
Abstract
Results from pharmacological studies have suggested that presynaptic N-type Ca2+ channels play an important role in regulating neuronal Ca2+ influx and transmitter nitric oxide (NO) release in isolated cerebral arteries. However, the presence of N-type Ca2+ channels in cerebral perivascular nerves has not been directly demonstrated. As a major source of cerebral perivascular NOergic innervation is the sphenopalatine ganglion (SPG), adult rat SPGs were cultured and examined by whole-cell patch-clamp technique. One week after growing in the culture medium, significant neurite outgrowth from the SPG neuronal cells was observed. Both soma and neurites of these cells were immunoreactive for N-type Ca2+ channels, transmitter-synthesizing enzymes (choline acetyltransferase and NO synthase), and several neuropeptides (vasoactive intestinal peptide, neuropeptide Y, calcitonin gene-related peptide, substance P, and pituitary adenylate cyclase-activating peptide-38) that had been found in cerebral perivascular nerves in whole-mount vascular preparations. In current-clamp recordings, injection of a small depolarizing current caused action potential firing. In voltage-clamp recordings, the fast inward currents were blocked by tetrodotoxin and outward currents by tetraethylammonium, which is typical for neurons. Most Ca2+ currents isolated by blockade of sodium and potassium currents were blocked by ω-conotoxin, indicating that N-type Ca2+ channels are the dominant voltage-dependent Ca2+ channels regulating Ca2+ influx during membrane depolarization of SPG neurons. The ability to culture postganglionic SPG neurons provides an opportunity to directly study the electrophysiological and pharmacological properties of these neurons.
Parasympathetic innervation has been shown to play a significant role in cerebral vasodilation under physiological and pathophysiological conditions (Seylaz et al., 1988; Suzuki and Hardebo, 1993; Kano et al., 1991; Morita-Tsuzuki et al., 1993; Branston, 1995; Faraci and Heistad, 1998). Parasympathetic nerves to cerebral circulation were classically defined as cholinergic in nature (Lee, 1994). Acetylcholine (ACh), which is released from these nerves, however, is not the transmitter that mediates cerebral neurogenic vasodilation (Lee et al., 1975; Lee, 1994). Several mediators other than ACh have been shown to be released from cerebrovascular parasympathetic nerves to relax vascular smooth muscle (Lee, 1994). These include nitric oxide (NO), vasoactive intestinal polypeptide (VIP), calcitonin gene-related peptide (CGRP), pituitary adenylate cyclase-activating peptide (PACAP), and substance P. In particular, NO is recognized as the major neurotransmitter that mediates cerebral neurogenic vasodilation in many species (Lee, 1994). Results from immunocytochemical studies have further indicated that choline acetyltransferase (ChAT) and NO synthase (NOS) and NOS and VIP are colocalized in the same perivascular nerves in cerebral blood vessels of the cat (Kimura et al., 1997) and pig (Yu et al., 1998). Cerebral perivascular NOergic nerves, therefore, have been classified as cholinergic-NOergic nerves and VIPergic-NOergic nerves (Kimura et al., 1997; Yu et al., 1998).
Results from anterograde and retrograde tracing experiments have indicated that one of the major origins of the cholinergic—NOergic innervation and VIPergic—NOergic innervation to cerebral blood vessels is the sphenopalatine ganglion (SPG). Other sources for NOergic nerves include the otic ganglion and internal carotid miniganglion (Suzuki and Hardebo, 1993; Hara et al., 1993; Minami et al., 1994; Okuno et al., 1994; Lee, 1994). Cytoplasmic ChAT, NOS, and VIP immunoreactivities, which are found in the cerebral perivascular nerves, are also shown to exist in neuronal cells of the SPG (Suzuki and Hardebo, 1993; Yu et al., 1998; Kimura et al., 1997).
Our previous report and those by others indicated that NO-mediated neurogenic vasodilation in the porcine cerebral arteries was inhibited by muscarinic receptor agonists and endogenous ACh (Toda and Ayajiki, 1990; Liu and Lee, 1999). These results suggested that endogenous ACh acts as a presynaptic transmitter or modulator (Liu and Lee, 1999) by binding to the presynaptic M2 receptors on the cerebral perivascular NOergic nerves, resulting in inhibition of NO release. The inhibition was suggested to be due primarily to a decreased Ca2+ influx through N-type Ca2+ channels (Liu and Lee, 1999). Direct evidence for the presence of functional N-type Ca2+ channels in these nerves, however, has not been presented.
Isolation and culture of the SPG neurons may offer several advantages for studying the parasympathetic innervation to the cerebral blood vessels. First, cultured neurons allow direct access for electrophysiological recording of membrane ionic currents, which are crucial in neurotransmitter release. Second, cultured neurons allow morphological studies of the distribution of neuronal elements such as neurotransmitters, enzymes, and receptors on the neuron soma and its neuronal extensions. Third, neuronal cultures allow continuous visual access for studies of morphological characteristics of neuritogenesis, connectivity, and toxicity and allow precise control of the environment that affects those processes (Brewer, 1997). Therefore, in the present study, we established a method of isolation and culture of the SPG neurons from adult rats. We demonstrated the presence of neuropeptides in these cultured neurons that have been found in whole-mount vascular preparations and examined the characteristics of calcium current and related electrophysiological properties in these neurons. To our knowledge, this is the first report of the culturing and patch-clamp recording of the ionic current of mammalian SPG neurons. The results indicate that the immunohistochemistry and pharmacology of these cultural neurons resemble those of isolated cerebral arterial preparations.
