Carrier-Mediated Delivery of Metabotropic Glutamate Receptor Ligands to the Central Nervous System: Structural Tolerance and Potential of the l -system Amino Acid Transporter at the Blood-Brain Barrier
Free accessResearch articleFirst published online January, 2000
Carrier-Mediated Delivery of Metabotropic Glutamate Receptor Ligands to the Central Nervous System: Structural Tolerance and Potential of the l -system Amino Acid Transporter at the Blood-Brain Barrier
The brain endothelial large neutral amino acid carrier (l-system) is well suited for facilitated drug transport to the brain because of its high transport capacity and relatively broad structural substrate tolerance. The authors have examined the potential of this transporter for central nervous system (CNS) delivery of a new family of compounds derived from the large neutral amino acid phenylglycine. These compounds are highly selective for specific isoforms of metabotropic glutamate receptors (mGluRs) but will only become effective therapeutics for CNS diseases such as ischemic disorders, stroke, and epilepsy if they can effectively cross the blood-brain barrier. Using the immortalized rat brain endothelial cell line RBE4 as in vitro blood-brain barrier model, the authors have studied the interaction of phenylglycine and selected derivatives with the l-system-mediated transport of l-[3H]-histidine. The transport of l-histidine was characteristic of the l-system in vivo with the following kinetic parameters: Km 135 ± 18 μmol/L, Vmax 15.3 ± 1.13 nmol/min/mg protein, and KD 2.38 ± 0.84 μL/min/mg protein. The affinities of the l-system for phenylglycine and the derivatives investigated increased in the order S-4-carboxy-phenylglycine (Ki = 16 mmol/L) < R-phenylglycine (2.2 mmol/L) < S-3-hydroxy-phenylglycine (48 μmol/L) < S-phenylglycine (34 μmol/L), suggesting that a negative charge at the side chain or R-configuration is detrimental for carrier recognition, whereas neutral side chain substituents are well tolerated. The authors have further shown (1) that the mode of interaction with the l-system of S-phenylglycine and S-3-hydroxy-phenylglycine is competitive, and (2) that the transporter carries these two agents into the cell as shown by high-performance liquid chromatography (HPLC) analysis of the RBE4 cell contents. The study provides the first evidence for the potential of S-phenylglycine derivatives for carrier-mediated delivery to the CNS and outlines the substrate specificity of the l-system at the blood-brain barrier for this class of mGluR ligands. As the affinities of S-phenylglycine and S-3-hydroxy-phenylglycine for the l-system carrier are even higher than those of some natural substrates, these agents should efficiently enter CNS via this route. Possible strategies for a synergistic optimization of phenylglycine-derived therapeutics with respect to desired activity at the CNS target combined with carrier-mediated delivery to overcome the blood-brain barrier are discussed.
Neurologic disorders such as ischemia, stroke, trauma, epilepsy, and schizophrenia, but also dementia, Parkinson's disease, and Alzheimer's disease have been associated with amino acid excitotoxicity (Lipton and Rosenberg, 1994). Consequently, current strategies to prevent the pathologic consequences of excess glutamate are focused on antagonists for glutamate receptors (GluRs), in particular those of the N-methyl-d-aspartate group (Boxer and Bigge, 1997). However, it is now appreciated that use of therapeutic antagonists to ionotropic GluRs may compromise fast excitatory synaptic transmission thereby complicating chronic therapy (Meldrum, 1994). Therefore, G protein-coupled metabotropic GluRs (mGluRs) that mediate excitatory synaptic transmission only under particular circumstances (Conn and Pin, 1997) are emerging as novel drug targets with the potential to overcome shortcomings associated with ionotropic GluRs (Knöpfel et al., 1995; Nicoletti et al., 1996).
The mGluR family comprises at least eight subtypes, which are grouped into three classes on the basis of sequence similarities, signal transduction mechanisms, and pharmacologic profile of activation (Conn and Pin, 1997). With respect to their effects on synaptic excitation it is expected that excitotoxicity will be prevented by class I (mGluR 1/5) antagonists, whereas agonists of class II (mGluR 2/3) or class III (mGluR 4/6/7/8) will be neuroprotective (Roberts, 1995). The discovery of the first definitive ligands of mGluRs which are derivatives of the large neutral amino acid phenylglycine not only improved understanding of the physiologic role of the mGluR subtypes but also revealed their involvement in excitotoxicity-induced neuronal degeneration (Bedingfield et al., 1995). Although several subtype-specific agents have been identified, the assessment of their neuroprotective potential is often hampered by insufficient in vivo activity, frequently related to failure to reach the central nervous system (CNS) target receptors (Sekiyama et al., 1996).
The clinical efficacy of treatments of CNS disorders depends greatly on the brain penetration of the therapeutic agent which is often poor because of the tightness of the blood-brain barrier (BBB) formed by the endothelial cells lining the cerebral microvasculature (Abbott and Romero, 1996). Many therapeutic molecules rely on lipid-mediated diffusion to cross the brain endothelium, and, therefore, drug lipidization has been a major strategy to enhance drug entry into brain. A more specific route into the brain is carrier-mediated drug delivery across the BBB, currently used successfully in Parkinson's patients treated with the large neutral amino acid l-DOPA (Smith, 1996). This drug utilizes the l-system amino acid transporter at the brain endothelium which exhibits a broad substrate specificity but prefers neutral alpha amino acids with large side chains (Smith et al., 1987).
