nevertheless, the higher lipophilicity of [123I]IBZM compared with [11C]raclopride should enable this ligand to better diffuse within the cell, if lipophilicity is the only factor involved in this diffusion. Since [123I]IBZM BP is affected by DA challenges in a manner comparable to [11C]raclopride, factors other than lipophilicity might be involved in the accessibility of internalized receptors. On the other hand, the lower binding, or lack thereof, of benzamides to internalized receptors might not be just a matter of diffusion and accessibility.
For example, the binding of benzamides is dependent on the presence of sodium, whereas binding of butyrophenones such as spiperone is not sodium dependent (Stefanini et al., 1980; Hall et al., 1990; Lepiku et al., 1996). Thus, shifting the receptors from an environment with high sodium concentration (extracellular compartment) to low sodium concentration (intracellular compartment) would result in a decrease of the affinity of raclopride and IBZM, but not of spiperone. Acidification of the receptor environment also is likely to differentially affect spiperone and raclopride binding. Spiperone binding is dependent on the protonization of one residue on the receptors with a pKa of 5.8, whereas raclopride is dependent on two ionizing groups with pKa of 6.7. Thus, as pH decreases during the acidification of the endosomes, raclopride affinity will be more affected than spiperone affinity (Williamson and Strange, 1990; Neve, 1991; Presland and Strange, 1991; D'Souza and Strange, 1995). Thus, as sodium concentration decreases and proton concentration increases during the endocytotic process, affinity of D2 receptors for benzamides will be significantly more affected than spiperone.
The D1 radiotracers tested so far behave like spiperone, that is, their binding is not reduced despite the significant internalization of the D1 receptor pool that follows agonist challenge (Dumartin et al., 1998). These tracers are lipophilic and should access internalized receptors. Yet, like the binding of [3H]raclopride, the binding of [3H]SCH 23390 is sodium and pH dependent (Schulz et al., 1985; Faedda et al., 1989; Gottberg et al., 1989; Hollis and Strange, 1992). Thus, internalization of D1 receptor in lower sodium and pH environment should decrease the affinity of SCH 23390 for D1 receptors, resulting in an apparent decrease in BP. Since this is not observed, the reduced affinity from increased pH and lower sodium concentration might be compensated for by a trapping mechanism similar to the one described for spiperone.
It might be concluded that the type of experiments reviewed in this section strongly support the importance of receptor cell trafficking (internalization and recycling) in affecting radiotracer binding. However, a comprehensive theory has not emerged, and additional work is warranted to document the relationship between receptor cellular localization and binding of PET and SPECT radiotracers in intact cells.
GENERAL DISCUSSION AND UNANSWERED QUESTIONS
The occupancy model provides an adequate framework for most observations reported with benzamides and D2 receptor agonists such as [3H]NPA. Challenges that increase synaptic DA decrease the BP of these radiotracers, and vice versa. The amphetamine-induced reductions of [11C]raclopride BP and [123I]IBZM BP have been especially well validated as a measure of amphetamine-induced DA release: the amphetamine effect is mediated by DA release, since it is blunted by cotreatments that are expected to block amphetamine-induced DA depletion (AMPT, reserpine, GBR 12909). The magnitude of the effect is proportional to the change in DA elicited in the extracellular fluids. Thus, the use of this challenge in clinical studies appears to be warranted. However, several other observations raise concerns about the general validity of the occupancy model.
The simple occupancy model could still be compatible with a lack of effect of DA manipulation on spiperone binding, if it is accepted that DA has a low potency to displace spiperone from its binding site on D2 receptors, and, with the lack of effect of DA manipulation on D1 receptors, if it is accepted that D1 receptors are located at sites too distant from the synapse to be affected by endogenous DA competition. At the limit, the model could survive the observation of the long-lasting effect of amphetamine on [11C]raclopride and [123I]IBZM BP, if the hypothesis is accepted that intrasynaptic DA remains elevated for several hours following amphetamine. However, the simple occupancy theory is difficult to reconcile with the paradoxical behavior of butyrophenones and D1 receptor ligands. These data clearly suggest that factors other than changes in DA occupancy might play a role in the modulation of receptor availability following changes in DA synaptic concentration.
The hypothesis that changes in radioligands BP observed in vivo following DA manipulations are mediated by receptor internalization and its consequences for radiotracer binding warrants further evaluation. Changes in receptor occupancy by DA are critical, since these changes will be responsible for the redistribution of the receptor pool between externalized and internalized compartments. Yet, the cellular redistribution of the receptor pool might be the mechanism that ultimately underlies the observed changes in BP. Experiments in cells expressing D2 receptors demonstrate that the redistribution of D2 receptors between externalized and internalized compartments could account for the opposite effects of DA agonists on raclopride and spiperone binding. This redistribution, although reversible, has a different temporal profile than the changes in receptor occupancy following transient changes in DA concentration, which might explain the long-lasting effects of amphetamine on [11C]raclopride and [123I]IBZM BP. Thus, although most observations are compatible with the classic occupancy model, the internalization model has the potential to account for additional observations that are difficult to reconcile with the occupancy model. Yet, although the internalization model is interesting, it remains speculative, and substantive work is needed to confirm the potential importance of receptor trafficking for the phenomena described in this review.
If confirmed, this hypothesis would not necessarily invalidate the conclusions drawn from clinical studies using these challenges, since binding of DA to D2 receptors is the first step in the cascade of events leading to D2 receptor internalization and changes in BP. To this extent, these challenges still would provide a noninvasive measure of D2 receptor stimulation by DA, even if internalization was the mechanism underlying the observed effect. However, as more intermediate steps are involved between the initial event (change in DA concentration) and observed outcome (reduction in BP), the more difficult becomes the interpretation of the data. For example, the exaggerated decreased in [11C]raclopride and [123I]IBZM BP observed in schizophrenic patients, a finding attributed to increased DA release (Laruelle et al., 1996; Breier et al., 1997; Abi-Dargham et al., 1998), might be caused by alterations in D2 receptor trafficking.
In addition, the elucidation of the exact mechanisms underlying this “benzamide effect” is important to the development of this field of brain imaging. Imaging studies of dynamic neurotransmission have been restricted to the study of DA transmission at the D2 receptors with substituted benzamides. Clinical results obtained with this benzamide effect highlight the promises of this technique for the field and motivate the extension of this technique to other receptor systems. Encouraging observations have been reported for the muscarinic (Dewey et al., 1990), nicotinic (Ding et al., 1998) and opiate systems (Frost, 1992), but these observations require considerable validation before maturing into clinical research tools. On the other hand, results obtained so far with D1, DAT, and serotonin 5HT1A ligands are disappointing (Gatley et al., 1995a, b ; Parsey et al., 1998; Abi-Dargham et al., 1999). A better understanding of the multiple factors that construe radiotracer binding vulnerability to changes in endogenous transmitter levels is essential to realize the unique potential of these techniques for measurement of synaptic transmission in the living human brain.
Footnotes
Acknowledgments
The author thanks Rikki N. Waterhouse, PhD, Yiyun Huang, PhD, and Steve Rayport, MD, PhD, for fruitful discussions.