
Abstract
Abnormalities in cerebrovascular reactivity or hemodynamic reserve are risk factors for stroke. The authors determined whether hemodynamic reserve is reduced in an experimental model of thromboembolic stroke. Nonocclusive common carotid artery thrombosis (CCAT) was produced in rats by a rose bengal-mediated photochemical insult, and moderate hypotension (60 mm Hg/30 min) was induced 1 hour later by hemorrhage. Alterations in local cerebral blood flow (lCBF) were assessed immediately after the hypotensive period by 14C-iodoantipyrine“ autoradiography, and histopathologic outcome was determined 3 days after CCAT. Compared to normotensive CCAT rats (n = 5), induced hypotension after CCAT (n = 7) led to enlarged regions of severe ischemia (i.e., mean lCBF < 0.24 mL/g/min) in the ipsilateral hemisphere. For example, induced hypotension increased the volume of severely ischemic sites from 16 ±4 mm3 (mean ± SD) to 126 ± 99 mm3 (P < 0.05). Histopathologic data also showed a larger volume of ischemic damage with secondary hypotension (n = 7) compared to normotension (22 ± 15 mm3 versus 5 ±5 mm3, P < .05). Both hypotension-induced decreases in lCBF and ischemic pathology were commonly detected within cortical anterior and posterior borderzone areas and within the ipsilateral striatum and hippocampus. In contrast to CCAT, mechanical ligation of the common carotid artery plus hypotension (n = 8) did not produce significant histopathologic damage. Nonocclusive CCAT with secondary hypotension therefore predisposes the post-thrombotic brain to hemodynamic stress and structural damage.
Carotid artery disease is believed to produce clinical stroke symptoms by two basic mechanisms (Ringelstein et al., 1983; Powers and Raichle, 1985; Powers, 1991). In the first, intracarotid occlusion from an embolus or from an anterograde extension of a thrombus into a major cerebral artery produces the ischemic insult. In the second case, perfusion failure due to inadequate collateral flow distal to the diseased vessel may also lead to ischemia. Persistent patterns of cerebral hypoperfusion have been detected in patients after a transient ischemic attack (TIA) and after acute stroke (Kempinsky et al., 1961; Skinhoj et al., 1970; Martin and Raichle, 1983; Hartman, 1985; Powers and Raichle, 1985; Powers, 1991; Bogousslavsky et al., 1990). Systemic hypotension, particularly when coupled with a local decrease in cerebral perfusion due to limited vasodilator reserve capacity, may also lead to a decrease in local cerebral blood flow (lCBF) and an increased risk of hemodynamically induced stroke (Carpenter et al., 1990; Herold et al., 1988; Leblanc et al., 1987; Torvik, 1984). In addition, a correlation between impaired CO2 reactivity and the incidence of ischemic stroke has been reported in patients with symptoms of cerebrovascular disease (Widder et al., 1986; Bullock et al., 1985; Herold et al., 1988; Ringelstein et al., 1988; Kleiser and Widder, 1992).
In an experimental model of nonocclusive common carotid artery thrombosis (CCAT), we have reported widespread reductions in lCBF (Dietrich et al., 1991; Stagliano et al., 1997b). Superimposed on moderate flow reductions were focal areas of severe ischemia, most likely a consequence of occlusive platelet embolization (Futrell et al., 1988; Dietrich et al., 1993a, b ; 1995; Stagliano et al., 1997). Transient hemodynamic abnormalities can also be induced in intact rats that received an intracarotid infusion of thrombogenically activated blood collected downstream from the forming carotid thrombus (Dietrich et al., 1991). Thus, humoral substances in addition to platelet emboli released during vascular thrombosis were hypothesized to have affected vascular tone in a time-dependent manner.
Widespread reductions in lCBF may signal limited vasodilator reserve capacity that could potentially promote ischemic damage under conditions of decreased systolic blood pressure. Transient hypotension occurs in patients who have cerebrovascular disease and may be an important risk factor for stroke (Torvik, 1984; Bogousslavsky and Regli, 1986). In addition, clinical studies have indicated that the anterior cerebral artery borderzone is susceptible to cerebral ischemia in patients with severe carotid stenosis (Leblanc et al., 1987). In this regard, recent experimental data indicate that the post-thrombotic brain is predisposed to widespread cerebral infarction with delayed transient global ischemia (Dietrich et al., 1998).
Although infarct volume after middle cerebral artery (MCA) occlusion is known to be extremely sensitive to hypotension during occlusion (Zhu and Auer, 1995), there is a lack of experimental data concerning the potential effects of mild hypotension after thromboembolic stroke. Thus, the purpose of this study was to determine whether a relatively brief period of hemorrhagic hypotension (60 mm Hg) induced 1 hour after CCAT would worsen outcome in a model of photochemically induced thromboembolic stroke. Quantitative histopathologic studies were conducted at 3 days to assess patterns of structural damage while autoradiographic lCBF measurements were made immediately after the hypotensive period to determine hemodynamic consequences. Finally, to test whether differences existed in the response of the brain to carotid thrombosis versus mechanical occlusion, transient occlusion of the common carotid artery (CCA) was also undertaken using a neurosurgical clip.
METHODS
Animal groups
Thirty-three male Wistar rats weighing between 250 and 300 g were randomized into three experimental groups. In the first group (n = 12), rats underwent nonocclusive CCA thrombosis (CCAT) followed by a 95-minute period of normotension. At the end of this period, five rats underwent the quantitative assessment of lCBF and seven rats were returned to their cages for 3 days before perfusion fixation. In group 2, 14 rats underwent CCAT followed 1 hour later by a 30-minute period of induced hypotension. In this group seven rats underwent lCBF studies at 95 minutes after CCAT and seven rats were perfusion fixed 3 days later. In group 3, eight rats underwent 1.5 hours of mechanical occlusion of the CCA combined with 30 minutes of induced hypotension initiated 1 hour after clip placement. All of these rats were perfusion fixed 3 days later.