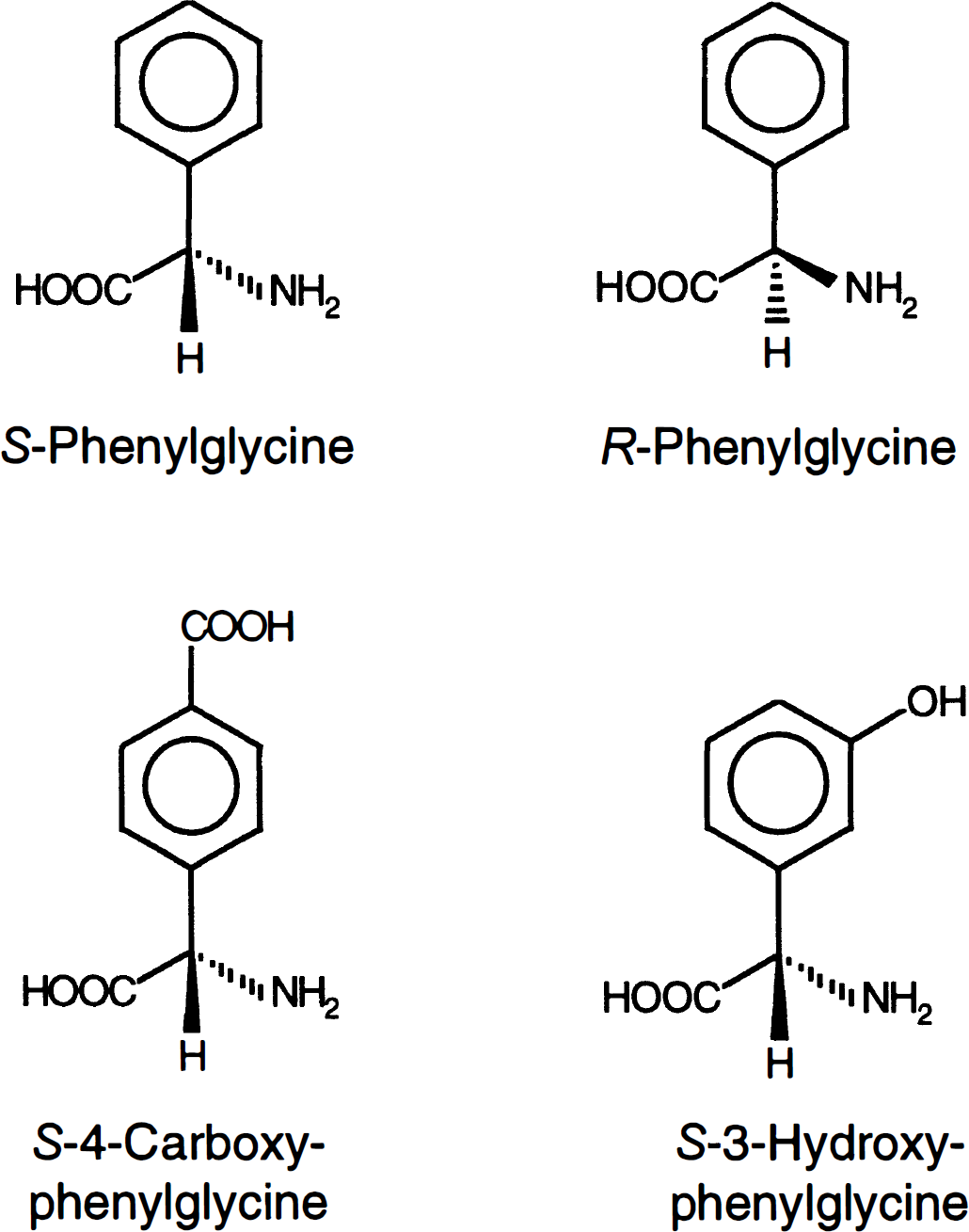
As these molecular characteristics of l-system substrates are shared by phenylglycine, we examined the potential of this transport system for carrier-mediated delivery, first of phenylglycine, and second of selected derivatives (Fig. 1) at the BBB in vitro. We have assessed the affinity of these agents for the l-system with respect to stereochemistry and structural changes in the side chain and confirmed carrier-mediated transport by high-performance liquid chromatography (HPLC) analysis of intracellular drug levels.
Chemical structures of phenylglycine and two derivatives.
The study provides further insight into structure-affinity relationships of the l-system amino acid transporter at the brain endothelium, suggesting that S-stereo-isomery and uncharged substituents on the phenyl ring of phenylglycine derivatives are essential structural requirements for efficient transport. Knowledge of substrate tolerance, together with the high transport capacity of the l-system, make this transporter highly suitable for the delivery of therapeutics by combining high affinity for BBB transport and for target receptors within the CNS.
METHODS
Cell culture
Immortalized rat brain endothelial cells (RBE4 cell line) were grown on rattail collagen-coated 96-well plates or tissue culture flasks in medium consisting of Ham's F10 and α-MEM (1:1, volume/volume) supplemented with 10% fetal calf serum, 1 ng/mL basic fibroblast growth factor, 300 μg/mL Geneticin, and 1 ng/mL glutamine. The medium was changed every other day, and when the cells reached confluence (after ≈4 days) the amounts of basic fibroblast growth factor and fetal calf serum in the medium were halved. The RBE4 cells were used between passages 40 and 50. Experiments were performed 6 to 7 days after seeding.
Transport kinetics
[3H]-histidine was used as model substrate for the l-system. Self-inhibition of [3H]-histidine uptake was assessed by incubating RBE4 cells with aliquots of the uptake medium (Hank's balanced salt solution) containing 10 mmol/L HEPES, pH 7.4) containing trace amounts of [3H]-histidine and [14C]-sucrose and increasing concentrations of unlabeled l-histidine (9 to 1,200 μmol/L). The incubation was carried out for 1 minute at 37°C. This incubation period was shown by time-course studies to be optimal, because uptake was linear with time and the amount of [3H]-histidine accumulated within the cells high enough to allow good resolution.
Estimation of inhibition constants
For studying the interaction of S-phenylglycine, R-phenylglycine, S-4-carboxy-phenylglycine, and S-3-hydroxy-phenylglycine with the l-system transporter, increasing concentrations (9 to 1,200 μmol/L) of the compounds were added to the incubation medium containing 33 nmol/L of [3H]-histidine (0.3 μCi/well) and [14C]-sucrose (0.04 μCi/well) to correct for nonspecific effects such as binding and trapping. The experiments were carried out at 37°C, and the incubation time was 1 minute. In addition, inhibition constants were estimated by adding a fixed concentration of the drug (100 μmol/L) to increasing concentrations of l-histidine (9 to 1,200 μmol/L).
