
Abstract
Lipopolysaccharide (LPS), administered 72 hours before middle cerebral artery (MCA) occlusion, confers significant protection against ischemic injury. For example, in the present study, LPS (0.9 mg/kg intravenously) induced a 31% reduction in infarct volume (compared with saline control) assessed 24 hours after permanent MCA occlusion. To determine whether LPS induces true tolerance to ischemia, or merely attenuates initial ischemic severity by augmenting collateral blood flow, local CBF was measured autoradiographically 15 minutes after MCA occlusion. Local CBF did not differ significantly between LPS- and saline-pretreated rats (e.g., 34 ± 10 and 29 ± 15 mL·100 g−1·min−1 for saline and LPS pretreatment in a representative region of ischemic cortex), indicating that the neuroprotective action of LPS is not attributable to an immediate reduction in the degree of ischemia induced by MCA occlusion, and that LPS does indeed induce a state of ischemic tolerance. In contrast to the similarity of the initial ischemic insult between tolerant (LPS-pretreated) and nontolerant (saline-pretreated) rats, microvascular perfusion assessed either 4 hours or 24 hours after MCA occlusion was preserved at significantly higher levels in the LPS-pretreated rats than in controls. Furthermore, the regions of preserved perfusion in tolerant animals were associated with regions of tissue sparing. These results suggest that LPS-induced tolerance to focal ischemia is at least partly dependent on the active maintenance of microvascular patency and hence the prevention of secondary ischemic injury.
Keywords
The brain can be rendered more tolerant to focal ischemia by preexposure to a stressful, but sublethal, stimulus such as brief transient ischemia, hypoxia, or low-dose lipopolysaccharide (LPS, a potent proinflammatory stimulant) (Chen and Simon, 1997; Tasaki et al., 1997). When the preconditioning stimulus is a brief period of ischemia, the improved outcome in tolerant animals is not because of acute changes in local CBF (Chen et al., 1996; Matsushima and Hakim, 1995), but instead has been attributed to alterations in the synthesis of specific neuroprotective proteins. These proteins remain to be definitively identified, but a growing list of candidates would include antioxidant enzymes, Ca2+ buffers, heat shock proteins, growth factors, and antiapoptotic gene products (Chen and Simon, 1997; Mattson, 1997). Alternatively, synthesis of injurious proteins such as N-methyl-
As an alternative approach, we have developed a different method for inducing tolerance to focal ischemia using LPS (bacterial endotoxin) as the preconditioning stimulus. Lipopolysaccharide has also been used to induce tolerance to myocardial ischemia (Rowland et al., 1996) and to higher (normally lethal) doses of LPS itself (Cavaillon, 1995). The physiologic responses to LPS are well documented (Cavaillon, 1995) and give a somewhat narrower field of search for determining which responses are important for the development of tolerance. For example, we have already determined that LPS-induced tolerance to focal ischemia can be blocked by coadministration of a tumor necrosis factor-α (TNF-α) binding protein, indicating that signaling pathways downstream from TNF receptors are critically involved in the process (Tasaki et al., 1997).
It is probable that tolerance to focal ischemia develops through common pathways, despite the disparate nature of the preconditioning stimuli used, because there is a striking similarity in the optimal interval between the preconditioning stimulus and the ischemic insult across models (e.g., for both ischemia-induced and LPS-induced tolerance the optimal interval for neuroprotection is 72 hours [Chen et al., 1996; Tasaki et al., 1997]). However, in contrast to ischemia-induced tolerance, we have not yet determined whether alterations in local CBF play a role in the development of LPS-induced neuroprotection. This possibility is of particular relevance to our model because LPS is a potent inducer of inducible nitric oxide (NO) synthase (iNOS) (Kifle et al., 1996), and the presence of high levels of vasodilatory NO at the time of middle cerebral artery (MCA) occlusion could conceivably reduce ischemia by augmenting collateral blood supply. Recent studies have shown that administration of LPS directly into the brain elicits cerebrova-sodilation and increased CBF, responses that are partly mediated by iNOS (Brian et al., 1995; Okamoto et al., 1997), whereas intravenous infusion of LPS (in certain models of septic shock) also results in higher CBF that can be attenuated by NO synthase inhibitors (Meyer et al., 1994; Offner et al., 1995). If, in our model, LPS pretreatment resulted in enhanced CBF at the time of MCA occlusion, then the apparent neuroprotective (tolerant) effect of LPS would instead be attributable to direct attenuation of ischemic severity.