MATERIALS AND METHODS
Cell culture
Sprague-Dawley rats (3 to 16 weeks old) were anesthetized with sodium pentobarbital (50 mg/kg intraperitoneally). The SPGs of both sides were dissected (Spencer et al., 1990) under a microscope and were placed in cold Hibernate A (Life Technologies, Grand Island, NY, U.S.A.) solution. After being cut into smaller pieces, the ganglia were transferred to Mg2+/Ca2+-free Hanks' balanced salt solution containing papain (1 U/mL; Sigma, St. Louis, MO, U.S.A.), collagenase D (0.6 mg/ml; Boehringer-Mannheim), and dispase (2.4 mg/ml; Gibco, Grand Island, NY, U.S.A.) and were incubated for 40 minutes at 37°C. Cells were released by gentle trituration at the end of the incubation. The cell suspension was centrifuged at 300 g for 5 minutes. The pellet was gently resuspended in Neurobasal culture medium (Life Technologies), containing B27 (1:50 dilution; Life Technologies),
Immunocytochemistry
Cultured cells were fixed in 4% paraformaldehyde for 20 to 60 minutes at room temperature or overnight at 4°C. After rinsing three times with phosphate-buffered saline (pH 7.4), cells were permeabilized and nonspecific sites were blocked with 5% normal goat serum in 0.2% Triton X-100/phosphate-buffered saline for 1 hour at room temperature. After rinsing once, the cells were incubated with the primary antibody. The following primary antibodies were used: anti-neurofilament 200 IgG (1:400, polyclonal; Sigma), anti-ChAT IgG (1:200, monoclonal; Chemicon), anti-brain isoform of NOS (anti-bNOS) IgG (1:200, monoclonal; Transduction Labs), anti-VIP (1:200; Peninsula Labs), anti-CGRP (1:200; Peninsula Labs), anti-galanin (1:10, polyclonal; Euro-Diagnostica), anti-PACAP-38 (1:200, polyclonal; Euro-Diagnostica), and anti-α1b subunit of voltage-gated Ca2+ channel (N-type Ca2+ channel, 1:200, polyclonal; Alomone Labs). Usually the first antibody incubation was at room temperature and overnight. Cells were then rinsed with phosphate-buffered saline three times before incubating them with secondary antibodies for 1 hour at room temperature. The secondary antibodies were fluorescein isothiocyanate (1:40 dilution; Vector Labs) or tetramethylrhodamine isothiocyanate (1:40 dilution; Jackson ImmunoResearch Labs) -conjugated antibody against the IgG of each species of the primary antibodies used. After incubation with second antibody, cells were rinsed with phosphate buffer (pH 8.2) three times and mounted in Vectashield mounting medium (Vector Labs). The stained cells were observed and photographed under a fluorescence microscope fitted with the proper filters (Olympus BX50 microscope). Negative controls were obtained by following the same incubation procedure with neutralized serum by corresponding antigen or without the primary antibody (Liu and Lee, 1999).
For simultaneous labeling of two antigens (double immunolabeling), the specimens were incubated with primary antibodies raised in different species at 4°C for 24 hours (Yu et al., 1998). Immunoreactivities were then revealed by applications of second antibodies in appropriate combinations, namely, fluorescein isothiocyanate-labeled goat anti-rabbit IgG (Vector Labs) in a dilution of 1:40 and tetramethylrhodamine isothiocyanate-conjugated donkey anti-mouse IgG (Jackson ImmunoResearch Labs) in a dilution of 1:40 for 2 hours at room temperature. The specimens were examined in an epifluorescence microscope fitted with proper filter settings for observation of fluorescein isothiocyanate and tetramethylrhodamine isothiocyanate.
Electrophysiological study
Solutions for patch-clamp recording. The extracellular solution for general current recording contained the following (in mmol/L): NaCl 153.7, KCl 5.36, MgCl2 0.812, NaHCO3 0.88, NaH2PO4 0.9, CaCl2 1.8, pyruvate 0.23, 3-[N-morpholino]propanesulfonic acid 10, and dextrose 25 (pH 7.25).
The intracellular solution for general current recording contained the following (in mmol/L): NaCl 5, potassium gluconate 128, CaCl2 0.5, ethylene glycol bis(β-aminoethylether)-N,N,N',N'-tetraacetic acid 5, 4-(2-hydroxyethyl)-
The extracellular solution for Ca2+ current recording contained the following (in mmol/L): NaCl 80.4, KCl 5.36, tetraethylammonium 35, BaCl2 5.0, MgCl2 0.812, NaHCO3 0.88, dextrose 25, NaH2PO4 0.9, CaCl2 1.8, pyruvate 0.23, and 3-[N-morpholino]propanesulfonic acid 10 (pH 7.25). Tetrodotoxin (1 μmol/L) was added in the extracellular medium before Ca2+ current recording.
The intracellular solution for Ca2+ current recording contained the following (in mmol/L): triethylammonium 5, cesium methanesulfonate 115.6, MgCl2 2, CaCl2 0.5, ethylene glycol bis(β-aminoethylether)-N,N,N',N'-tetraacetic acid 5, and 4-(2-hydroxyethyl)-
Cell recording. For recording electrophysiological properties, a glass coverslip containing neurons was transferred from the growth medium to a 35-mm plastic petri dish containing the extracellular recording solution at 37°C. The dish was allowed to cool to room temperature and then transferred to the stage of an inverted microscope for further study.
Cells were studied using the tight-seal whole-cell recording technique (Hamill et al., 1981). Electrodes were pulled from 1.5-mm outer diameter, 1.0-mm inner diameter capillary glass (World Precision Instruments PG52151-4), and the tips were then slightly fire-polished. After filling with intracellular solution, electrode impedance in the extracellular recording solution was 4 to 6 MΩ. This component of the series resistance was fully compensated with the bridge balance control of the Axoclamp 2B (Axon Instruments) used for recording. The electrode tip potential was also subtracted while in bridge mode. Tight seals of at least 2 GΩ, but usually 5 to 10 GΩ, were obtained by light suction. Electrode series resistance increased to 8 to 10 MΩ after entry into whole-cell mode but was not further compensated.
