
Abstract
Delayed treatment with aminoguanidine (AG), a relatively selective inhibitor of inducible nitric oxide synthase, ameliorates brain damage produced by occlusion of the rat's middle cerebral artery (MCA). We investigated whether the protection exerted by AG is dose-dependent and whether it is associated with improved neurologic outcome. We also studied the effect of the timing of administration of AG relative to the induction of cerebral ischemia. Halothane-anesthetized spontaneously hypertensive rats underwent permanent MCA occlusion distal to the lenticulostriate branches. Neurologic deficits were assessed daily by the postural reflex test and beam balance test. Infarct volume was determined in thionin- stained sections 96 hours after ischemia and values corrected for swelling. Treatment with AG (intraperitoneally, twice daily), starting 24 hours after MCA occlusion, decreased neocortical infarct volume in comparison to vehicle-treated rats. After correction for swelling, the decrease was 8 ± 12% at 50 mg/kg (n = 8; P > .05; analysis of variance), 25 ± 13% at 100 mg/kg (n = 7; P < .05), 30 ± 16% at 200 mg/kg (n = 7; P < .05) and 32 ± 9% at 400 mg/kg (n = 5; P < .05). Twenty-four hours after induction of ischemia neurologic deficits scores did not differ between treated and untreated rats (P > .05). However, from 48 to 96 hours after ischemia, neurologic deficits improved significantly in rats treated with AG (100 to 400 mg/kg) compared to rats in which vehicle was administered (P < .05). The decrease in neocortical infarct volume was greatest when AG (100 mg/kg; twice daily) was administered 12 (26 ± 17%; n = 9) or 24 hours (25 ± 13; n = 7) after MCA occlusion. The findings show that AG decreases ischemic brain damage dose-dependently and improves neurologic recovery. Delayed treatment with AG may be a therapeutic strategy to selectively target the evolution of ischemic damage that occurs in the post-ischemic period.
We have previously shown that administration of aminoguanidine (AG), a compound that inhibits inducible nitric oxide synthase (iNOS), decreases the infarct resulting from transient or permanent occlusion of the middle cerebral artery (MCA) in rats (Iadecola et al., 1996; Iadecola et al., 1995a; Zhang et al., 1996). AG blocks the increase in iNOS activity that follows focal cerebral ischemia (Iadecola, et al., 1995a). Furthermore, its protective effect is counteracted by the nitric oxide precursor L-arginine but not by D-arginine (Iadecola, et al., 1995a). These observations indicate that the decrease in ischemic damage by AG is, at least in part, related to iNOS inhibition.
The decrease in ischemic damage afforded by AG is notable in that it occurs even when the drug is administered 24 hours after MCA occlusion (Iadecola, et al., 1995a). This finding indicates that brain tissue can be rescued from infarction many hours after induction of ischemia. Therefore, treatment with AG may be effective in preventing the delayed evolution of the damage that occurs in the post-ischemic period. Considering that most stroke patients reach medical attention many hours after the onset of symptoms (Alberts et al., 1990), AG would be a welcome addition to current therapeutic options available for cerebral ischemia. However, several important questions regarding the protective effect of AG remain to be addressed. The concentration of AG resulting in maximal protection and the optimal timing of administration of AG relative to the induction of ischemia have not been established. In addition, it remains to be determined whether the decrease in brain damage, assessed histologically, is associated with an improvement in the neurologic outcome. Therefore, considering the potential therapeutic value of AG in stroke, it would be important to obtain data to clarify these issues.
In the present study we used a rat model of focal cerebral ischemia to better characterize the protective effect exerted by AG. First, we sought to identify the dose of AG resulting maximal protection. Second, we tested the hypothesis that the protective effect of AG is associated with a better neurologic recovery after stroke. Third, we sought to determine the optimal time for AG administration after cerebral ischemia. We found that administration of AG results in a dose-related decrease in cerebral ischemic damage that is associated with a corresponding improvement of neurologic deficits. The greatest decrease in ischemic damage was observed when AG was administered 12 to 24 hours after ischemia. The data show that delayed administration of AG improves both histologic damage and neurologic outcome after cerebral ischemia and provide additional evidence in support of the hypothesis that AG is a potentially useful agent for the treatment of the late stages of cerebral ischemia.
METHODS
Methods for MCA occlusion with monitoring of physiologic parameters and for determination of infarct size have been described in detail in previous publications (Iadecola, et al., 1995a,1995b; Zhang and Iadecola, 1992) and will only be summarized.
General surgical procedures
Studies were approved by the Institutional Animal Care and Use Committee and were conducted on 60 male spontaneously hypertensive rats (SHRs; Harland) weighing 300 to 350 g. SHRs were used because of the well-known reproducibility of focal cerebral ischemic damage in this strain (Ginsberg and Busto, 1989). Under halothane anesthesia (induction: 5%; maintenance: 1%, in an oxygen-nitrogen mixture), the left femoral artery was cannulated. Body temperature was maintained at 37 ± 0.5°C by a thermostatically controlled infrared lamp (YSI, mod. 73A-TA, Yellow Springs, OH). The arterial catheter was connected to a pressure transducer for recording of mean arterial pressure. Serum glucose was measured by a glucose analyzer (Beckman) and arterial blood gases were monitored by a blood gas analyzer (CIBA-Corning, mod. 178, Medfield, MA). The arterial catheter was tunneled under the skin and exteriorized at the level of the tail. The catheter was used for recording of arterial pressure and for determination of serum glucose, hematocrit, and arterial blood gases at different times after MCA occlusion.