Therefore, to ascertain whether LPS induced true ischemic tolerance or simply reduced the severity of ischemia induced by MCA occlusion via an immediate enhancement of collateral blood flow, the first aim of the present study was to measure local CBF in the immediate postischemic period in LPS-pretreated (“tolerant”) and control (“nontolerant”) rats to verify whether the primary ischemic insults were equivalent for the two groups. Having verified ischemic equivalency, the second aim of the study was to assess cerebrovascular perfusion several hours after the onset of MCA occlusion to determine whether a secondary disparity in the pathologic response to ischemia developed between tolerant and nontolerant animals. There has been a tendency in the literature to emphasize the role of neuroprotective proteins in the development of tolerance (i.e., proteins that prevent damage to neurons while essentially leaving their blood supply unaltered), but we hypothesize that there may also be a vascular component to the process. A progressive impairment in microvascular perfusion normally begins to develop several hours after the onset of focal ischemia (Dawson et al., 1997; Garcia et al., 1994b) and may contribute to the evolution of ischemic injury (Siesjo et al., 1995). We aimed to determine whether the tolerant state is associated with attenuation of this secondary microvascular impairment and hence preservation of cerebrovascular perfusion. In other words, we sought to establish whether the different outcome in LPS and control rats was because of different secondary responses to ischemia, and not simply differences in the severity of the primary ischemic insult.
METHODS
Adult male spontaneously hypertensive rats (SHR; 274 to 338 g) were used in all experiments (n = 5 to 6 per group). Previous studies have established the most effective dose (0.9 mg/kg intravenously) and time (72 hours before MCA occlusion) for development of LPS-induced tolerance (Tasaki et al., 1997), and these parameters were used in the present study. The study consisted of two experiments: (1) autoradiographic measurement of local CBF 15 minutes after MCA occlusion, and (2) fluorescent assessment of microvascular perfusion 4 hours or 24 hours after MCA occlusion. In all cases body weight and rectal temperature were recorded on day 1 (before LPS or saline administration), day 4 (before MCA occlusion), and, if applicable, day 5 (24 hours after MCA occlusion). All experiments were approved by the National Institute of Neurological Disorders and Stroke Animal Care and Use Committee.
Administration of LPS to induce tolerance
On day 1, animals were anesthetized with halothane in nitrous oxide-oxygen (70:30). The right femoral artery and vein were cannulated with polyethylene catheters. Baseline arterial blood samples were withdrawn for measurement of plasma levels of nitrates and nitrites and differential white blood cell counts. Animals were treated with 0.9 mg/kg LPS (from Escherichia coli 0111:B4 phenol extract; Sigma, St. Louis, MO, U.S.A.) or an equivalent volume (1 mg/kg) of vehicle (sterile saline) administered intravenously. Both femoral vessels were ligated, the cannulas removed, and the incision site sutured. Anesthetics were withdrawn and animals returned to their home cages.
Induction of focal cerebral ischemia
On day 4 (i.e., 72 hours after LPS or saline injection), the rats were anesthetized again. The femoral vessels were cannulated for arterial blood sampling and intravenous infusion as appropriate. Arterial blood pressure and blood gases were monitored throughout the surgical procedure (approximately 0.5 hours duration). Body temperature was maintained at approximately 37°C by means of a heating pad. The left MCA was exposed via a subtemporal approach and occluded between the inferior cerebral vein and lateral olfactory tract. This model of permanent focal ischemia differs from that used in the original study (distal MCA occlusion in tandem with bilateral carotid artery ligation; Tasaki et al., 1997). Preliminary studies (data not shown) verified that the degree of tolerance achieved was essentially the same despite the two different methods of MCA occlusion performed by two different surgeons.
Measurement of local cerebral blood flow
Under halothane anesthesia the left MCA was permanently occluded as described above. Anesthetics were then withdrawn, and animals restrained in loose-fitting plaster casts. Fifteen minutes after MCA occlusion, local CBF measurements were performed using the [14C]iodoantipyrine technique (Sakurada et al., 1978). A ramped infusion of [14C]iodoantipyrine was administered intravenously for 60 seconds while 18 timed samples of arterial blood were collected. At the end of the infusion the animal was decapitated, and the brain was removed, frozen in 2-methylbutane (–42°C), and sectioned on a cryostat. Sections were apposed to [14C]-sensitive film together with a set of precalibrated [14C] standards. Optical densities of the resultant autoradiograms were measured by means of an image analysis program (NIH Image) and converted to [14C]iodoantipyrine concentrations by reference to the standards. Cerebral blood flow was measured in six predefined regions of cortex at four levels distributed throughout the territory of the MCA (illustrated in Fig. 1). Blood sample [14C]iodoantipyrine concentrations were determined by liquid scintillation counting. Cerebral blood flow values were then calculated on the basis of a blood—brain partition coefficient of 0.8.