Current and voltage traces were low-pass filtered at 3 kHz and digitized at 20 kHz. Membrane input resistance was measured by observing the currents in response to a series of 100-millisecond duration hyperpolarizing and depolarizing voltage pulses from a holding potential of −65 mV. The steady-state current response was measured at a 90-millisecond latency from onset of the voltage pulse. A straight line was fitted to the linear portion of the voltage-clamp current-voltage (I – V) curve. Input resistance was taken as the reciprocal of the linear portion of the curve. For measurement of voltage-sensitive responses, the resistive component of the leak current was then subtracted from the measured response. For measurement of calcium currents, we used a P/4 procedure to subtract both the resistive and the capacitative component of the measured currents (Evans et al., 1998). Data were expressed as means ± SD.
RESULTS
In vitro growth of adult rat sphenopalatine ganglion neurons
Isolated SPG cells started to adhere to the poly-
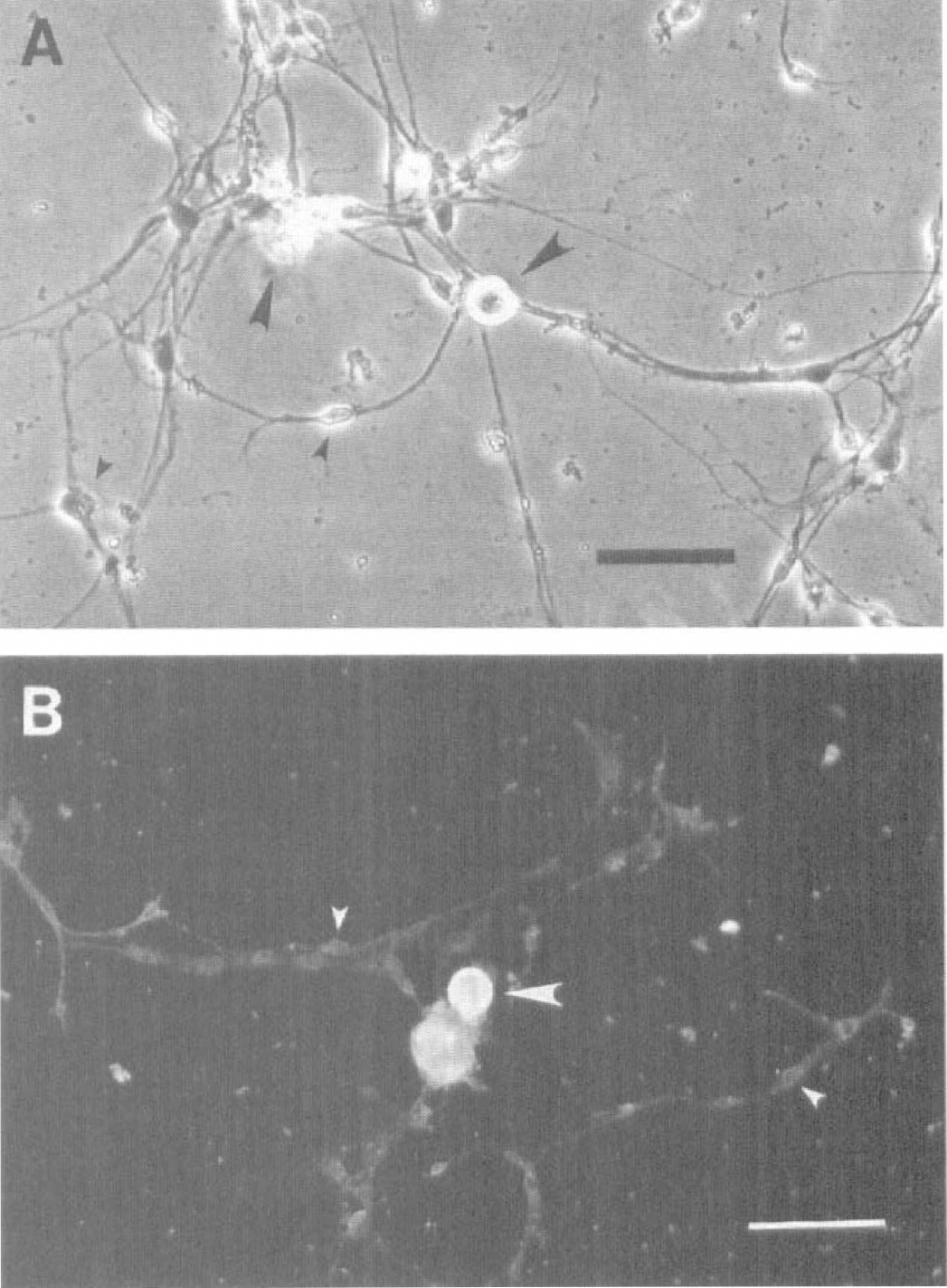
Phase-contrast image of adult rat sphenopalatine ganglion (SPG) neurons in culture (15th day).
Neuron identification
Immunocytochemical and electrophysiological approaches were applied to further distinguish the small and the large soma cells. Strong immunoreactivity to neurofilament 200, a neuron marker, was shown in the large soma cells (Fig. 1B). The same staining was relatively weak in the small soma cells (Fig. 1B).
Most of the large soma cells (8 of 10) fired action potentials (APs) in response to a small depolarizing current injected during patch-clamp recording (see below). No small soma cells (0 of 15) fired AP, nor did they exhibit any active membrane current. Therefore, only the cells with large soma were considered as the SPG neurons, and further experiments were carried out in these cells.
Immunocytochemistry
The cultured SPG neuronal cells were immunoreactive for ChAT, bNOS, VIP, CGRP, neuropeptide Y, substance P, and PACAP (Fig. 2). For negative controls, immunoreactivities of these enzymes and peptides were not observed by omitting primary antibodies (data not shown). The cultured SPG neuronal cells were immunonegative for tyrosine hydroxylase and galanin (data not shown).