CCAT
The method of inducing nonocclusive CCAT using the photochemical rose bengal insult has previously been described in detail (Watson et al., 1987; Dietrich et al., 1993; Stagliano et al., 1997a, b ). Rats were anesthetized with 3% halothane for 3 to 5 minutes during intubation procedures. Rats were then mechanically ventilated and maintained on 1% halothane and a mixture of 70% nitrous oxide and 30% oxygen. Femoral arterial and venous catheters were inserted for the measurement of arterial blood pressure, blood gases, and for fluid administration. Brain temperature was indirectly monitored by a thermocouple probe placed in the temporalis muscle and maintained at normothermic (37°C) levels using a feedback heating lamp. Rats were then placed on their backs and the right CCAT exposed under a Zeiss operating microscope.
The beam of a tunable argon-dye laser (562 nm; continuous-wave power, 325 mW) was focused to an intensity of approximately 25 W/cm2 onto the saline-immersed CCA with a 61-cm focal length spherical lens. The photosensitizing dye, rose bengal (15 mg/mL in 0.9% saline), was injected at a dose of 20 mg/kg (1.33 mL/kg) and the CCA irradiated for 10 minutes. Published data indicate that this protocol leads to a 50% to 75% stenosis of the irradiated carotid segment (Wester et al., 1992). At 1 hour after CCAT (or mechanical occlusion), systemic blood pressure was reduced to 60 mm Hg by the gradual withdrawal of blood into a heparinized syringe. After a 30-minute period, shed blood was reinfused to restore mean arterial blood pressure to 100 to 120 mm Hg.
For mechanical occlusion of the carotid artery (n = 8), a neurosurgical clip was placed over the CCA to interrupt the flow of blood. After a 1.5 hour period, the clip was removed and the return of blood flow through the previously occluded carotid segment visually assessed. For chronic survival studies, rats were administered antibiotics and returned to their cages.
Regional CBF
CBF changes were assessed in thrombosed rats with and without secondary hypotension by using 14C-iodoantipyrine autoradiographic procedures (Sakurada et al., 1978; Dietrich et al., 1991; Back et al., 1995). At 5 minutes after the termination of the hypotensive insult, thrombosed animals were injected with 20 uCi of the radiotracer 14C-iodoantipyrine (specific activity 40 uCi/mmol, New England Nuclear, dissolved in isotonic saline) intraveneously at a constant rate over 45 seconds via a Harvard infusion pump. Arterial blood was sampled from the tip of a freely flowing femoral artery catheter at approximately 2-second intervals. Studies were terminated by decapitation. Coronal sections were prepared in a cryostat, and these sections, together with calibrated 14C-methylmethacrylate standards, were exposed to Kodak film.
Densitometry of ipsilateral and contralateral hemispheres was performed by means of an automated digitizing densitometer (Optronics Colorscan) interfaced to a MicroVAX computer system (Digital Equipment Corp.); the autoradiographic films were converted to digital images of optical density. As previously described by Back et al. (1995), a thresholding approach was used that allowed us to produce selective sections depicting only those pixels lying within predetermined thresholds for lCBF. In the rat, the threshold for ischemic damage has been reported to be 0.25 mL/g/min (Bolander et al., 1989; Tyson et al., 1984; Back et al., 1995). This value was therefore used in this investigation as an indicator of severe ischemia. The volume of severe (0 to 0.24 mL/g/min) and moderate (0.24 to 0.48 mL/g/min) flow reductions were computed by calculating the specific areas at 16 sections of interest (i.e., 4.7, 3.7, 3.2, 2.2, 1.7, 1.2, 0.2, −0.8, −2.3, −3.3, −3.8, −4.8, −5.8, −6.8, −7.8 and −9.3 mm to bregma (Zilles, 1985). Once areas were calculated, volumes of severe or moderate ischemia were determined by numerical integration of sequential areas.
Histopathologic analysis
At 3 days after CCAT, rats were perfusion-fixed for histopathologic analysis. Rats were deeply anesthetized with halothane and perfused transcardially with physiological saline (5 minutes) and then with FAM (a mixture of 40% formaldehyde, glacial acetic acid and methanol; 1:1:8 by volume) for 20 minutes at a pressure of 120 mm Hg. Brain sections 10-µm thick were then prepared at 50-µm intervals and stained with hematoxylin and eosin. For the determination of infarct areas and volumes, six coronal sections (i.e., 4.2, 0.7, −1.3, −3.3, −5.3, and −7.3 mm to bregma) were selected and each section was viewed at low (10X) power. Areas of ischemic damage were then traced onto paper using a camera lucida microscope attachment by an investigator (W.D.D.) blinded to the experimental protocol. Because hypotension might aggravate the frequency of ischemic neuronal injury in addition to infarction, regional patterns of selective neuronal necrosis and infarcted areas were included in the drawings. Each drawing was then retraced onto a digitizing tablet interfaced to a computer which calculated histopathologically defined areas at each coronal level.
Statistical analysis
Statistical analysis data are expressed as mean ± standard deviation (SD). Areas and volumes of severe ischemia and ischemic damage were analyzed by one or two-way analysis of variance (ANOVA) and statistical significance was assessed by Scheffe's test and Dunn's multiple comparison procedure.
RESULTS
Physiologic data from sham and thrombosed rats are summarized in Table 1. Physiologic values were within normal ranges before and 1 hour after carotid thrombosis or mechanical occlusion.