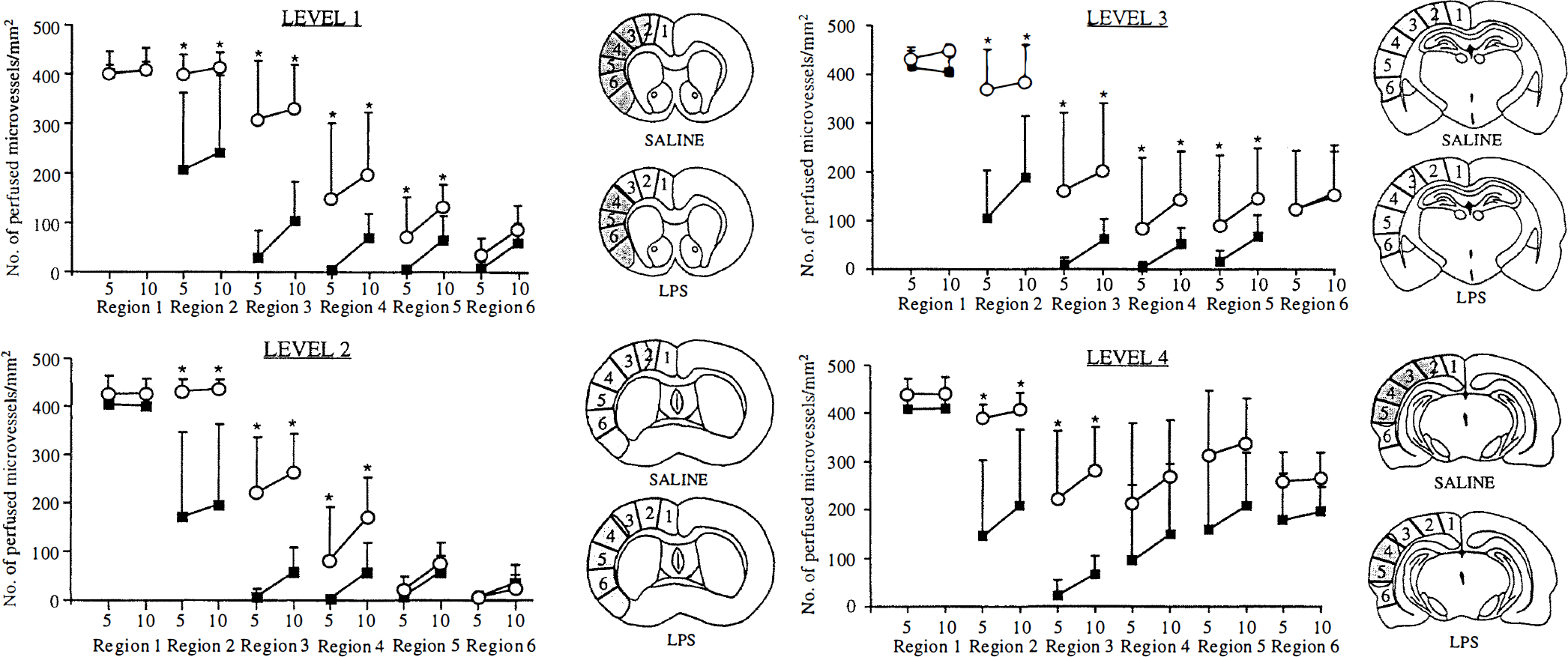
Microvascular perfusion and infarct area measured 24 hours after MCA occlusion in rats pretreated 72 hours previously with LPS (open circles) or saline (filled squares). The mean (± SD) number of microvessels perfused within the 5- and 10-second circulation periods (indicated by “5” and “10” on the x-axis) were measured in the same cortical regions used for the local CBF measurements in Fig. 2. Lipopolysaccharide pretreatment resulted in significantly higher numbers of perfused microvessels compared with saline control (* P < 0.05 Tukey's multiple comparison test after three-way analysis of variance). The shaded portions on the line drawings indicate the median infarct areas for each group. Infarcts were consistently smaller for the LPS-pretreated group, and regions with well preserved perfusion (e.g., region 2, levels 1 through 4) were associated with noninfarcted tissue. For further details see text.
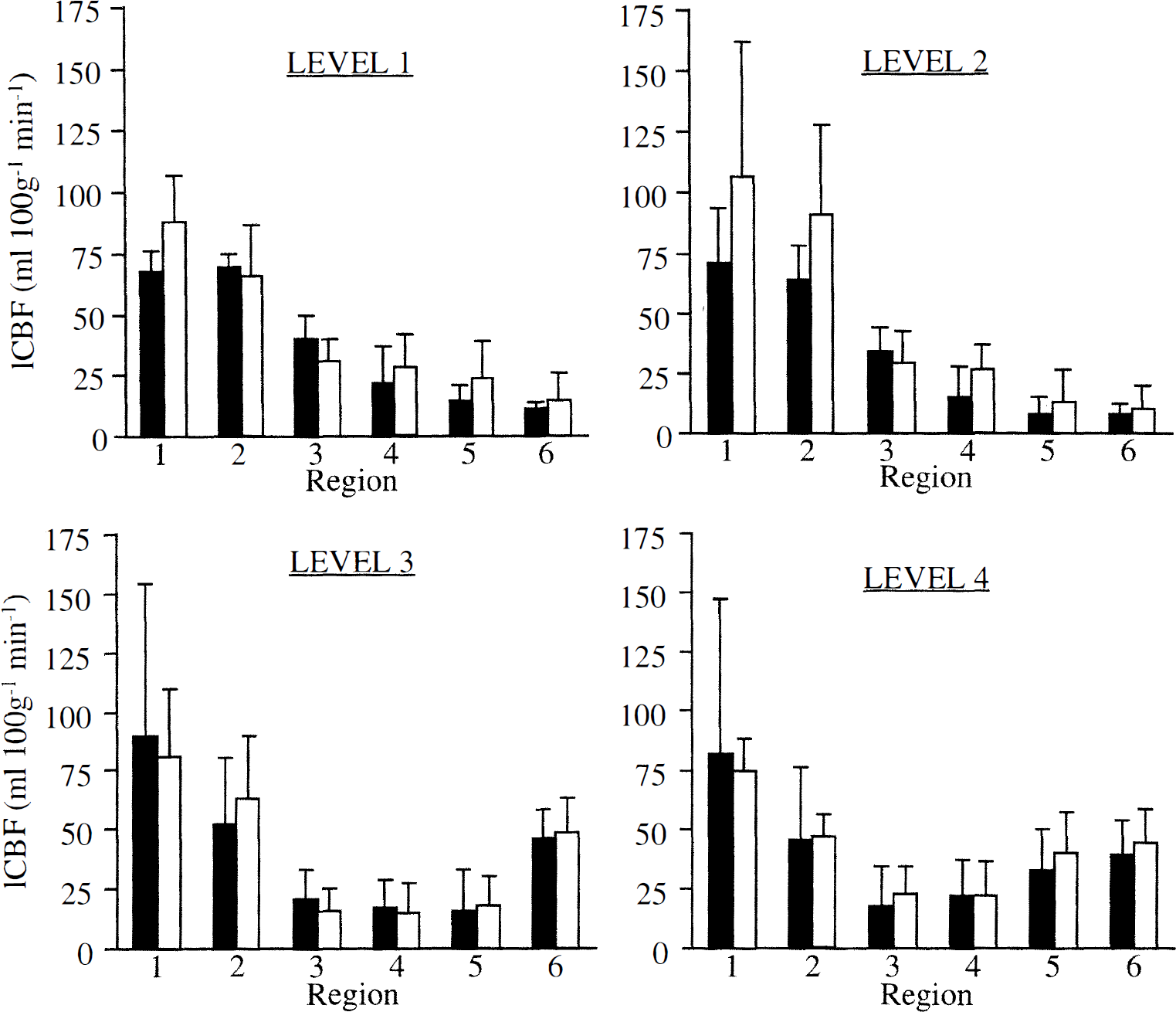
Local CBF (mean ± SD), measured 15 minutes after MCA occlusion, in regions of cortex ipsilateral to MCA occlusion in rats pretreated 72 hours previously with LPS (0.9 mg/kg intravenously, open columns) or saline (filled columns). Measurements were made in six areas of cortex (regions 1 through 6) at each of four coronal levels. These regions of interest are illustrated in the line drawings in Fig. 1. No significant differences were found between the LPS and saline groups (P > 0.05, two-way analysis of variance).
During preliminary experiments it was discovered that blood pressure had a tendency to fall progressively during collection of the 18 timed blood samples, in some instances reaching the lower limit of autoregulation for the SHR (approximately 80 mm Hg; Dimagl and Pulsinelli, 1990). Hypotension does not develop when the same protocol is routinely used with normotensive rat strains. This phenomenon probably reflects the fact that SHR are less able to compensate for hemorrhage than normotensive strains (Bond and Johnson, 1985). Because cerebral autoregulation is impaired after ischemia (Dimagl and Pulsinelli, 1990), even moderate reductions in blood pressure are likely to influence CBF