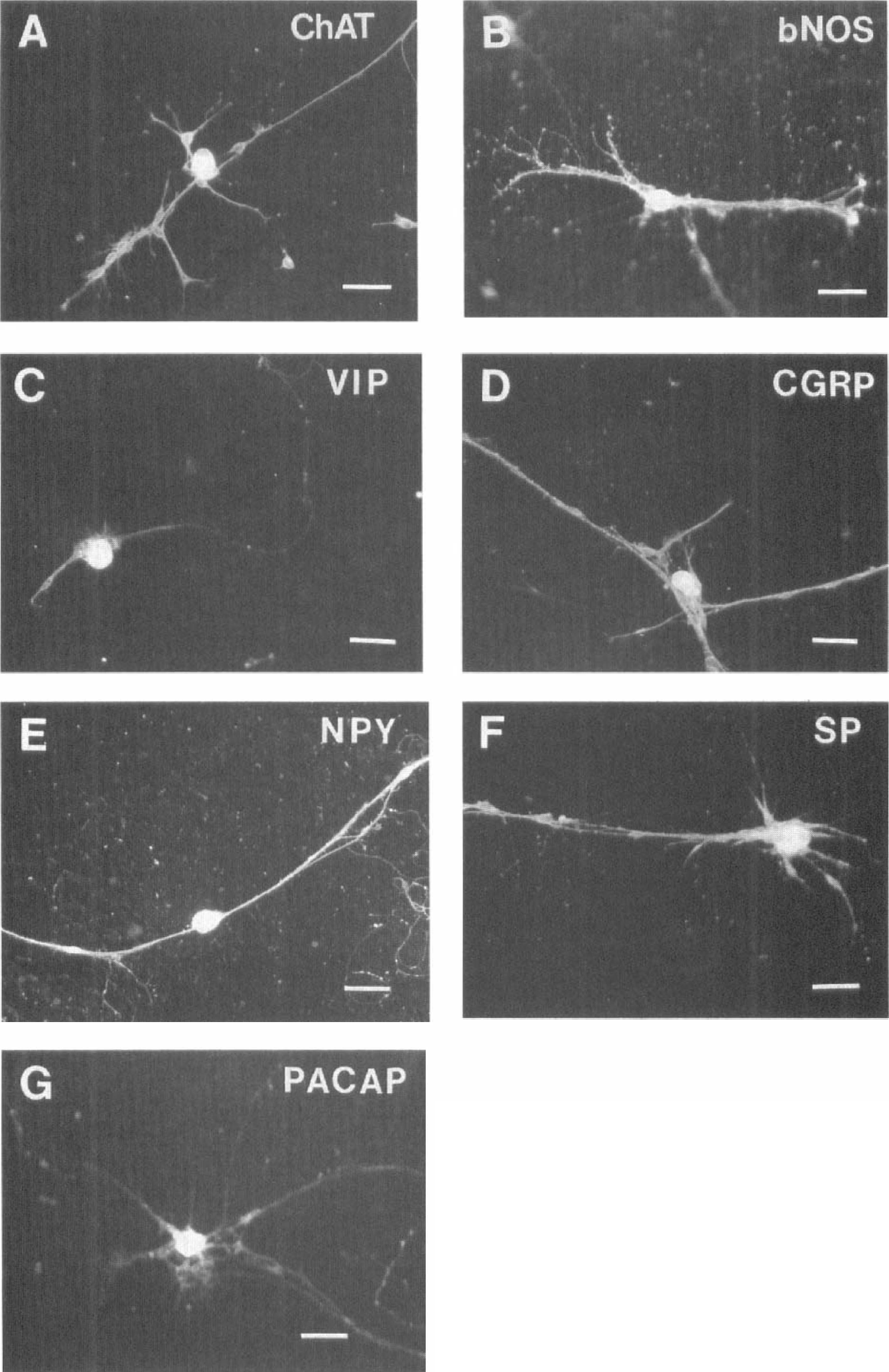
Cultured adult rat sphenopalatine ganglion neurons are immunoreactive for choline acetyltransferase (ChAT;
Passive membrane properties and action potential
The resting membrane potential of the cultured SPG neurons was measured immediately after entry into the whole-cell recording configuration (Fig. 3). The resting membrane potential was variable and ranged from −32 to −66 mV (−45.9 ± 10.9 mV; n = 10). The (I – V) curves were usually linear from −40 to −70 mV. Input resistance calculated from the linear portion of the curve was variable, ranging from 221 to 2,000 MΩ (773.7 ± 653.8 MΩ; n = 7). The membrane capacitance of the SPG neurons ranged from 26.9 to 68 pF (40.6 ± 15.9 pF; n = 7). Capacitance was estimated by injecting a small hyperpolarizing voltage pulse, subtracting the resistive component of the current, integrating the capacitative component, and dividing by the magnitude of the voltage pulse. Although cultured SPGs were not spontaneously active, depolarizing current injection elicited APs in most SPG neurons (8 of 10 examined). Of the recorded sampled neurons, the mean threshold of APs was −35.1 ± 1.6 mV (n = 4), the amplitude of APs was 68.9 ± 5.7 mV (n = 4), and the duration of APs at half the maximum AP amplitude was 3.6 ± 0.8 ms (n = 4) (Fig. 3A). In the presence of tetrodotoxin (1 μmol/L), none of the SPG neurons fired APs (n = 21).