Physiologic variables
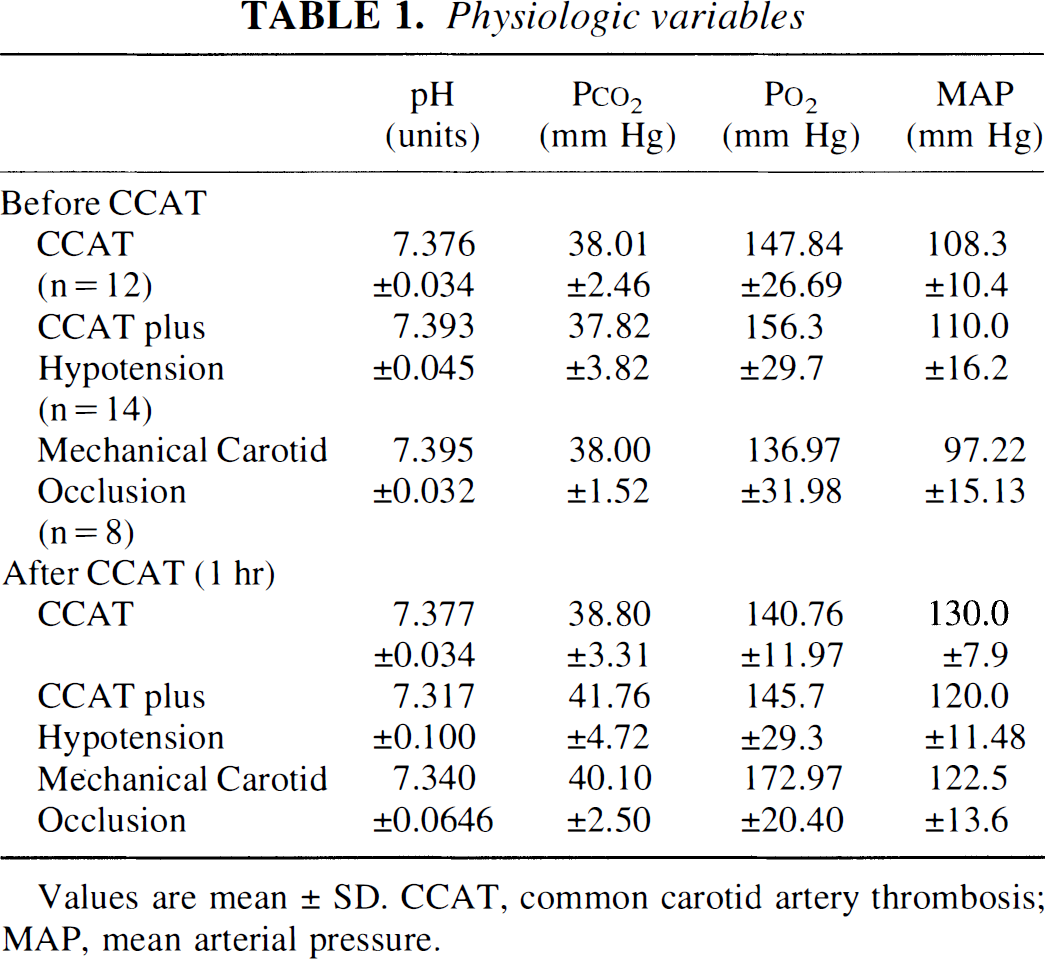
Values are mean ± SD. CCAT, common carotid artery thrombosis; MAP, mean arterial pressure.
In normotensive CCAT rats, moderate reductions in lCBF (i.e., 0.24 to 0.48 mL/g/min) were found within the right and left cerebral cortex 95 minutes after CCAT (Fig. 1). Superimposed on moderate flow reductions were infrequent focal areas of severe ischemia. Focal ischemic areas were commonly detected within the forelimb and hindlimb regions of the ipsilateral cerebral cortex (anterior borderzone area) and occipital cortex (posterior borderzone area). Less common sites included the ipsilateral striatum and thalamus.
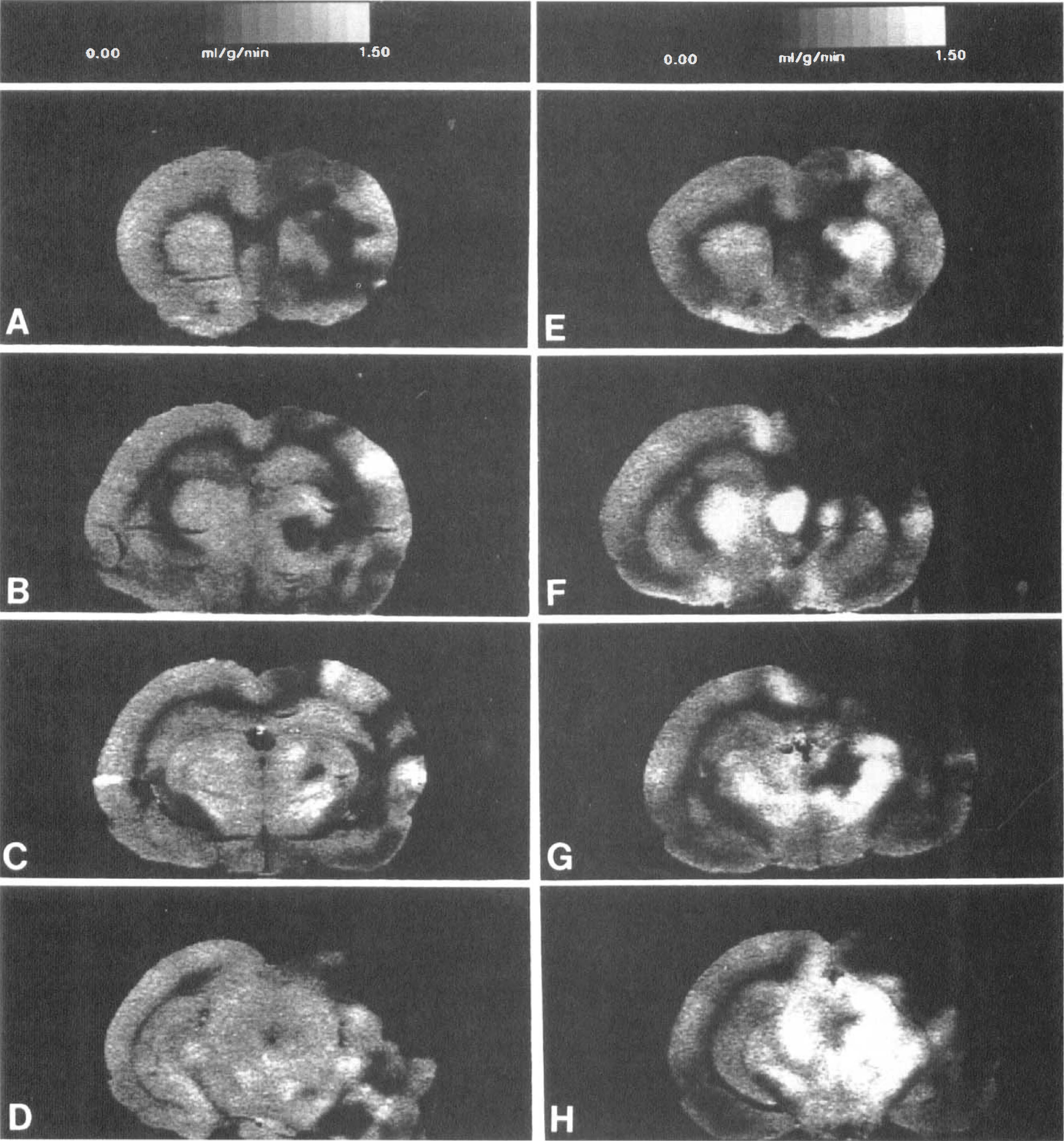
Autoradiographic images of flow reductions 95 min after common carotid artery thrombosis (CCAT) in normotensive
Compared to normotensive CCAT rats, induced hypotension 1 hour after CCAT dramatically increased areas of severe ischemia in ipsilateral and sometimes contralateral regions (Figs. 1 and 2). Areas of focal ischemia were detected within cortical borderzone areas of the anterior and MCA territories. Hypotension-induced ischemia