measurement. Therefore, to compensate for the blood loss during arterial sampling, saline was infused intravenously (through the opposite femoral vein of that used for [14C]iodoantipyrine administration) at approximately the rate of blood loss through the arterial cannula (1.5 mL/min). Saline infusion successfully maintained blood pressure at the pre-CBF measurement level throughout the 60-second sampling period.
Assessment of microvascular perfusion
After MCA occlusion the arterial cannula was removed, incision sites sutured, and anesthetics withdrawn. The animal was kept on a heating pad until it fully regained consciousness. Four hours or 24 hours after MCA occlusion rats were anesthetized again, a tracheostomy was performed, and rats were artificially ventilated. The femoral vessels were recannulated. Arterial blood samples were collected as before, and blood pressure and blood gases monitored. Microvascular perfusion was measured using a double-label intravascular fluorescent tracer technique previously described (Dawson et al., 1997). Two fluorescent tracers—Evans blue (2% in saline; 2 mL/kg body weight; Sigma) and FITC-dextran (10% in saline; 2 mL/kg body weight; 71,000 molecular weight; Sigma)—were used to obtain two measurements of microvascular perfusion within each animal. After 4 or 24 hours of MCA occlusion, the tracers were sequentially administered into separate femoral veins 10 seconds (FITC-dextran) and 5 seconds (Evans blue) before decapitation. The brain was then removed, frozen in 2-methylbutane (–42°C), and sectioned (6 µm) at −21°C. To quantify the number of perfused microvessels (i.e., vessels containing fluorescent tracer), sections were examined by fluorescence microscopy, and images were acquired and processed using the Metamorph image processing system (Universal Imaging, West Chester, PA, U.S.A.). Images were acquired from the same predefined regions of cortex used for local CBF measurement. The images were manually thresholded before automatic calculation of the number of perfused microvessels (<20 µm diameter) per field of view. Final results are expressed as the mean (± SD) number of perfused microvessels per mm2 for each region of interest.