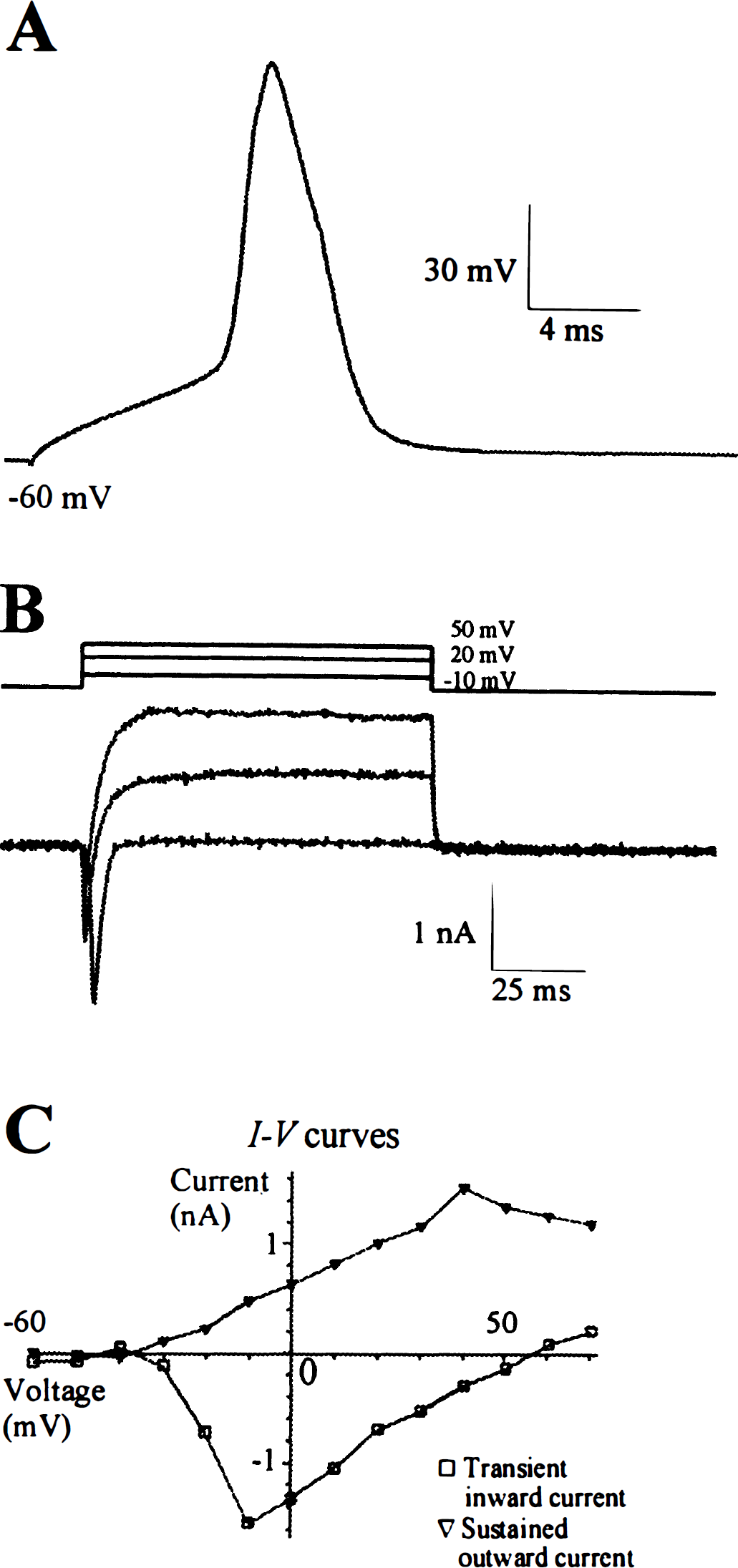
Action potentials (APs) and voltage-sensitive membrane currents in a cultured adult rat sphenopalatine ganglion (SPG) neurons (20th day in culture).
Voltage-sensitive membrane current
Depolarizing voltage steps from a holding potential of −60 mV activated transient inward currents and sustained outward currents in cultured SPG neurons (Fig. 3B). The transient inward currents were voltage sensitive and always activated at steps −20 to −30 mV, peaked at −10 to 0 mV, and reversed at +55 to +60 mV (Fig. 3C). The currents lasted no longer than 10 milliseconds. These inward currents were blocked by tetrodotoxin (1 μmol/L; n = 4, data not shown), suggesting that the main charge carrier of this transient inward current comprises sodium ions. The outward currents, which followed behind the transient inward currents, were sustained and did not substantially inactivate during a 100-millisecond voltage pulse (Fig. 3B). The outward currents were voltage sensitive also. They were activated at −20 to −10 mV and increased linearly with further depolarization (Fig. 3C). The reversal potential of the outward current was measured in some neurons, and it was close to −70 mV (n = 4, data not shown). Cells examined in the presence of extracellular triethylammonium (35 mmol/L) and intracellular cesium completely lacked outward currents (Fig. 4A), suggesting that potassium ions were the major charge carrier for the sustained outward currents.
Calcium currents in cultured adult rat sphenopalatine ganglion neurons (15th day). Sodium currents were blocked with tetrodotoxin (1 μmol/L), and potassium currents were blocked with tetraethylammonium (35 mmol/L) and cesium. Barium (Ba2+, 5 mmol/L) was added to the extracellular solution as the charge carrier.