overlying the occipital cortex, representing the borderzone area between the MCA and posterior cerebral artery (PCA) territories, was also commonly observed (Fig. 1). In addition to these cortical responses, areas of severe ischemia were also found within the ipsilateral striatum, hippocampus, and thalamus. Evidence of hypotension-induced hemodynamic depression within the contralateral (left) hemisphere was also present in four of seven rats (Fig. 2). In these rats, hypoperfusion was found within the cerebral cortex, striatum, and hippocampus.
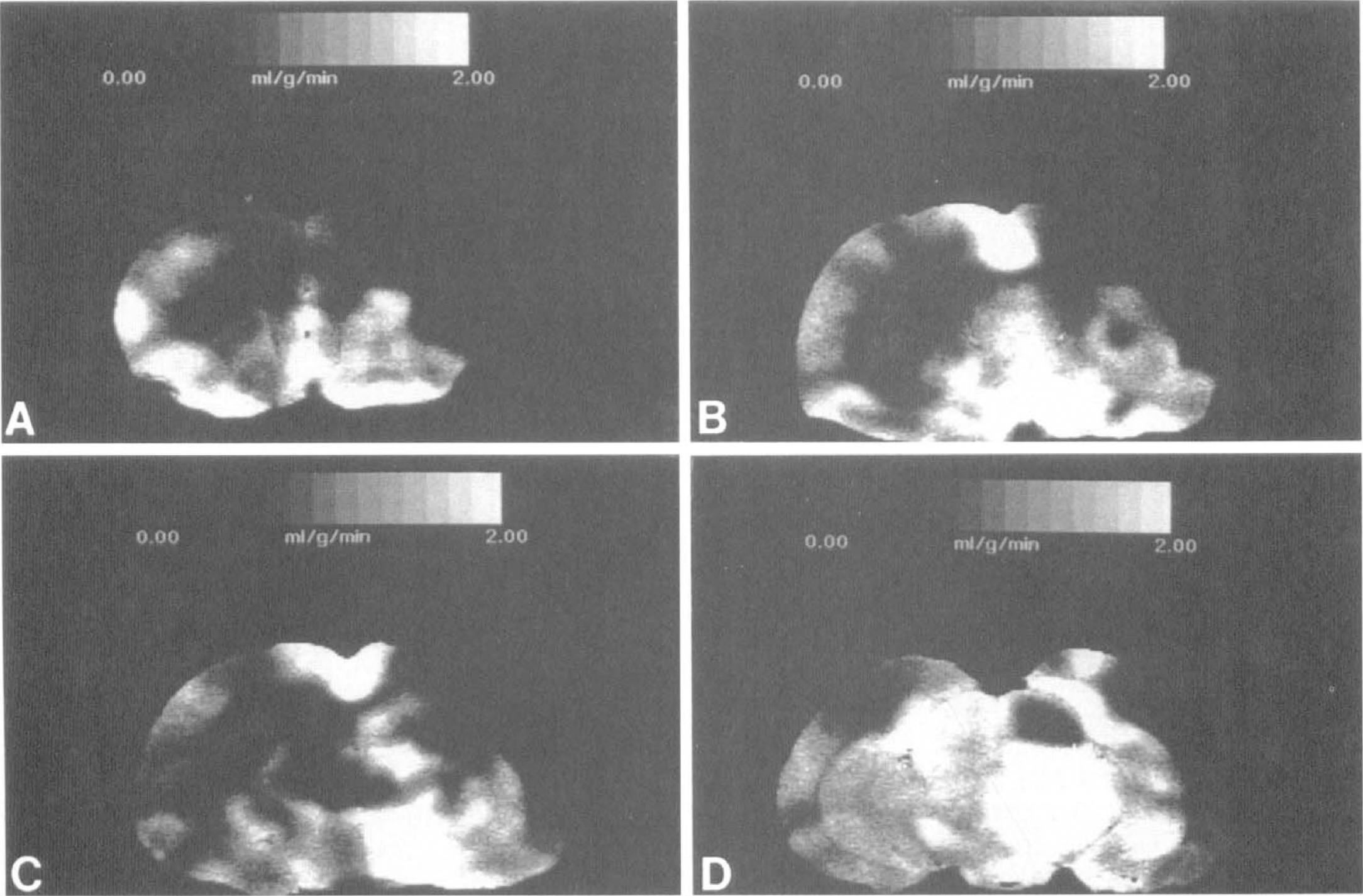
Autoradiographic images of flow changes in a rat that underwent common carotid artery thrombosis and hypotension. In contrast to Figure 1 E-H, regions of severe ischemia are also present in contralateral brain regions including the cerebral cortex
In addition to these focal regions of ischemia, areas of relative hyperemia were found in CCAT rats (Figs. 1 and 2). Induced hypotension appeared to enlarge these areas of hyperemia compared to normotensive CCAT rats (Fig. 1). In most cases, the hyperemic regions were found adjacent to sites of severe ischemia.