Intracellular uptake of compounds
RBE4 cells grown to confluence in 6-well plastic plates were exposed to Hank's balanced salt solution containing 10 mmol/L HEPES (pH 7.4) and 6.4 mmol/L S-phenylglycine or S-3-hydroxy-phenylglycine, respectively, for 1 minute at 37°C. The uptake of S-phenylglycine and S-3-hydroxy-phenylglycine was also examined in the presence of 10 mmol/L l-histidine. The monolayer was thoroughly washed after terminating the uptake experiment and cells lysed in 1 mL methanol per well. The lysate was centrifuged for 5 minutes to remove precipitated protein and the supernatant frozen until HPLC analysis.
High-performance liquid chromatography analysis of intracellular compounds
Because S-phenylglycine, S-3-hydroxy-phenylglycine, and l-histidine are all α-amino acids, a previously developed HPLC protocol for detection of free primary amino acids in biologic fluids was used (Begley et al., 1994). The analysis was performed using a two-buffer HPLC system and fluorimetric detection by precolumn derivatization of primary amino acids with o-phthaldialdehyde/β-mercaptoethanol.
The HPLC used (A) 0.1 mol/L sodium acetate buffer containing 5% tetrahydrofuran, adjusted to pH 7.2 with glacial acetic acid, and (B) methanol containing 5% tetrahydrofuran. Separation was performed on a 50- × 4.6-mm inner diameter column of Spherisorb 5 octadecylsilyl (ODS) preceded by a guard column 10- × 3.0-mm ID 5 ODS at a flow rate of 1.5 mL/min at 43°C.
Free primary amino acids (including the compounds in question) were derivatized to their fluorescent isoindoles by allowing aliquots (10 μL) of deproteinized cell lysate and 5 μL of fluoraldehyde (Pierce) to react for 1 minute at room temperature. The reaction was stopped by adding 100 μL of starting buffer (A).
Aliquots (20 μL) of the reaction mixture were injected onto the column and separated with a linear gradient from A:B (90:10, vol/vol) to 100% B within 13 minutes. Fluorescence was detected with a fluoromonitor (excitation: 330 nm; emission: 450 nm) and chromatograms were computed using DATA SYSTEM MT2 software (Kontron Instruments).
S-phenylglycine, S-hydroxy-phenylglycine, and l-histidine peaks were identified using (1) 100 μmol/L standard solutions of the respective compound and (2) by spiking cell lysates of untreated RBE4 cells.
Liquid scintillation counting and protein assay
Aliquots (100 μL) of the cell lysate were mixed with 1 mL of isopropanol and 4 mL of scintillation cocktail (Cocktail-T). The radioactivity was determined by liquid scintillation spectrometry (TRICARB 1600TR, Packard) using a quench correction optimized for [3H]/[14C] dual labels. Background activity was determined by measuring the radioactivity of an equivalent volume of Triton X-100 alone.
The protein content of the cell lysates was measured using the BCA protein assay kit and compared to bovine serum albumin protein standards. The absorbance of the samples was measured using a microplate reader (MR 500, Dynatech) with Triton X-100 as the reference sample. The protein content in a 96-well plate at confluency was ≈10 μg per well.
Data analysis and statistics
Cellular accumulation of the tracers has been calculated as distribution volume (Vd, μL/mg protein) and was expressed as a ratio of the cellular radioactivity per milligram protein over the radioactivity in the uptake medium per microliter of uptake medium. Distribution volumes for [3H]-histidine were corrected for nonspecific binding and surface trapping by subtracting the respective Vd values for [14C]-sucrose, a nonpermeant extracellular marker.
Kinetic parameters of the histidine transport were estimated using nonlinear regression analysis of the concentration (C) dependence of the [3H]-histidine influx (J) based on conventional Michaelis-Menten kinetics, i.e., J = (Vmax * C)/(Km + C) + KD * C, with Km as the half-saturation concentration, Vmax the maximum transport capacity, and KD the constant of passive diffusion. Eadie-Hofstee plots (J versus J/C) were used to derive estimates of the kinetic parameters required for the nonlinear regression analysis and to assess the number of transport systems involved.
The interaction of phenylglycine derivatives with the l-system was examined by measuring the effect of increasing concentrations (9 to 1,200 μmol/L) of the respective compound on [3H]-histidine uptake by RBE4 cells. Inhibition constants (Ki) were estimated by adding a fixed concentration of the putative inhibitor ([i] = 100 μmol/L) to increasing concentrations of l-histidine and measurement of the unidirectional influx of [3H]-histidine. The resulting apparent transport affinity (Kapp) was used to calculate Ki according to Ki = (Km * [i])/(Kapp – Km). Lineweaver-Burk plots (1/J versus 1/C) were used to distinguish between competitive and noncompetitive modes of inhibition.
All results are expressed as means ± SD, and significant differences were calculated using two-tailed Student's t test or ANOVA followed by Tukey's protected t test. Kinetic parameters of the [3H]-histidine transport are given with standard errors.
Materials
Tissue culture plastics were purchased from Falcon and culture media and supplements from Gibco. All other chemicals were from Sigma (St. Louis, MO), including [14C]-sucrose (specific activity, 12.6 GBq/mmol). l-[3H]-histidine (specific activity, 1.48 TBq/mmol) was purchased from Amersham. The BCA protein assay kit was supplied by Pierce and Cocktail-T by BDH. All HPLC reagents were purchased from Fisons.
RESULTS
The interaction of phenylglycine and its derivatives with the l-system amino acid transporter at the brain endothelium has been studied using l-[3H]-histidine as substrate for the carrier system and the RBE4 cell line as an in vitro model of the BBB.