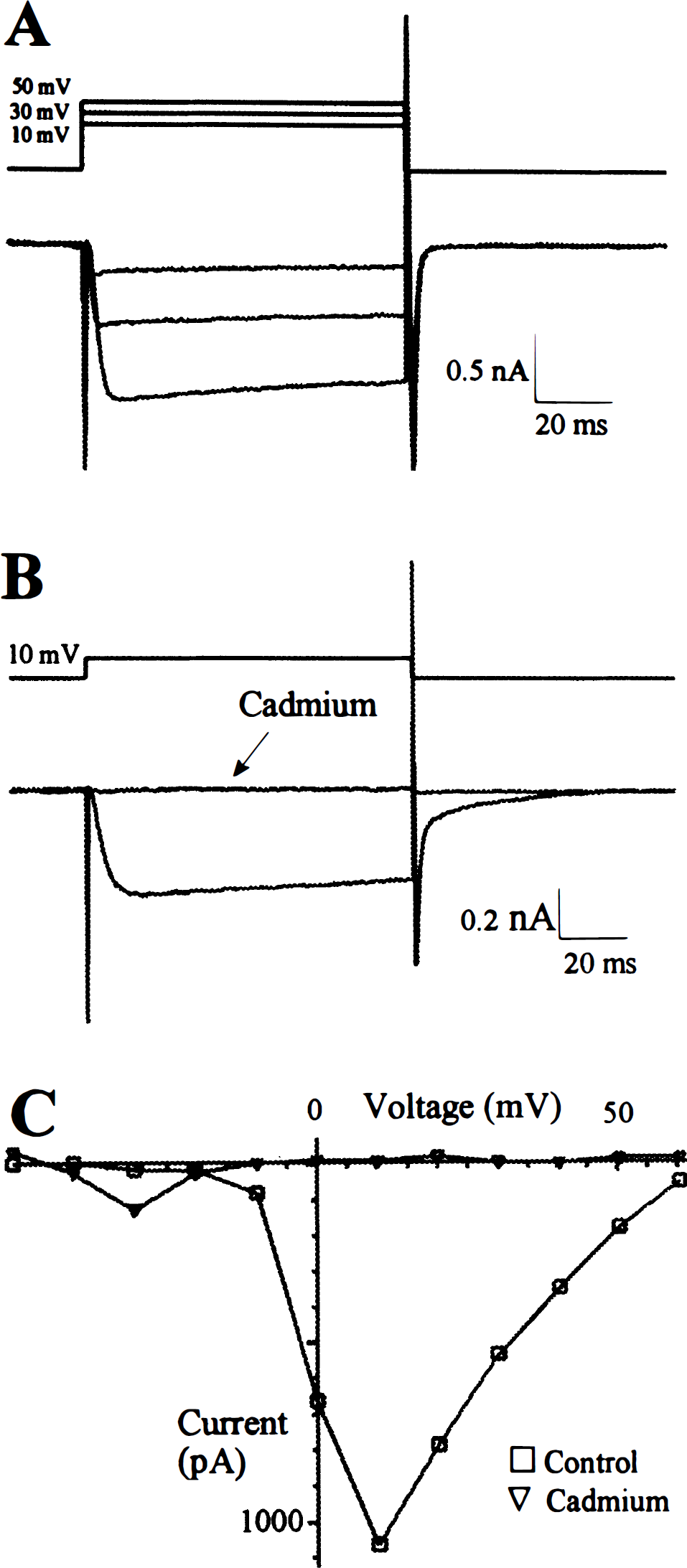
Calcium current
In the presence of tetrodotoxin (1 μmol/L), triethylammonium (35 mmol/L), and barium (5 mmol/L) in the extracellular recording solution and cesium (115.6 mmol/L) in the intracellular solution, both sustained outward currents and transient inward currents were absent, but a slowly activating and inactivating inward current was still present in SPG neurons (Fig. 4A). These inward currents were voltage sensitive, activating about −20 mV with a maximum around 10 mV (Fig. 4C). The currents were blocked by cadmium (0.01 mol/L; Figs. 4B and 4C), suggesting that this voltage-sensitive inward current was a Ca2+ current.
The specific subtype of Ca2+ channels on the SPG neuron was further characterized. ω-Conotoxin GVIA (0.1 μmol/L), a selective N-type Ca2+ channel blocker, significantly inhibited the Ca2+ current on the SPG neurons (Fig. 5; n = 3). On the other hand, nifedipine (0.1 mmol/L), a selective
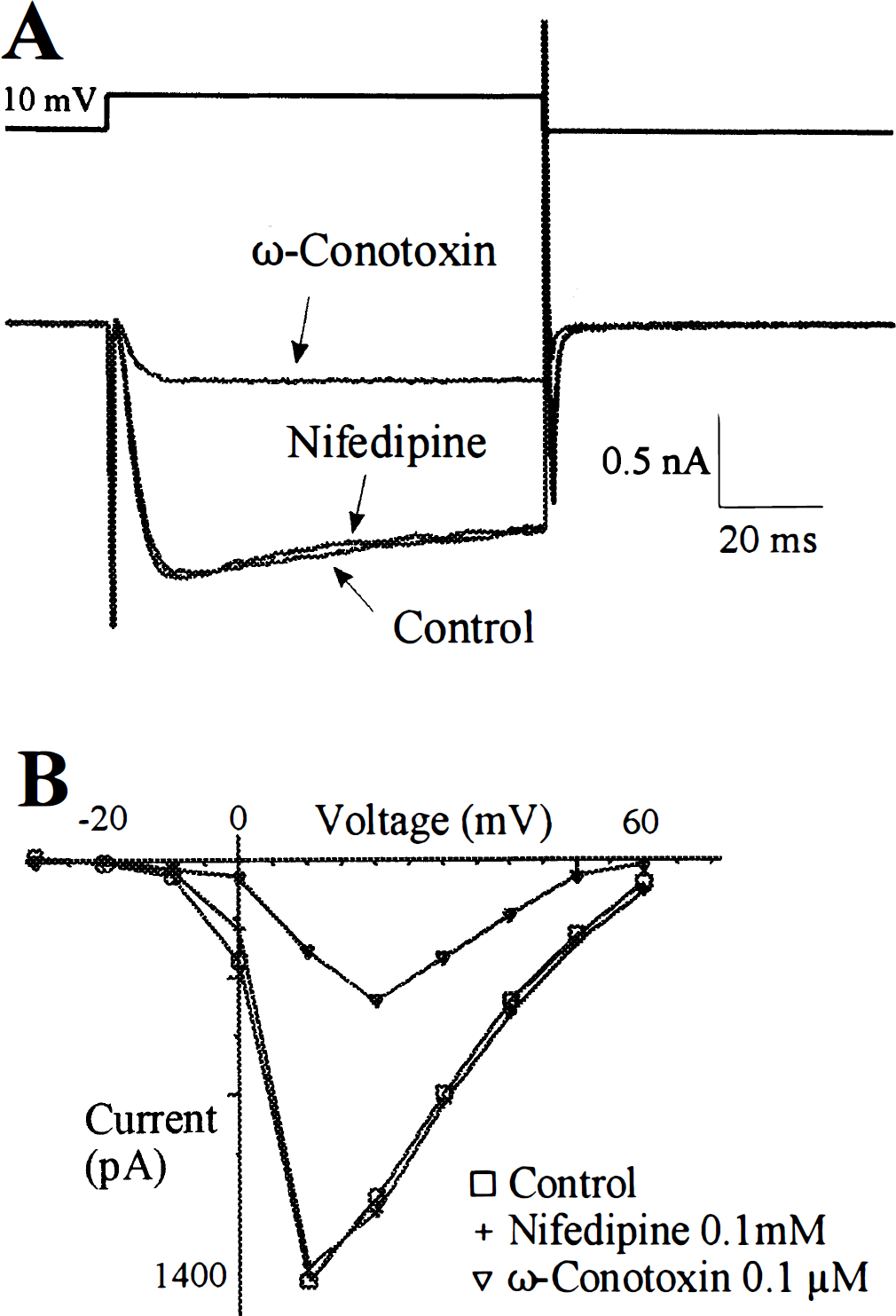
Characterization of Ca2+ channels on adult rat sphenopalatine ganglion neurons (15th day).