Compared to normotensive CCAT rats, induced hypotension after CCAT increased the aggregate volume of severe ischemia in both the ipsilateral and contralateral hemispheres (Fig. 3, Table 2). Within the ipsilateral cortex, the volume of severe ischemia (i.e., 0 to 0.24 mL g/min) was significantly increased from 16 ± 4 mm3 (mean ± SD) in normotensive CCAT rats to 126 ± 99 mm3 after hypotension (P < 0.02, ANOVA). Although not significant, induced hypotension also increased the volume of severe ischemia within the contralateral hemisphere from 15 + 3 mm3 to 36 + 23 mm3. In contrast, hypotension did not increase the volume of moderate ischemia (i.e., 0.24 to 0.48 mL/g/min).
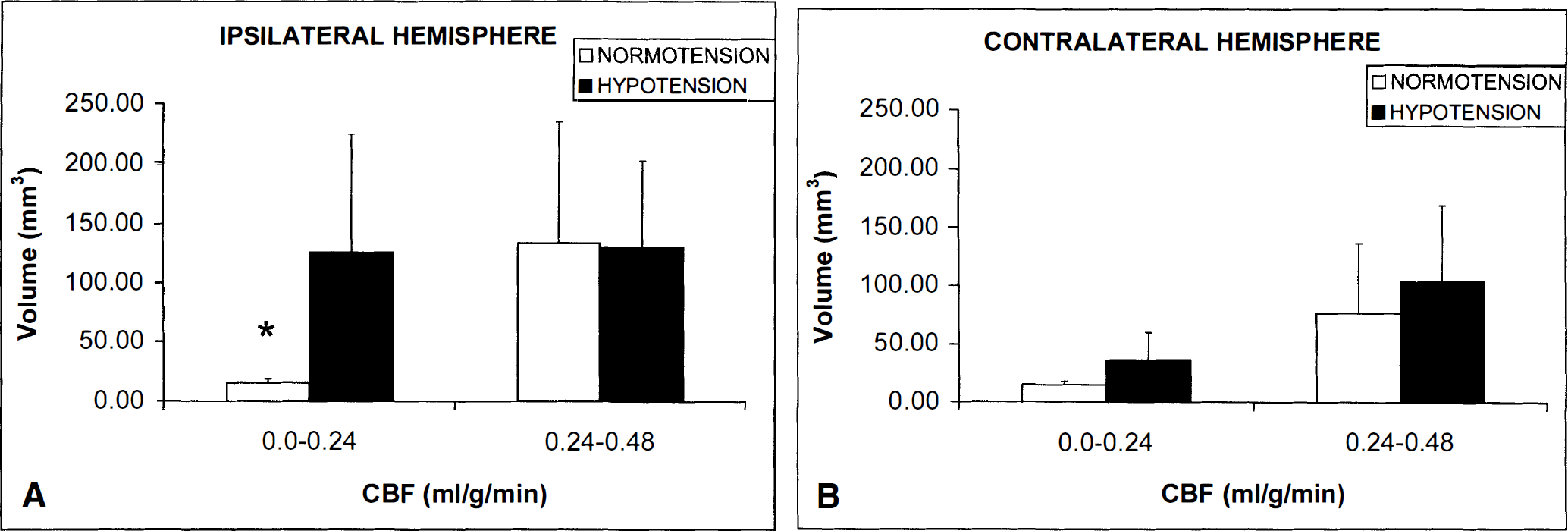
Bar graphs showing volumes of severe (0 to 0.24 mL/g/min) and moderate (0.24 to 0.48 mL/g/min) ischemia in ipsilateral
Volumes of ischemic damage and severe* hemodynamic depression
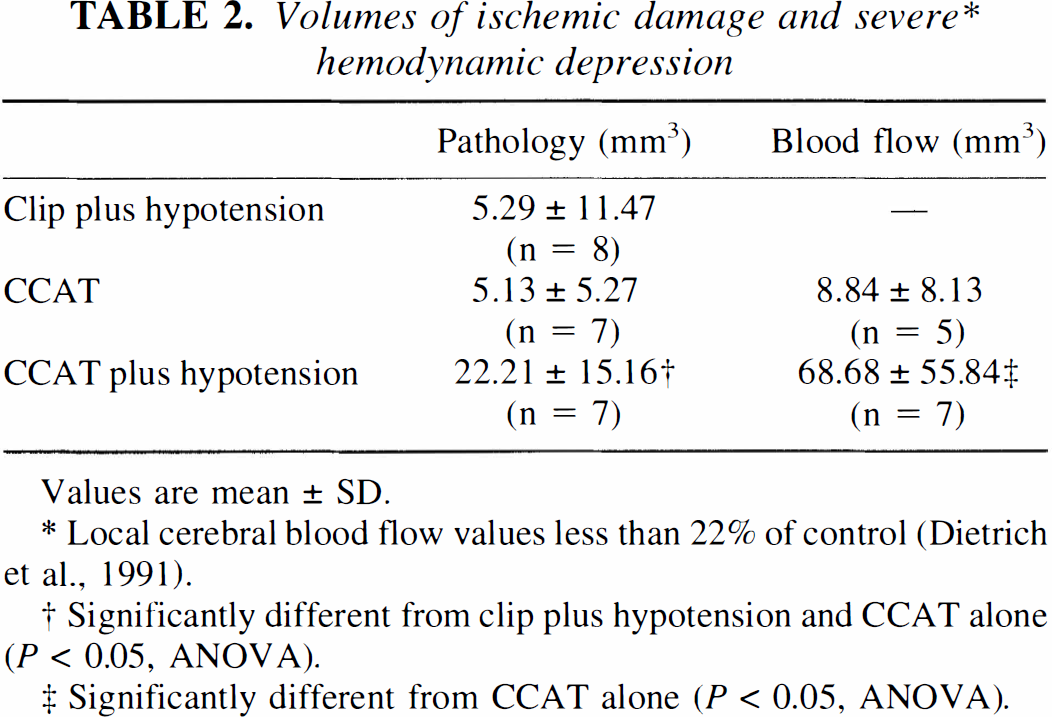
Values are mean ± SD.