Transport of l-[3H]-histidine in RBE4 cells
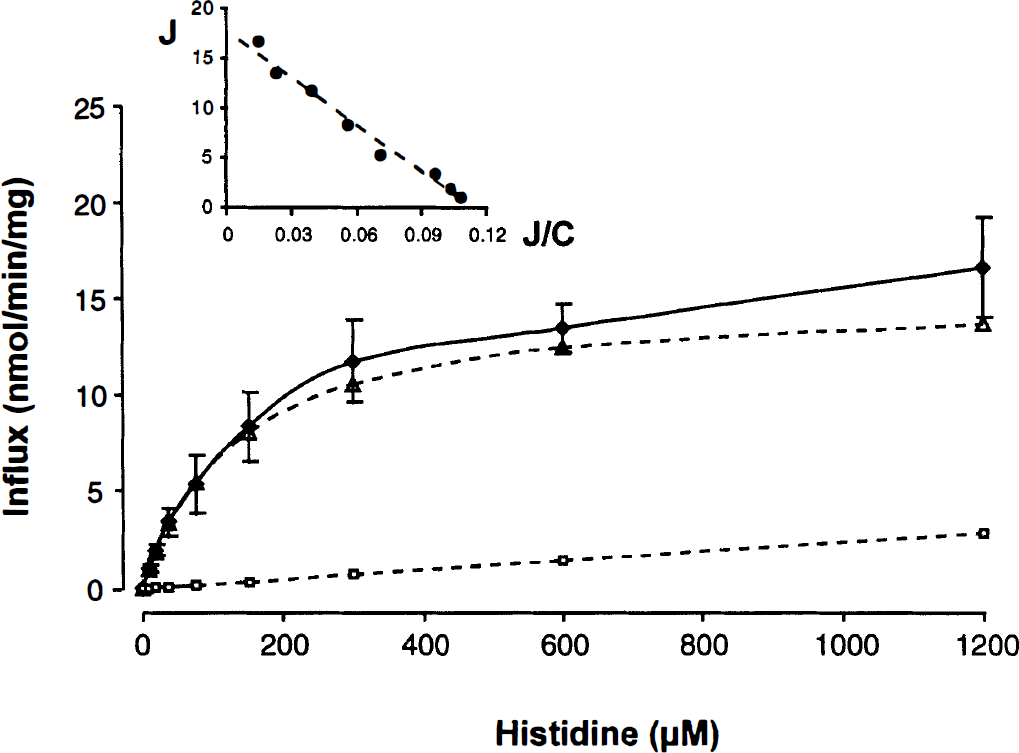
The uptake of l-[3H]-histidine by RBE4 cells was time-dependent with an initial rate of 5.11 ± 0.098 nmol/ min/mg protein and was significantly inhibited by the l-system substrates l-leucine (51%) and 2-aminobicyclo[2.2.1]-heptanecarboxylic acid (43%) but not by the A-system substrate 2-methyl-amino-isobutyric acid (300 μmol/L, 5-minute incubation period). There was some inhibition (19%) after incubation of the cells with ouabain (20-minute preincubation, 5-minute incubation period). The transport of l-[3H]-histidine decreased with increasing concentrations of l-histidine in the incubation medium, showing 92.2% inhibition at a concentration of 1.2 mmol/L (data not shown). The unidirectional influx of l-[3H]-histidine into the RBE4 cells was saturable, reaching a plateau at concentrations above 300 μmol/L (Fig. 2). The transport followed single-carrier Michaelis-Menten kinetics with a Km of 135 ± 18 μmol/L, a Vmax of 15.3 ± 1.13 nmol/min/mg protein, and a KD of 2.38 ± 0.84 μL/min/mg protein. The linear Eadie-Hofstee plot (inset, Fig. 2) suggests that transport of histidine is mediated by one major carrier system.
Saturability of the unidirectional influx of l-[3H]-histidine in RBE4 cells. The incubation time was 1 minute. (Mean ± SD, n = 6, Vd corrected for sucrose). Dotted lines represent the saturable and nonsaturable component of histidine transport as calculated from total influx (J) with J = (Vmax * C)/(Km + C) + KD * C with the kinetic parameters as given in the text and C as the concentration of histidine. The inset shows the Eadie-Hofstee plot (J versus J/C) based on the total flux of histidine.
Effect of phenylglycine and its derivatives on l-[3H]-histidine transport
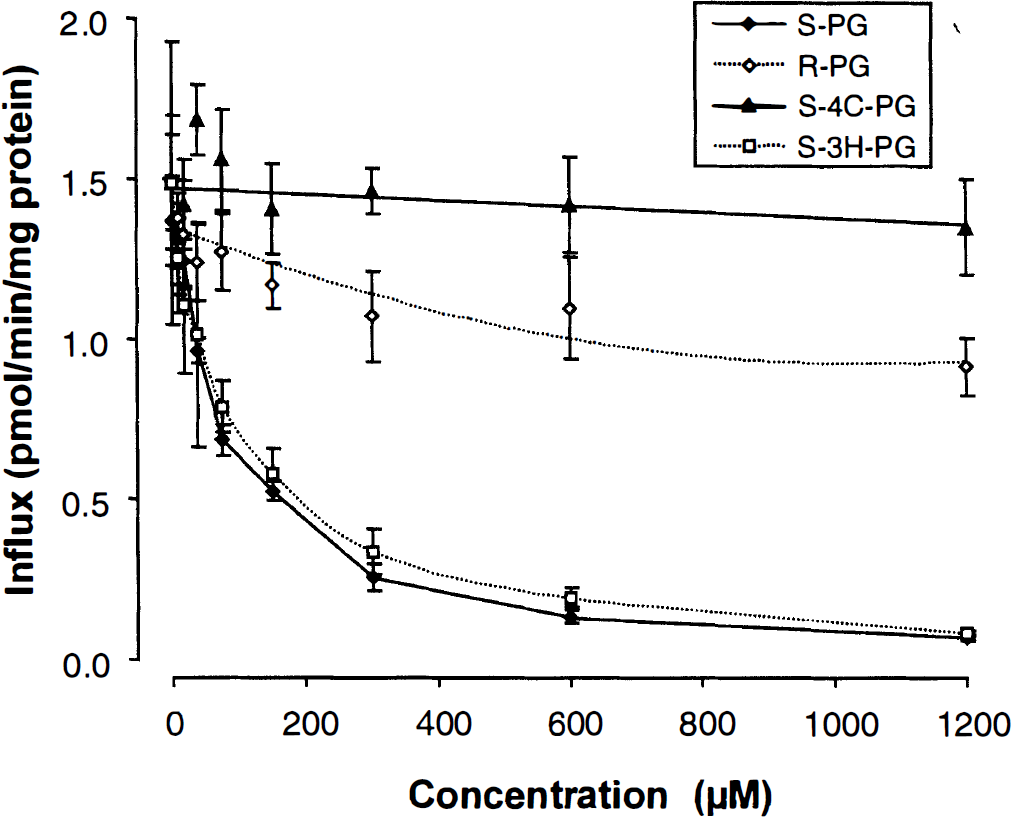
The uptake of l-[3H]-histidine was significantly inhibited by increasing concentrations of the l-form of phenylglycine (S-phenylglycine) but to a much lesser degree by the d-form (R-phenylglycine) (Fig. 3). High concentrations (1.2 mmol/L) of S-phenylglycine led to a virtually complete inhibition of the l-[3H]-histidine transport into RBE4 cells (95%), whereas the same concentration of R-phenylglycine only produced a decrease of 39%. S-4-carboxy-phenylglycine did not affect uptake of l-[3H]-histidine by RBE4 cells up to concentrations of 1.2 mmol/L, whereas increasing concentrations of S-3-hydroxy-phenylglycine significantly decreased l-[3H]-histidine uptake resulting in a 95% reduction of the influx at 1.2 mmol/L (Fig. 3). The respective inhibition constants Ki were estimated as 34.2 ± 3.33 μmol/L (S-phenylglycine), 2.21 ± 0.15 mmol/L (R-phenylglycine), 15.9 ± 4.42 mmol/L (S-4-carboxy-phenylglycine), and 48.6 ± 9.67 μmol/L (S-3-hydroxy-phenylglycine).