All cultured SPG neurons (both soma and fine dendrites) were immunoreactive for N-type Ca2+ channels (Fig. 6A). These N-type Ca2+ channels also were found to be completely colocalized with NOS immunoreactivities (Figs. 6B and 6C). When incubated with antibodies against N-type Ca2+ channels preabsorbed with excess antigens, no N-type Ca2+ channel reactivities were found (data not shown).
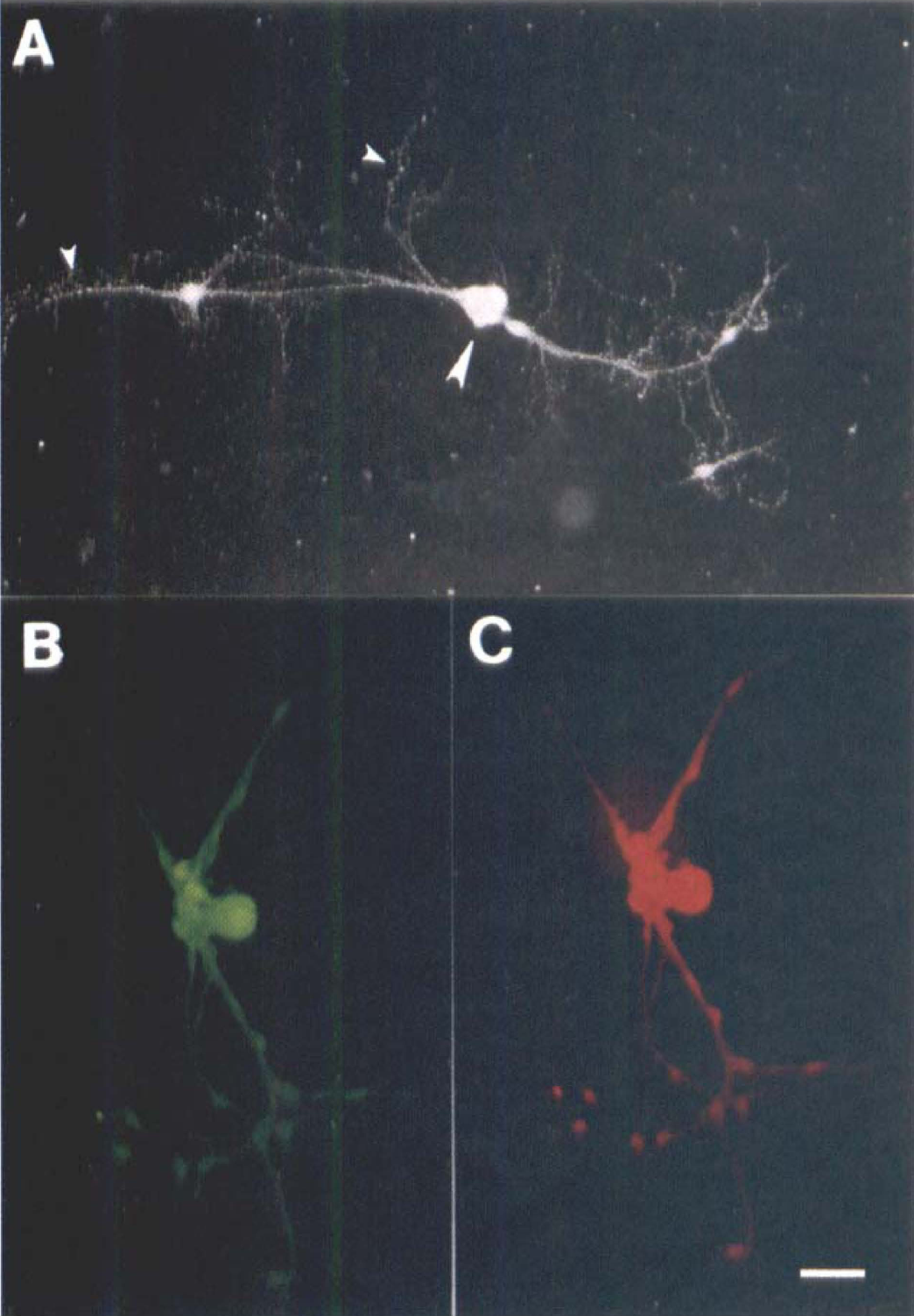
Immunofluorescence labeling of N-type Ca2+ channels in a cultured sphenopalatine ganglion neuron (10th day). The N-type Ca2+ channel immunoreactivities were found in the soma (large arrowhead) and fine fibers (small arrowheads)
DISCUSSION
We demonstrated in the present study that neuronal cells isolated from the adult rat SPG were morphologically similar to other cultured peripheral neurons (Fukuda, 1985; Kelly et al., 1995). By immunocytochemistry, they are shown to possess transmitter substances that have been found in perivascular nerves of cerebral blood vessels, and by electrophysiology they exhibit characteristics of typical neurons. To our knowledge, this is the first report of the culture and patch-clamp recording of adult mammalian SPG neurons and demonstration of the presence of N-type Ca2+ channels on these neurons.