Local cerebral blood flow values less than 22% of control (Dietrich et al., 1991).
Significantly different from clip plus hypotension and CCAT alone (P < 0.05, ANOVA).
Significantly different from CCAT alone (P < 0.05, ANOVA).
Histopathologic examination of normotensive CCAT rats at 3 days showed small infarcts within five of the seven animals (Fig. 4). A significantly larger volume of ischemic damage, including zones of infarction and also selective neuronal necrosis, was found with secondary hypotension 3 days after CCAT (Table 2). Infarcts were well demarcated and restricted to the thrombosed hemisphere. In addition to increased areas of tissue necrosis, widespread areas of selective neuronal necrosis were detected in cortical and subcortical areas (Fig. 4). For example, selective neuronal damage within the ipsilateral striatum was found in three of the rats with hypotension. Widespread neuronal damage to the ipsilateral CA1 hippocampus was also detected in two rats. Thus, the volume of ischemic damage was significantly increased from 5.1 ± 5.2 mm3 in normotensive CCAT rats to 22.2 ±15.1 mm3 (mean ± SD) with induced hypotension (P < 0.05, ANOVA). Significant differences between areas of damage in normotensive versus hypotensive rats were documented at four of the six coronal levels investigated (Fig. 5).
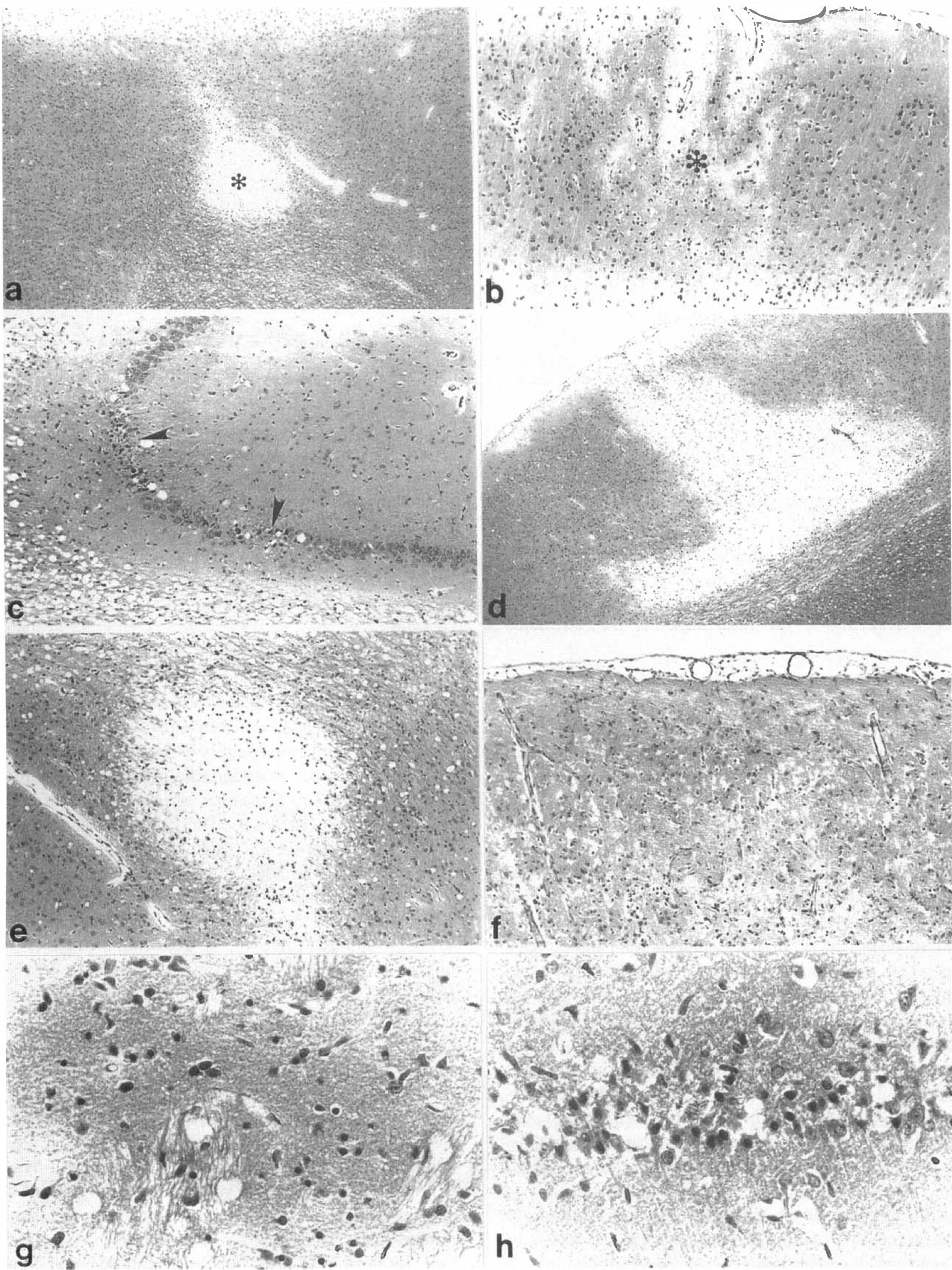
Paraffin sections stained with hematoxylin and eosin 3 days after common carotid artery thrombosis (CCAT)
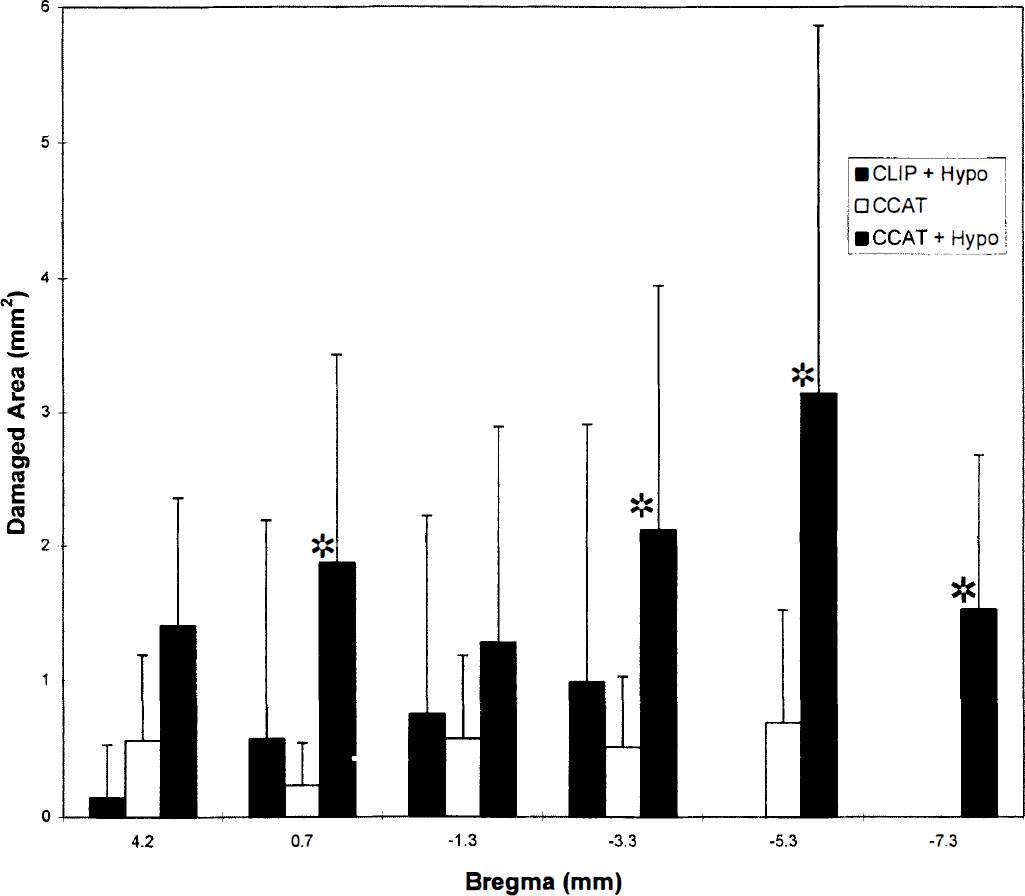
Bar graph showing areas of ischemic damage (infarction plus ischemic cell change) at six coronal levels. Values are mean + SD. Striped bars are clip occlusion plus hypotension, clear bars are common carotid artery thrombosis (CCAT), and closed bars are CCAT plus hypotension. *Significantly different from clip occlusion plus hypotension and CCAT alone (P < 0.05, analysis of variance).
In contrast to these findings with CCAT, mechanical ligation of the right CCA for 1.5 hours plus hypotension did not produce significant histopathologic damage. The volume of histopathologic damage found after clip occlusion plus hypotension was significantly smaller that that found after CCAT plus hypotension (5.3 ± 11.5 mm3 versus 22.2 ± 15.1 mm3, P < 0.05, ANOVA). In this setting, pathologic damage consisted of selective neuronal necrosis and not infarction.
DISCUSSION
The present findings indicate that a brief period of delayed systemic hypotension significantly aggravates the histopathologic consequences of nonocclusive CCAT. In addition to increased volumes of infarction, hypotension also led to widespread zones of selective neuronal necrosis. These histopathologic findings were associated with patterns of hypotension-induced hemodynamic stress. Nonocclusive carotid thrombosis therefore predisposes the brain to secondary ischemia and widespread tissue injury in the case of delayed hypotension. Patients