Additional cryostat sections (16 µm) were stained with hematoxylin and eosin for volumetric assessment of ischemic lesion volume. Ischemic regions were transcribed from the sections onto scale drawings at eight predefined stereotactic levels (Osborne et al., 1987). Lesion areas were then measured using an image analyzer (NIH Image) and converted to total volume of ischemic damage. The use of scale drawings circumvents the confounding influence of edema on lesion measurement, and negates the use of an edema correction factor.
Estimation of plasma nitric oxide
Heparinized blood samples were immediately centrifuged at 3,500 rpm for 10 minutes at 4°C. The plasma was removed and stored at −20°C until subsequent assay. Nitric oxide levels were estimated by assaying for plasma nitrates and nitrites using a commercial Griess reagent kit (Alexis Corporation, San Diego, CA, U.S.A.).
Statistical analysis
Cerebral blood flow and microvascular perfusion data were compared at each brain level by two-way and three-way repeated measures analysis of variance, respectively. Post-hoc comparisons were by Tukey's multiple comparison test. Physiologic variables (blood gases, MABP, rectal temperature, body weight), ischemic lesion volume, plasma nitrate and nitrite concentration, and differential white blood cell counts were each compared by unpaired Student's t tests.
RESULTS
Local CBF 15 minutes after MCA occlusion
Mean arterial blood pressures at the start and end of the iodoantipyrine protocol were not statistically different between the saline and LPS groups (128 ± 24 mm Hg and 128 ± 24 mm Hg for the saline-pretreated group; 138 ± 18 mm Hg and 129 ± 19 mm Hg for the LPS-pretreated group). Blood gases (data not shown) were also not significantly different between the two groups.
Local CBF in cortical regions ipsilateral to the MCA occlusion are illustrated in Fig. 2. There were no significant differences between the local CBF values for the saline- and LPS-pretreated groups at each of the four brain levels studied (P values for levels 1 through 4, respectively, were 0.40, 0.13, 0.99, and 0.84), indicating that the severity of the initial ischemic insult was similar in both groups.
Ischemic lesion volume and microvascular perfusion 4 hours and 24 hours after MCA occlusion
Body temperature was unaffected by LPS treatment (Table 1). However, LPS did cause a small, but significant, reduction in body weight before MCA occlusion (Table 1). Plasma glucose was not measured in this study, but was previously found to be unaffected by LPS pretreatment (Tasaki et al., 1997). Plasma nitrate and nitrite levels were significantly increased in the LPS-pretreated group on day 4 before MCA occlusion (Table 1). Plasma nitrate and nitrite levels also increased in the saline group but only 