Effect of increasing concentrations of the two phenylglycine stereoisomers and two derivatives on the transport of l-[3H]-histidine in RBE4 cells. The incubation time was 1 minute. (Mean ± SD, n = 6, Vd corrected for sucrose). S-PG and R-PG, S- and R-phenylglycine; S-4C-PG, S-4-carboxy-phenylglycine; S-3H-PG, S-3-hydroxy-phenylglycine.
Mode of interaction of S-phenylglycine and S-3-hydroxy-phenylglycine with the l-system transporter
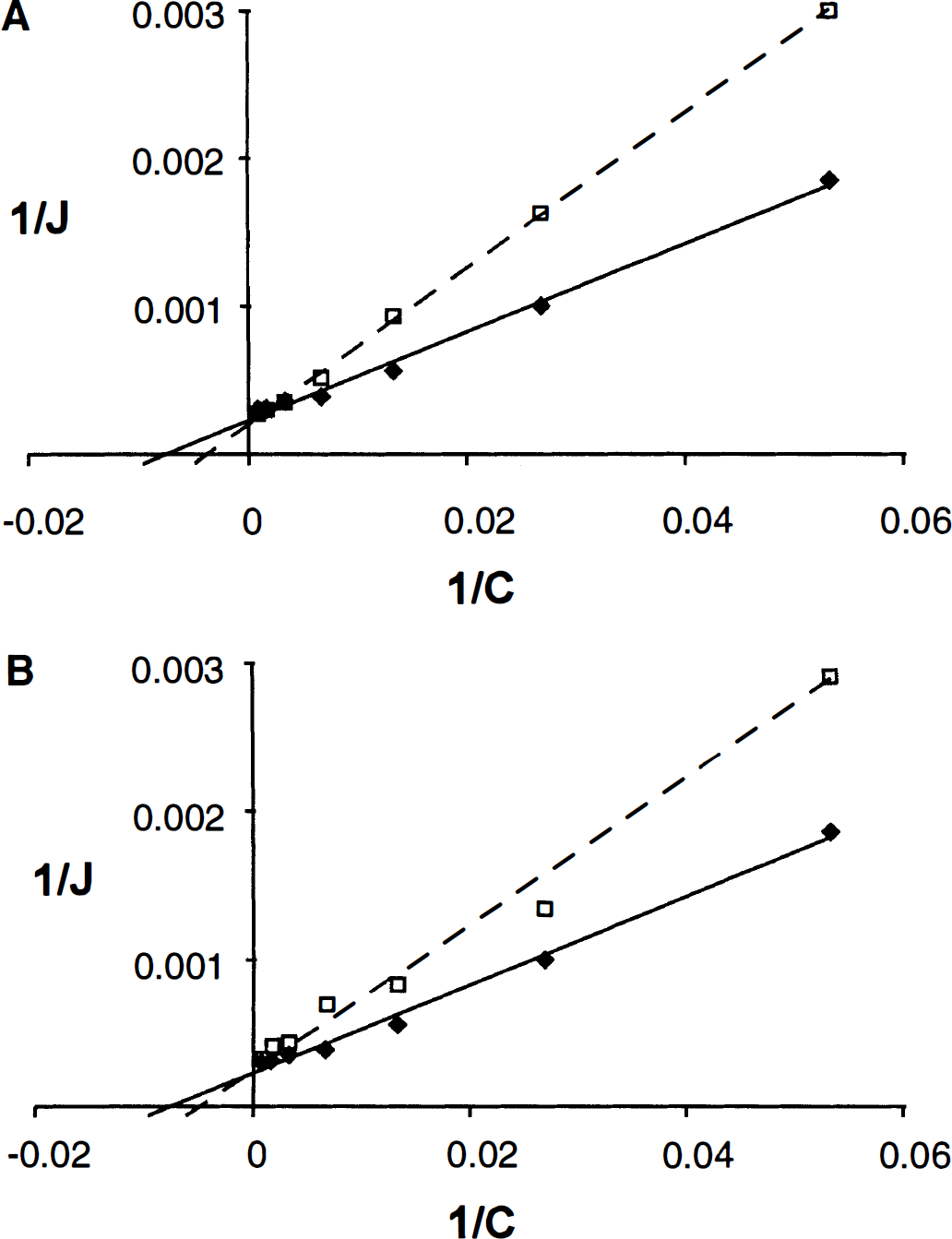
To determine whether S-phenylglycine and S-3-hydroxy phenylglycine act as competitive or noncompetitive inhibitors of l-[3H]-histidine transport in RBE4 cells, the kinetics of l-[3H]-histidine transport were also examined in the absence and presence of a fixed concentration (100 μmol/L) of the respective compound. The data were plotted as double reciprocal (Lineweaver-Burk) plots. As Fig. 4 shows, the regression lines intercept the ordinate at the same point (= 1/Vmax) suggesting a competitive mode of inhibition for both S-phenylglycine and S-3-hydroxy-phenylglycine.