with cerebrovascular disease can experience periods of acute hypotension resulting in watershed or borderzone infarcts (Torvik, 1994; Bogousslavsky and Regli, 1986). The present experimental model may therefore be useful in (1) investigating why the post-thrombotic brain is predisposed to infarction with delayed hypotension, and (2) the development of therapeutic strategies to protect the post-thrombotic brain against secondary injury.
The histopathologic, hemodynamic and neurochemical consequences of photochemically induced CCAT have been described in a previous publication (Dietrich et al., 1994). The photochemical insult leads to acute endothelial damage and robust platelet recruitment at the irradiated carotid site (Watson et al., 1987; Futrell et al., 1988; Wester et al., 1992; Dietrich et al., 1993a). Based on previous studies, the carotid injury induced in the present study led to a 50% to 75% stenosis of the carotid artery (Wester et al., 1992). Thus, it is important to note that stenosis and not occlusion was investigated in this study. Previous reports have shown that CCAT leads to acute platelet embolization to the brain (Dietrich et al., 1993a, b , 1995; Futrell and Riddle, 1993; Stagliano et al., 1997b). Thus, the present model incorporates an important characteristic of clinical stroke, namely, platelet embolization leading to embolic stroke.
The relative importance of hemodynamic and embolic factors in the pathogenesis of ischemic stroke has been difficult to determine from clinical studies (Powers et al., 1989). This is an important issue because treatment strategies directed at hemodynamic versus embolic mechanisms might differ. We have previously described the early hemodynamic consequences of CCAT and thrombogenically activated blood (Dietrich et al., 1991; Stagliano et al., 1997b). Double-label studies using indium-labeled platelets and 14C-iodoantipyrine autoradiography have identified aggregates of platelet emboli associated with focal ischemic sites but not associated with the more widespread hemodynamic perturbations (Stagliano et al., 1997b). This thromboembolic model therefore appears to induce hemodynamic disturbances by multiple mechanisms, including occlusive platelet embolization and the probable release of vasoactive humoral factors (Wester et al., 1992). Thrombotic processes might also lead to disturbances in the synthesis of endothelium-dependent vasodilating factors such as nitric oxide, resulting in limited vasodilator capacity of collateral vessels (Heistad et al., 1990; Rosenblum et al., 1997). In this regard, treatment with the nitric oxide synthase inhibitor nitro-L-arginine methyl ester (L-NAME) immediately after CCAT increases numbers of indium-labeled platelets in the thrombosed hemisphere, significantly depresses lCBF, and exacerbates water maze retention deficits compared to nontreated thrombosed rats (Stagliano et al., 1997a, b ).