24 hours after the MCA occlusion (Table 1), at which time they were not significantly different from the value for the LPS-pretreated group. Differential white blood cell counts showed a significant increase in the number of circulating monocytes measured 72 hours after LPS treatment compared with control. For both groups, circulating neutrophil counts were increased at 24 hours after MCA occlusion, but the increase was significantly smaller for LPS-pretreated rats than for the control group.
Physiologic parameters in rats before treatment with saline or LPS (baseline), 72 hours later (pre-MCAO), and 24 hours after MCAO
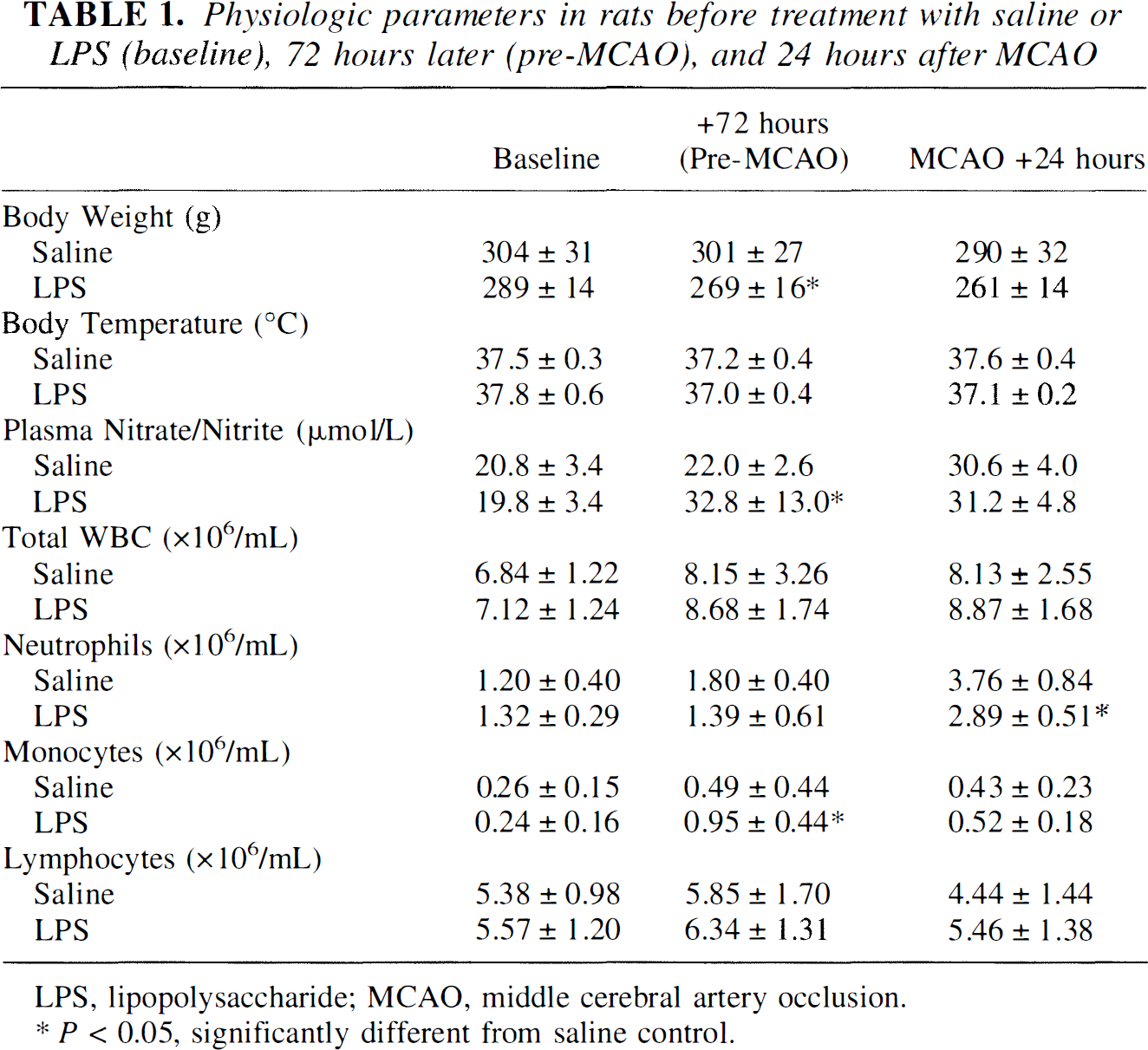
LPS, lipopolysaccharide; MCAO, middle cerebral artery occlusion.
P < 0.05, significantly different from saline control.
Blood gases and blood pressure were within acceptable limits and did not differ between the saline- and LPS-pretreated groups either at the time of MCA occlusion or at the time of measurement of microvascular perfusion (data not shown).
Microvascular perfusion 24 hours after MCA occlusion is illustrated in Fig. 1 (similar results were found at 4 hours, but are not shown to conserve space). Overall, perfusion was significantly higher in the LPS-pretreated group compared with the saline group at each of the four brain levels studied (P values for levels 1 through 4, respectively, were 0.0005, 0.001, 0.04, and 0.003). Post-hoc comparisons of microvascular perfusion in individual cortical regions revealed a consistent pattern of statistically significant differences between the saline and LPS groups (Fig. I). Perfusion in region 1 was unaffected by MCA occlusion and represents the normal range for the number of perfused microvessels per square millimeter. For the saline control group, microvascular perfusion tended to be moderately reduced in region 2 and severely reduced in regions 3 through 6 (except for level 4, located toward the border of MCA territory, in which the reductions in perfusion were less severe). In contrast, LPS pretreatment was associated with higher levels of microvascular perfusion, particularly in regions 2 and 3, in which significant differences were observed between the control and LPS groups. The shaded areas of the line diagrams in Fig. 1 represent the median ischemic lesion from each group of animals. It can be seen that, for the LPS-pretreated group, the improved perfusion in regions 2 and 3 is associated with sparing of tissue in these regions, whereas all of region 3 and part of region 2 were included in the infarct for the saline group. There was also significant partial preservation of perfusion in region 4 for the LPS group, but the magnitude of the effect was less than that observed for regions 2 and 3, and tissue in region 4 was still infarcted.