Lineweaver-Burk plots (1/J versus 1/C) of the effect of increasing concentrations of unlabeled histidine on the transport of l-[3H]-histidine in RBE4 cells without (solid line) and with (broken line) a fixed concentration (100 μmol/L) of S-phenylglycine (A) or S-3-hydroxy-phenylglycine (B).
Effect of histidine on intracellular levels of phenylglycine and its derivatives
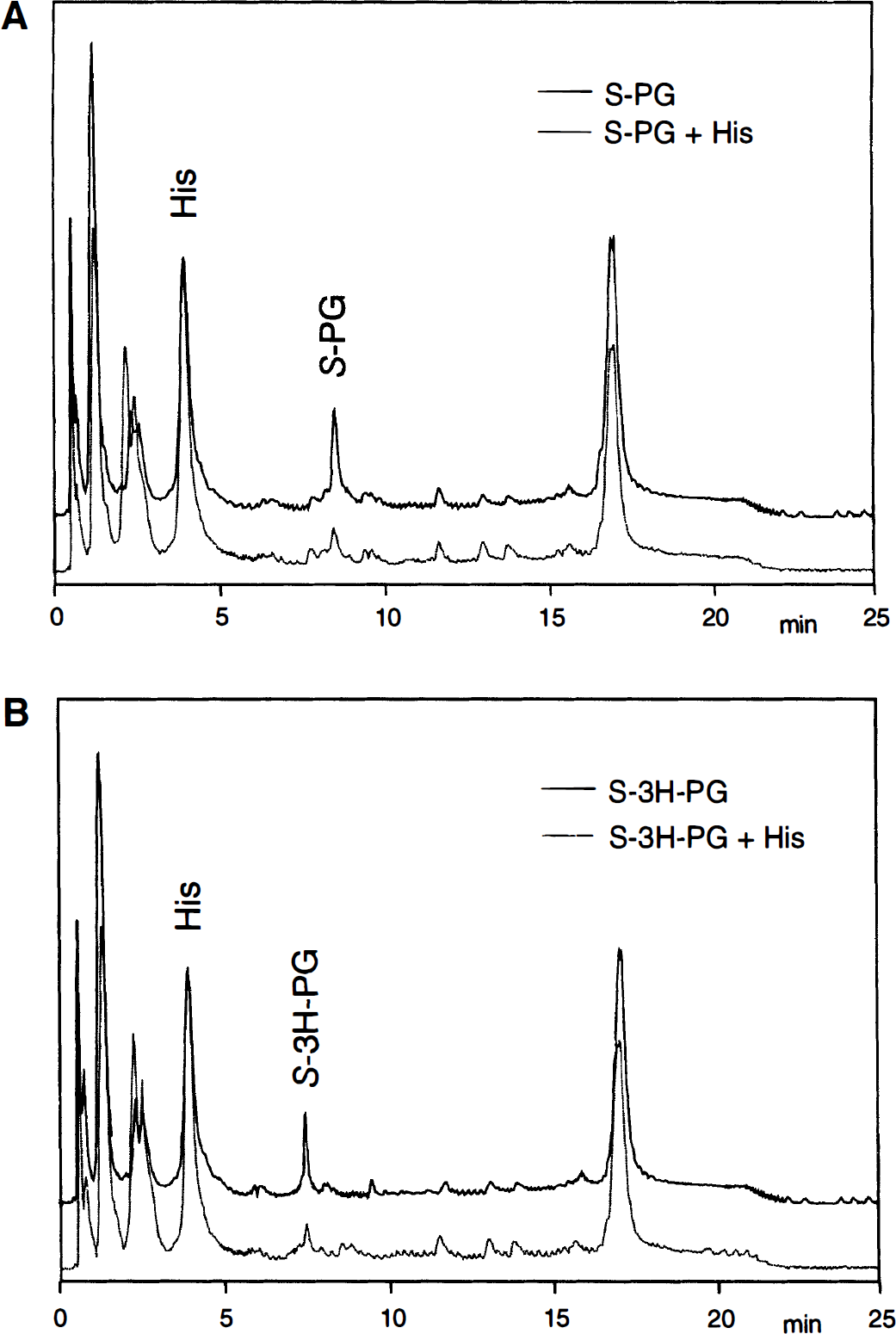
Using the HPLC conditions described, S-phenylglycine, S-3-hydroxy-phenylglycine, and l-histidine were separated with retention times of 8.48, 7.48, and 3.92 minutes, respectively. Variations in the retention times were negligible because of the use of an autosampler for sample injection. The cellular uptake of S-phenylglycine and S-3-hydroxy-phenylglycine was initially investigated after a 1-minute exposure of RBE4 cells to concentrations of 500 μmol/L of the respective drug in Hank's balanced salt solution. The resulting cellular levels of the compounds were however below the detection limit of the HPLC system; therefore, higher concentrations of the drugs in the incubation medium were chosen for subsequent experiments. Figure 5 shows chromatograms representing the cellular amino acid content of RBE4 cells treated with 6.4 mmol/L of S-phenylglycine or S-3-hydroxy-phenylglycine alone and in the presence of 10 mmol/L l-histidine. Under these conditions the peak areas for both S-phenylglycine and S-3-hydroxy-phenylglycine were reduced by 83% and 75%, respectively. These results show (1) that both compounds are capable of entering RBE4 cells and (2) that the entry of both drugs is blocked by an excess of l-histidine.
Overlay of elution profiles of free primary amino acids of RBE4 cell lysates. RBE4 cells were exposed for 1 minute at 37°C to Hank's balanced salt solution (10 mmol/L HEPES, pH 7.4) containing 6.4 mmol/L S-phenylglycine (A, upper trace) or S-3H-phenylglycine (B, upper trace) and in the presence of 10 mmol/L l-histidine (lower traces). The high-performance liquid chromatography (HPLC) method was essentially as given by Begley et al. (1994).