Although clinical data suggests no strong relationship between cerebral hemodynamic abnormalities and subsequent risk of stroke (Powers et al., 1989), limitations in hemodynamic reserve or vascular reactivity may predispose patients with carotid stenosis or cerebrovascular disease to TIA or stroke (Ringelstein et al., 1983; Bullock et al., 1985; Widder et al., 1986; Herold et al., 1988). Previous retrospective studies have found a close correlation between an impaired CO2 reactivity measured by Doppler CO2 testing and the incidence of ipsilateral ischemic stroke (Widder et al., 1986; Ringelstein et al., 1988; Kleiser and Widder, 1992). By means of transcranial Doppler ultrasonography during various degrees of hypercapnia, Ringelstein et al. (1988) reported a striking association between low-flow infarctions, hypostatic TIA, and reduced vasomotor reactivity. Thus, it has been suggested that asymptomatic patients with carotid artery occlusions and reduced vasomotor reactivity may be stroke-prone and vulnerable to challenges to their marginal cerebral blood supply (Ringelstein et al., 1988). The occurrence of ischemic deficits in patients with internal carotid artery occlusions after a decrease in blood pressure supports such a contention (Kleiser and Widder, 1992). Our experimental findings indicate that CCAT affects collateral circulation in borderzone areas in such a way as to predispose the brain to hypotension-induced histopathologic damage.
Focal regions of hyperemia were found in all CCAT rats. In our previous blood flow study, areas of hyperemia were found after CCAT and after the intracarotid infusion of thrombogenically activated blood (Dietrich et al., 1991). Different types of hyperemia or “luxury perfusion,” including borderzone, postischemic, and remote have been described in the clinical and experimental literature (Lassen, 1966; Cronqvist and Laroche, 1967; Hoedt-Rasmussen et al., 1967; Sundt and Waltz, 1971; Olsen et al., 1981). In the present study, zones of hyperemia appeared to be more widespread after induced hypotension. Because blood flow studies were conducted 5 minutes after blood pressure normalization, this hyperemic response may have resulted from the generation of vasodilative substances from ischemic zones causing borderzone hyperemia after blood pressure normalization (Winn et al., 1981; Wahl et al., 1988; Kontos, 1989).
In normotensive CCAT rats, small infarcts were found in cortical and subcortical regions ipsilateral to the injured carotid artery. Frequent sites of infarction were the cortical forelimb region and occipital cortex. In previous CCAT studies, these cortical regions were also shown to accumulate large numbers of platelet emboli (Dietrich et al., 1993a, 1995; Stagliano et al., 1997b). In rats, these regions represent the borderzone areas between the MCA territory and either the anterior (ACA) or posterior (PCA) cerebral artery territories (Coyle and Heistad, 1987; Coyle, 1987). Because of the transient nature of the majority of the platelet emboli, (Dietrich et al., 1993a), only a small percentage of the occlusive events lead to embolic infarcts under normotensive conditions. Multiple microvascular events including permanent and transient platelet accumulation, lethal and sublethal endothelial damage, or platelet-derived vasoactive substances, combine to increase the risk of the post-thrombotic brain to hypotension-induced secondary ischemic damage.
The morphologic presentation of low-flow infarcts occurring in watershed areas and those produced by thrombotic or embolic occlusion of a cerebral artery are considered to be different (Weiller et al., 1991). While territorial infarcts are restricted to brain regions supplied by the occluded vessel, hemodynamic infarcts may occur between neighboring arterial territories (Ringelstein et al., 1983). In our study, induced hypotension after CCAT led to large regions of severely reduced lCBF and histopathologic damage within borderzone regions. In contrast, induced hypotension after mechanical occlusion of the CCA did not lead to significant histopathology. It therefore appears that CCA injury with subsequent platelet activation and embolization leads to reduced hemodynamic reserve within borderzone areas. Cerebral autoregulation, i.e., the relative constancy of blood flow during alterations in perfusion pressure (Edvinsson and MacKenzie, 1977), is impaired in humans with cerebral ischemia (Fieschi et al., 1968; Meyer et al., 1973). In experimental ischemia studies involving mechanical occlusion of the MCA, autoregulation is attenuated in ischemic brain regions with lCBF of less than 30% control, while autoregulation is lost at lower CBF levels (Dirnagl and Pulsinelli, 1990). In the present study, we compared CCAT rats to rats that underwent complete carotid occlusion for 1.5 hours by a mechanical clip. Because the thrombotic carotid artery was not completely occluded, one might question whether complete occlusion by mechanical means is a good control. Nevertheless, the present findings underscore the importance of model selection when investigating the pathobiology of clinical stroke (Watson and Dietrich, 1990).
In summary, nonocclusive CCAT has been shown to produce a complex pattern of hemodynamic abnormalities that may result from multiple mechanisms. Induced hypotension led to aggravation of hemodynamic and histopathologic damage in specific brain regions known to be vulnerable in this thromboembolic stroke model. The lack of a similar hypotensive consequence after mechanical occlusion of the carotid artery indicates that mechanisms underlying autoregulatory dysfunction after CCAT may not be a dominant feature of conventional focal ischemia models. Therapeutic strategies directed at impaired cerebral hemodynamics after carotid thrombosis may provide protection in patients at risk for stroke.
Footnotes
Acknowledgments
The authors thank Isabel Valdes, Susan Kraydieh, and Raul Busto for their superb technical assistance, and Helen Valkowitz for typing the manuscript.