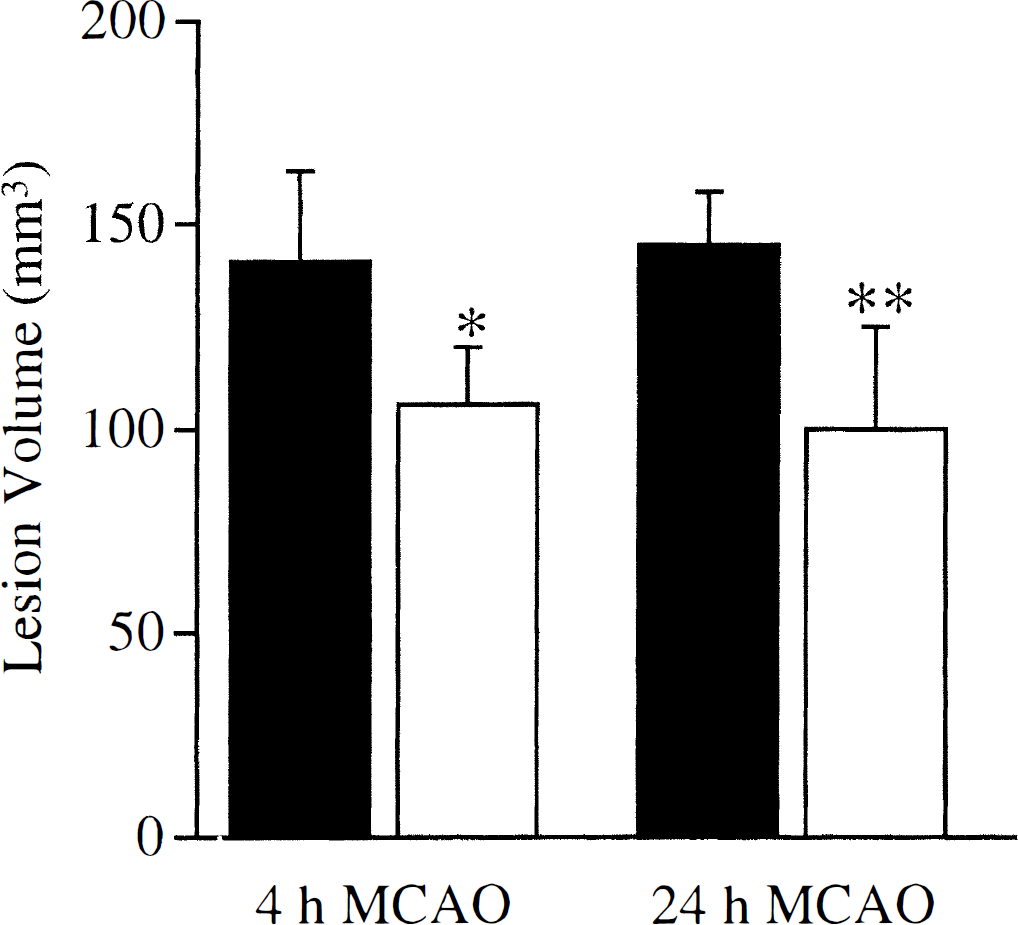
Concomitant with the preservation of microvascular perfusion, LPS pretreatment also significantly reduced ischemic lesion volume (Fig. 3). The magnitude of effect was similar at both 4 and 24 hours after MCA occlusion and represented a 25% to 31 % reduction in lesion volume.
Ischemic lesion volumes (mean ± SD) measured 4 hours or 24 hours after permanent MCA occlusion, in rats pretreated 72 hours previously with LPS (open columns) or saline (filled columns). Lesion volume was significantly smaller for LPS-pretreated animals at both points (* P < 0.05, ** P < 0.01, unpaired t tests).
DISCUSSION
In this study and previous work (Tasaki et al., 1997), we have demonstrated that LPS pretreatment (72 hours before permanent MCA occlusion) renders the brain more tolerant to focal ischemia, with a consistent reduction in lesion volume of 25 % to 31 %, This magnitude of protection is particularly striking given that it is achieved in a model of permanent ischemia using a strain of rat (SHR) that is more resistant to conventional pharmacotherapy (e.g., MK-801) than normotensive strains because of poor collateral circulation (Roussel et al., 1992).
The present study provides further characterization of this model of tolerance. First, we have demonstrated that CBF during the first 15 minutes of MCA occlusion does not differ significantly between tolerant and nontolerant animals, i.e., the protective effect of LPS pretreatment can not be attributed to direct attenuation of the ischemic insult. It was important to establish this fact because LPS is a potent inducer of iNOS, and higher levels of NO at the time of MCA occlusion could potentially have enhanced collateral blood flow in a similar way to the NO precursor
Although there was no significant difference in ischemic severity between LPS-treated and control animals, the dose of LPS used to induce tolerance does appear to have stimulated NO synthesis because increased plasma levels of NO metabolites (nitrates and nitrites) were observed 72 hours after LPS treatment (immediately before MCA occlusion). However it is not possible to determine from this data when or where the NO was produced that resulted in the increased concentration of metabolites. Peripheral administration of LPS (as used in our model) induces no, or only minimal, expression of iNOS protein within the brain (although there is widespread mRNA expression; Knowles et al., 1990; Suzuki et al., 1997; Van Dam et al., 1995), suggesting that the major sources of NO production are likely to be the peripheral vasculature and activated monocytes-macrophages. It is probable that LPS would have to be directly administered into the brain to induce central expression of iNOS and alter CBF (Okamoto et al., 1997). The increases in CBF reported in certain models of septic shock after LPS intravenous infusion appear to reflect an increased cardiac output and are only observed in models in which high volumes of fluid are infused (Meyer et al., 1994; Offner et al., 1995); in the absence of such aggressive resuscitation, CBF does not increase, and may even decrease, after LPS treatment (Odnopozov et al., 1996).