DISCUSSION
The present study investigates the interaction of phenylglycine derivatives with the l-system amino acid transporter at the brain endothelium. l-[3H]-histidine was chosen as the marker substrate because at pH 7.4 it is transported into the brain predominantly by the l-system (Oldendorf et al., 1988). The results confirm that histidine flux was mediated mainly by a single-carrier system with properties characteristic of the l-system. Although we also observed some effect of ouabain, which is in agreement with the presence of transport by the sodium-dependent system N as reported by (Yamakami et al. 1998), the contribution of this carrier to overall histidine flux was only minor (see Fig. 2).
The RBE4 cell line is a well-established in vitro model of the BBB derived from rat brain cortical capillary endothelial cells immortalized with the plasmid pE1A-neo (Abbott et al., 1995). The cell line is well characterized with respect to transport systems such as the l-system amino acid carrier (Gomes and Soares-da-Silva, 1999), nucleoside transporters (Chishty et al., 1997), the GLUT-1 glucose transporter (Regina et al., 1997), and the drug efflux pump P-glycoprotein (Begley et al., 1996). We have demonstrated that the functional characteristics of the l-system amino acid transporter expressed in RBE4 cells closely resemble those found at the BBB in vivo indicating the validity of the model for the present study and the relevance of the findings with respect to drug design for carrier-mediated delivery to the CNS by means of the l-system amino acid transporter (Reichel et al., unpublished data).
The present findings demonstrate that phenylglycine, the parent compound of the congeneric group of specific ligands for mGluRs, is a substrate for carrier-mediated transport at the brain endothelium. The strong dose dependency of the inhibition of the l-[3H]-histidine transport is indicative of a potent interaction of S-phenylglycine with the l-system transporter, and the low Ki value of 34 μmol/L for S-phenylglycine suggests a high transport affinity. The study also agrees with the high stereoselectivity of the l-system (Pardridge, 1983), because the R-form of phenylglycine showed a much weaker interaction with l-[3H]-histidine transport (Ki of 2.21 mmol/L). Poor substrate properties of R stereoisomers were also found for N-methyl-d-aspartate receptor antagonists (Li et al., 1995) and thus should be considered when applying compounds as racemates. Because S-phenylglycine was efficiently recognized by the l-system carrier at the brain endothelium, we also investigated the extent to which the carrier tolerates the addition of different functional groups to the phenyl side chain. Presence of a carboxylic acid group on the phenyl ring (S-4-carboxy-phenylglycine) resulted in complete loss of interaction with the l-[3H]-histidine transport, with the respective Ki value being as high as 16 mmol/L. By contrast, the addition of a hydroxyl group to the phenyl ring (S-3-hydroxy-phenylglycine) only slightly diminished the interaction of S-phenylglycine with the l-[3H]-histidine transport with a Ki value of 49 μmol/L as compared to 34 μmol/L for S-phenylglycine.
The results further suggest that the interaction of S-phenylglycine and S-3-hydroxy-phenylglycine with the l-system transporter is competitive. The comparatively low inhibition constants which are in the lower range of the Km values of natural substrates for the l-system transporter at the BBB in situ (10 to 900 μmol/L) (Smith et al., 1987) suggest a high affinity for the substrate-binding site of the transporter. This is not unexpected, because these two compounds differ only by a single CH2 group from phenylalanine and tyrosine, two natural substrates of the l-system carrier with relatively high affinity: Km = 11 and 64 μmol/L, respectively, at the brain endothelium (Smith et al., 1987). High-performance liquid chromatography analysis of RBE4 cell contents confirmed that both compounds indeed enter RBE4 cells by means of the l-system transporter, inasmuch as their uptake was abolished by an excess of l-histidine. The low intracellular content of S-phenylglycine and S-3-hydroxy-phenylglycine when the transporter was inhibited also suggests that entry by simple diffusion is negligible, which is in agreement with the poor lipophilicity of the compounds, e.g., S-phenylglycine has an octanol/water partition coefficient, log Poct of −1.70 (Hansch and Leo, 1979).
The findings agree with the proposed structural requirements for affinity for the l-system transporter (Smith, 1996), i.e., the carrier prefers α-amino acids with neutral side chains. Polar substituents at the side chain, although reducing the affinity for the transporter, are tolerated when they are uncharged at physiologic pH (e.g., hydroxyl groups). The positive relationship between the affinity for the l-system transporter and the side chain lipophilicity of the substrate (Smith et al., 1987) suggests that the substrate binding site of the transporter is close to or buried within the plasma membrane, a hypothesis which may also explain why charged α-amino acids (e.g., S-4-carboxy-phenylglycine) do not interact with the carrier.
The derivatives examined in the present study are representative of the phenylglycine family of mGluR ligands, mainly varying in side chain substitutions by hydroxyl, carboxylic acid, and methyl groups (Watkins and Collingridge, 1994). The results therefore permit some more general conclusions concerning the tolerance of the l-system carrier for phenylglycine derivatives: (1) Phenylglycine derivatives with carboxylic acid groups will be poor l-system substrates as demonstrated for S-4-carboxy-phenylglycine, whereas phosphonic acid groups, e.g., might be less detrimental because of lower acidity. (2) Phenylglycine derivatives with hydroxyl groups will be efficiently transported by the carrier, with the transport affinity decreasing with the number of hydroxy groups, e.g., S-phenylglycine > S-3H-phenylglycine or Phe > Tyr > l-DOPA (Smith et al., 1987). (3) Phenylglycine derivatives with methyl or ethyl groups at the α-carbon atom will not be tolerated by the l-system transporter (Matthews and Zand, 1977). By contrast, addition of alkyl groups to the phenyl ring will increase side chain lipophilicity and thereby improve the affinity for the l-system transporter.
The present findings indicate that phenylglycine derivatives can use the l-system transporter at the BBB to gain access to the CNS when the compound is in the S-stereoisoform and carries uncharged substituents on the phenyl ring. To translate this knowledge into the design of neuroprotective drugs, a synergistic optimization of both affinity for mGluRs within the CNS and for the l-system at the BBB could be attempted to overcome the often observed lack of in vivo efficacy of mGluR ligands (Boxer and Bigge, 1997). Because not only substrate recognition by the l-system transporter but also the agonist/antagonist profile at mGluRs depends on the side chain substituents of phenylglycine derivatives (Sekiyama et al., 1996), the synergistic optimization of the nature, number, and relative position of the substituents with respect to both beneficial CNS target activity and efficient BBB transport appears a very promising way forward in the search for orally active mGluR agents. Recently, some orally active mGluR antagonists have been reported (Toms et al., 1996; Monn et al., 1997; Gasparini et al., 1999), although the mechanism of brain entry remains to be elucidated.