The medium used in culturing SPG, B27-supplemented Neurobasal, is a novel serum-free neuron culture medium. It contains vitamins, essential fatty acids, hormones, and antioxidants (Brewer, 1997). It has been shown that B27-supplemented Neurobasal has several advantages over the serum-containing medium in the primary culture of rat hippocampal neurons. It significantly increases neuron survival in low-density culture. It promotes the development of processes of neurons but tends not to support glial cells in long-term culture (Brewer, 1997). In addition, this medium has been successfully used in the primary culture of the neurons from other sources such as hippocampus, neocortex, striatum, substantia nigra, cerebellum, and dentate gyrus (Brewer, 1995). Moreover, it has been successfully used in culturing adult human cortical neurons (Brewer, 1997). This medium, to our knowledge, was for the first time used successfully in culturing peripheral neurons.
A study in the adult rat SPG has shown that there are ~5,000 neurons in each ganglion (Motosugi et al., 1992). In the present study, the surviving neurons from each SPG in the culture usually numbered below 100. Although the yield of SPG cells was low, the low density of cells allowed good patch-clamp recording and immunocytochemical labeling.
A major source of parasympathetic innervation to cerebral vessels in many species including the rat is the SPG (Uddman et al., 1993; Suzuki and Hardebo, 1993; Hara et al., 1993; Kimura et al., 1997). Several neurotransmitter substances that have been found in cerebral perivascular nerves originating in the SPGs in several species have also been demonstrated in the cultured SPGs in the present study. These include ACh (using acetylcholinesterase/ChAT as markers; Hara et al., 1989; Suzuki and Hardebo, 1993), NO (using NOS as marker; Nozaki et al., 1993; Suzuki et al., 1994; Minami et al., 1994; Kadota et al., 1996), VIP (Uddman et al., 1993; Suzuki and Hardebo, 1993; Motosugi et al., 1992; Nozaki et al., 1993), and neuropeptide Y (Leblanc and Landis, 1988; Suzuki and Hardebo, 1993; Motosugi et al., 1992). Our present results further support the hypothesis that the postganglionic neurons in the SPG are the source of these neurons innervating the cerebral circulation. This is consistent with the findings that these cultured neurons showed no immunoreactivities to tyrosine hydroxylase, a key enzyme for norepinephrine formation in sympathetic nerves. Thus, cultured SPG neurons appear to have immunoreactivities identical to those of nerve fibers in whole-mount vascular preparations.
Calcitonin gene-related peptide and substance P have also been found in cerebral perivascular sensory nerves in several species including the rat, but these nerves originate mainly in the trigeminal ganglion (Suzuki and Hardebo, 1993; Edvinsson et al., 1998). The presence of these two neuropeptides in ganglionic cells in the SPG remains undetermined (Edvinsson et al., 1989; Motosugi et al., 1992). Calcitonin gene-related peptide-I fibers, however, have been shown to innervate the NOS-I ganglionic cells in the SPG (Suzuki and Hardebo, 1993; Yu et al., 1998). The demonstration of CGRP or substance P immunoreactivities in the cultured neurons despite low populations suggests that some CGRP and substance P neurons may originate in the SPG. Whether these sensory nerves distribute to the cerebral vessels remains undetermined.
Furthermore, we found that the cultured SPG neurons were immunoreactive to PACAP-38, suggesting that the PACAP-containing nerves found in cerebral vessels of some species such as the cat (Uddman et al., 1993) and dog (Seki et al., 1995) originate in the SPG. The negative galanin immunoreactivity found in the cultured SPG neurons is consistent with a report that the sparse galanin-I fibers to cerebral blood vessels may originate in the trigeminal ganglion (Suzuki et al., 1989).
The SPG neurons were easily distinguished from non-neuronal cells under the phase-contract microscope by their larger cell soma and brighter phase appearance. This judgment was confirmed by electrophysiological study. Most large soma (8 of 10) fired AP or showed voltage-sensitive Ca2+ current. On the contrary, none of the small soma cells, although they also had well developed processes, fired AP or showed any active membrane current. The identity and characteristics of these small soma cells remain to be determined.
The membrane properties of the SPG neurons are, in general, similar to those of other peripheral neurons. However, the resting membrane potential of the cultured SPG neuron (−45.9 ± 10.9 mV) is slightly less negative than that found in the adult rat parasympathetic ciliary ganglion neuron (−50 ± 8 mV; Kelly et al., 1995), the neonatal rat parasympathetic intracardiac neuron (−52 ± 2.1 mV; Xu and Adams, 1992), and the rat sympathetic superior cervical ganglion neuron (−54.9 ± 4.6 mV; Schofield and Ikeda, 1988). The reason for this difference in findings is not clear.
In examining the ionic currents in the cultured SPG neurons by whole-cell voltage-clamp experiments, voltage-sensitive Na+, K+, and Ca2+ currents were isolated and identified based on electrophysiological and pharmacological characteristics. As our interest has been focused on the mechanism of neurotransmitter release in cerebral blood vessels, and it is well established that Ca2+ current, especially N-type Ca2+ current, plays a critical role in triggering neurotransmitter release (Bleakman and Miller, 1995), we therefore further characterized the Ca2+ channels on the cultured SPG neurons. The results indicated that the major component of Ca2+ current was ω-conotoxin sensitive, suggesting that N-type Ca2+ channels were dominant on SPG neuron soma. On the other hand,
In summary, the present immunocytochemical studies demonstrated the presence of transmitter substances in cultured SPG that are similar to those reported in whole-mount cerebrovascular preparations. Electrophysiological and immunocytochemical studies provided further evidence for the presence of functional N-type Ca2+ channels on the cultured neurons, which appear to be the predominant type of Ca2+ channels and have been shown to play an important role in transmitter NO release in cerebral perivascular nerves of isolated vascular preparations. Cultured postganglionic SPG neurons therefore offer an opportunity to directly study the electrophysiological and pharmacological properties of these neurons and provide important information in understanding the neurogenic control of cerebral circulation.