Although the initial ischemic insult was essentially the same for tolerant and nontolerant animals, there does appear to be a vascular component to this model of tolerance because the secondary deficits in microvascular perfusion that developed in nontolerant controls were significantly attenuated in the LPS-pretreated animals. Two consistent patterns for attenuation of the perfusion deficits were observed: (1) preserved perfusion in region 2 associated with the downward shift of the lesion border and sparing of tissue in the same region, and (2) a more limited preservation of perfusion within core ischemic areas, without prevention of ischemic injury (e.g., region 4, levels 1 and 2). Although a causal relationship cannot be definitively inferred between the maintained perfusion in region 2 and neuroprotection within this region, the fact that perfusion was also moderately spared within core ischemic areas does suggest that improved microcirculatory perfusion was the primary effect, helping to preserve tissue in regions of moderate ischemia (e.g., region 2), but being insufficient to maintain tissue viability in areas of more dense ischemia (e.g., region 4).
In our laboratories we have instigated in vivo and in vitro studies to further investigate the mediators of LPS-dependent microvascular preservation and tolerance. We have initially focused our research on events occurring at the blood—endothelial interface because such events can have a profound impact on ischemic outcome. Therapeutic strategies aimed at preventing leukocyte adhesion to endothelium (by leukocyte depletion, administration of antibodies against adhesion molecules, or inhibition of proinflammatory cytokines) can significantly reduce ischemic injury (Hartl et al., 1996; Dawson et al., 1996). Therefore, it is plausible that endogenous downregulation of leukocyte—endothelial interactions could also act to limit ischemic injury and could, by facilitating the maintenance of a patent microvasculature, be a major component of the tolerant state.
Downregulation of leukocyte—endothelial interactions could be achieved by modulation of the transcription factor nuclear factor-κB. Nuclear factor-κB is normally activated by LPS, but remains inactive in the LPS-tolerant state (Blackwell et al., 1997; Kohler and Joly, 1997), preventing the expression of chemokines, proinflammatory cytokines, and adhesion molecules such as intercellular adhesion molecule-1 (Blackwell et al., 1997; Takasuka et al., 1995). In vitro studies in our laboratory have demonstrated that expression of intercellular adhesion molecule-1 is inhibited in tolerant cells (Ginis et al., 1998), suggesting that downregulation of nuclear factor-κB-mediated inflammation could be a factor in cerebral ischemic tolerance. Furthermore, inhibition of persistent activation of nuclear factor-κB is also associated with drug-induced neuroprotection after cerebral ischemia (Clemens et al., 1998).
The attenuated increase in circulating neutrophils 24 hours after MCA occlusion observed in tolerant animals in our study may also be a consequence of the downregulation of the inflammatory response and could also serve to limit leukocyte-dependent injury. However, it is equally possible that the increase in circulating neutrophils 24 hours after MCA occlusion is a secondary response to brain injury, and that the smaller number of neutrophils in the tolerant animals reflects their smaller infarct volumes and not vice versa. The small increase in circulating monocytes observed 72 hours after LPS treatment is probably accounted for by the fact that LPS is an inducer of colony stimulating factor-1 (Roth et al., 1997). Changes in monocyte counts are not likely to have had a significant impact on the outcome of the present study (maximum at 24 hours after MCA occlusion) because monocyte infiltration into ischemic tissue occurs much later than for neutrophils (Garcia et al., 1994a).
As an alternative (or parallel) pathway to downregulation of inflammation, tolerance could also be mediated by upregulation of endogenous protective mechanisms such as increased expression of antioxidant enzymes or Ca2+-buffer proteins. Lipopolysaccharide upregulates manganese-superoxide dismutase (Kifle et al., 1996), an enzyme that has been specifically implicated in the protective mechanism of ischemic preconditioning (Kato et al., 1995; Toyoda et al., 1997). Furthermore TNF-α, which appears to be a mediator of LPS-induced tolerance to ischemia (Tasaki et al., 1997) and can directly confer protection against focal ischemia and excitotoxicity (Nawashiro et al., 1997; Cheng et al., 1994), also enhances antioxidant activity and increases expression of calbindin (Cheng et al., 1994; Mattson, 1997). These protective effects of TNF-α pretreatment are in contrast to the detrimental effect of TNF-α synthesized during ischemia (Dawson et al., 1996; Barone et al., 1997). Upregulation of NOS within the first few hours of the onset of ischemia may also be protective (Hudetz et al., 1997). Indeed, tolerance to myocardial ischemia is associated with NO-dependent inhibition of neutrophil infiltration (Zhao et al., 1997), whereas tolerance to LPS is also mediated by NO (Zingarelli et al., 1995). However, similar to postischemic TNF-α production, delayed induction of iNOS within the brain (at least in nontolerant animals) can also have detrimental effects on ischemic outcome (Iadecola et al., 1995).
In summary, we have demonstrated that pretreatment with the proinflammatory stimulant LPS induces a state of tolerance to subsequent focal ischemia. Lipopolysaccharide pretreatment did not increase CBF immediately after MCA occlusion, but did attenuate the development of secondary microperfusion deficits. This sparing of the microvasculature may be one of the mechanisms by which LPS confers its neuroprotective effect and could be mediated by either active downregulation of the inflammatory response or upregulation of protective genes.