In conclusion, the present study outlines the potential of the l-system amino acid transporter at the BBB to carry specific mGluR ligands based on phenylglycine derivatives into the CNS. The study demonstrates the usefulness of in vitro BBB models in identifying structural features that contribute to poor CNS penetration and hence lack of in vivo efficacy and suggests a strategy toward a more rational drug design through parallel optimization of CNS target activity and BBB transport efficacy.
Footnotes
Acknowledgments
The authors thank P.-O. Couraud and F. Roux (Institut Cochin de Génétique Moléculaire, Paris) for the supply of the RBE4 cell line.
Abbreviations used
References
1.
AbbottNJRomeroIA (1996) Transporting therapeutics across the blood—brain barrier. Mol Med Today2:106–113
2.
AbbottNJRouxFCouraudPOBegleyDJ (1995) Studies on an immortalized brain endothelial cell line: characterization, permeability and transport. In: New Concepts of a Blood-Brain Barrier (GreenwoodJBegleyDJSegalMB, eds), New York, Plenum Press, pp 239–249
3.
BedingfieldJSKempMCJaneDETseHWRobertsPJWatkinsJC (1995) Structure-activity relationships for a series of phenylglycine derivates acting at metabotropic glutamate receptors (mGluRs). Br J Pharmacol116:3323–3329
4.
BegleyDJReichelAErmischA (1994) Simple HPLC analysis of free primary amino acid concentrations in rat plasma and cisternal cerebrospinal fluid. J Chromatogr B657:185–191
5.
BegleyDJLechardeurDChenZDRollinsonCBardoulMRouxFSchermanDAbbottNJ (1996) Functional expression of P-glycoprotein in an immortalized cell line of rat brain endothelial cells, RBE4. J Neurochem67:988–995
6.
BoxerPABiggeCF (1997) Mechanisms of neuronal cell injury/death and targets for drug intervention. Drug Disc Today2:219–228
7.
ChishtyMReichelABegleyDJAbbottNJ (1997) Characterization of nucleoside transporters in RBE4, an immortalized rat brain endothelial cell line. J Physiol501:31P
8.
ConnPJPinJP (1997) Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol37:205–237
9.
GaspariniFBrunoVBattagliaGLukicSLeonhardtTInderbitzinWLaurieDSommerBVarneyMAHessSDJohnsonECKuhnRUrwylerSSauerDPortetCSchmutzMNicolettiFFlorJ (1999) (R,S)-4-Phosphonophenylglycine, a potent and selective group III metabotropic glutamate receptor agonist, is anticonvulsive and neuroprotective in vivo. J Pharmacol Exp Ther289:1678–1687
10.
GomesPSoares-da-SilvaP (1999) l-DOPA transport properties in an immortalized cell line of rat capillary cerebral endothelial cells, RBE4. Brain Res829:143–150
11.
HanschCLeoA (1979) Substituent Constants for Correlation Analysis in Chemistry and Biology, New York, Wiley & Sons, pp 169–330
12.
KnöpfelTKuhnRAllgeierH (1995) Metabotropic glutamate receptors: novel targets for drug development. J Med Chem38:1417–1426
13.
LiJHBiggeCFWilliamsonRMBoroskySAVatanianMGOrtwineDF (1995) Potent, orally active, competitive N-methyl-d-aspartate (NMDA) receptor antagonists are substrates for a neutral amino acid uptake system in Chinese hamster ovary cells. J Med Chem38:1955–1965
14.
LiptonSARosenbergPA (1994) Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med330: 613–622
15.
MatthewsRHZandR (1977) Basis for substrate preference of amino acid transport system L over amino acid transport system A. Biochemistry16:3820–3824
16.
MeldrumBS (1994) The role of glutamate in epilepsy and other CNS disorders. Neurology44:14–23
17.
MonnJAValliMJMasseySMWrightRASalhoffCRJohnsonBGHoweTAltCARhodesGARobeyRLGriffeyKRTizzanoJPKallmanMJHeltonDRSchoeppDD (1997) Design, synthesis, and pharmacological characterization of (+)-2-aminobicyclo-[3.1.0]hexane-2,6dicarboxylic acid (LY354740): a potent, selective, and orally active group 2 metabotropic glutamate receptor agonist possessing anticonvulsant and anxiolytic properties. J Med Chem40:528–537
18.
NicolettiFBrunoVCopaniACasabonaGKnöpfelT (1996) Metabotropic glutamate receptors: a new target for the therapy of neurodegenerative disorders?Trends Neurosci19:267–271
19.
OldendorfWHCranePDBraunLDGosschalkEADiamondJM (1988) pH dependence of histidine affinity for blood-brain barrier carrier transport systems for neutral and cationic amino acids. J Neurochem50:857–861
20.
PardridgeWM (1983) Brain metabolism: a perspective from the blood-brain barrier. Physiol Rev63:1481–1535
21.
ReginaARouxFRevestPA (1997) Glucose transport in immortalized rat brain capillary endothelial cells in vitro: transport activity and GLUT1 expression. Biochim Biophys Acta1335:135–143
22.
RobertsPJ (1995) Pharmacological tools for the investigation of metabotropic glutamate receptors: phenylglycine derivatives and other selective antagonists—an update. Neuropharmacology34: 813–819
23.
SekiyamaNHayashiYNakanishiSJaneDETseHWBirseEFWatkinsJC (1996) Structure-activity relationships of new agonists and antagonists of different metabotropic glutamate receptor subtypes. Br J Pharmacol177:1493–1503
24.
SmithQR (1996) Carrier-mediated drug transport at the blood-brain barrier and the potential for drug targeting to the brain. In: New Concepts of a Blood-Brain Barrier (GreenwoodJBegleyDJSegalMB, eds), New York, Plenum Press, pp 265–276
25.
SmithQRMommaSAoyagiMRapoportSI (1987) Kinetics of neutral amino acid transport across the blood-brain barrier. J Neurochem49:1651–1658