Middle cerebral artery occlusion and measurement of infarct volume
Procedures for MCA occlusion and for determination of infarct volume are similar to those published previously (Zhang and Iadecola, 1992). Briefly, a 2-mm hole was drilled at a site superior and lateral to the left foramen ovale to expose the left MCA. The MCA was elevated and cauterized distal to the origin of the lenticulostriate arteries and medial to the inferior cerebral vein (Zhang and Iadecola, 1992). Animals were then returned to their cages and monitored until they recovered consciousness. All rats regained consciousness rapidly and did not exhibit clinical evidence of seizures. After consciousness was regained, rats did not exhibit behavioral evidence of pain or discomfort.
Rats were killed 96 hours after induction of ischemia and their brains were removed and processed for determination of infarct volume. The forebrain was frozen in cooled isopentane (−30°C). Coronal forebrain sections (thickness 30 μm) were cut serially in a cryostat. One section every 300 μm was collected and stained with thionin. As described in detail elsewhere (Zhang and Iadecola, 1992), infarct volume was determined using an image analyzer (Imaging Research Inc., MCID, St. Catharines, Ontario, Canada). Infarct volume in cerebral cortex was corrected for swelling according to the method of Lin et al. (Lin et al., 1993), as previously described (Iadecola, et al., 1995a; Zhang and Iadecola, 1994).
Neurologic evaluation
Examinations were performed before MCA occlusion and every day thereafter up to 96 hours after MCA occlusion. The examiner was blinded to the treatment groups. Postural reflexes were tested as previously described (Bederson et al., 1986). Rats were suspended by the tail 1 m above the floor and slowly lowered while observing their posture. Rats were then placed on a flat surface and gently pushed from side to side. Deficits were scored as follows: 0, no deficit; 1, forelimb flexion while suspended by the tail; 2, decreased resistance to lateral push. Rats were also evaluated by the beam balance test (Feeney et al., 1982). Animals were required to walk on a narrow beam (width: 3/4″; length: 10″) for 60 seconds. Deficits were graded based on the scale by Clifton et al. (Clifton et al., 1991): 1, steady posture with paws on top of beam; 2, paws on side of beam or wavering; 3, one or two limbs slip off beam; 4, three limbs slip off beam; 5, rat attempts to balance with paws on beam but falls; 6, rat drapes over beam, then falls; 7, rat falls off the beam without attempting to stay on.
Experimental protocol
Rats were returned to their cages after insertion of the femoral arterial catheter and, when they fully recovered from the anesthesia, baseline arterial pressure, blood gases, serum glucose, rectal temperature, and hematocrit were measured. Rats were then re-anesthetized for MCA occlusion.
Data analysis
Data in the text, table, and figures are expressed as means ± SD. Comparisons among multiple groups were evaluated by one-way analysis of variance followed by the Tukey-Kramer honestly significant difference (HSD) test as a post hoc multiple comparison procedure (Systat Inc.)(Kirk, 1982). Neurologic deficits were statistically evaluated by Kruskal-Wallis test followed by post hoc Tukey-Kramer HSD test (Sokal and Rohlf, 1994). Differences were considered significant at P < .05.
RESULTS
Effect of AG dose on infarct volume and neurologic deficits
In these experiments we studied the relationship between dose of AG (50 to 400 mg/kg) and the corresponding decrease in infarct volume. Treatments were started 24 hours after MCA occlusion. Arterial pressure, rectal temperature, and serum glucose did not differ among the groups studied (P > .05, analysis of variance and Tukey-Kramer HSD test) (Fig. 1), except for the 400-mg/kg group in which AG produced a mild increase in arterial pressure and a decrease in rectal temperature (32 to 74 hours after MCA occlusion) (Fig. 1). Baseline pCO2, pO2, pH, and hematocrit in the vehicle group were, respectively, 37 ± 10 mm Hg, 85 ± 5 mm Hg, 7.44 ± 0.04 and 49.6 ± 1.4%. No differences in these variables were noted among the various treatment groups during the course of the study (P > .05). In vehicle-treated rats (n = 9), distal MCA occlusion produced infarcts involving almost exclusively the neocortex (Table 1). The volume of the infarct and its regional distribution were similar to those previously reported after distal MCA occlusion (Iadecola, et al., 1995a). Administration of AG decreased neocortical infarct volume dose-dependently (Table 1). After correction for ischemic swelling, the decrease in neocortical infarct volume was 8 ± 12% at 50 mg/kg (P > .05 from vehicle; n = 8), 25 ± 13% at 100 mg/kg (P < .05; n = 7), 30 ± 16% at 200 mg/kg (P < .05; n = 7), and 32 ± 9% at 400 mg/kg (P < .05; n = 5). The volume of tissue swelling did not differ among the groups studied (P > .05) (Table 1).
Effect of aminoguanidine dose on infarct volume after middle cerebral artery occlusion

P < 0.05 from vehicle, analysis of variance and Tukey-Kramer HSD test.
P < 0.05 from aminoguanidine 50 mg/kg.
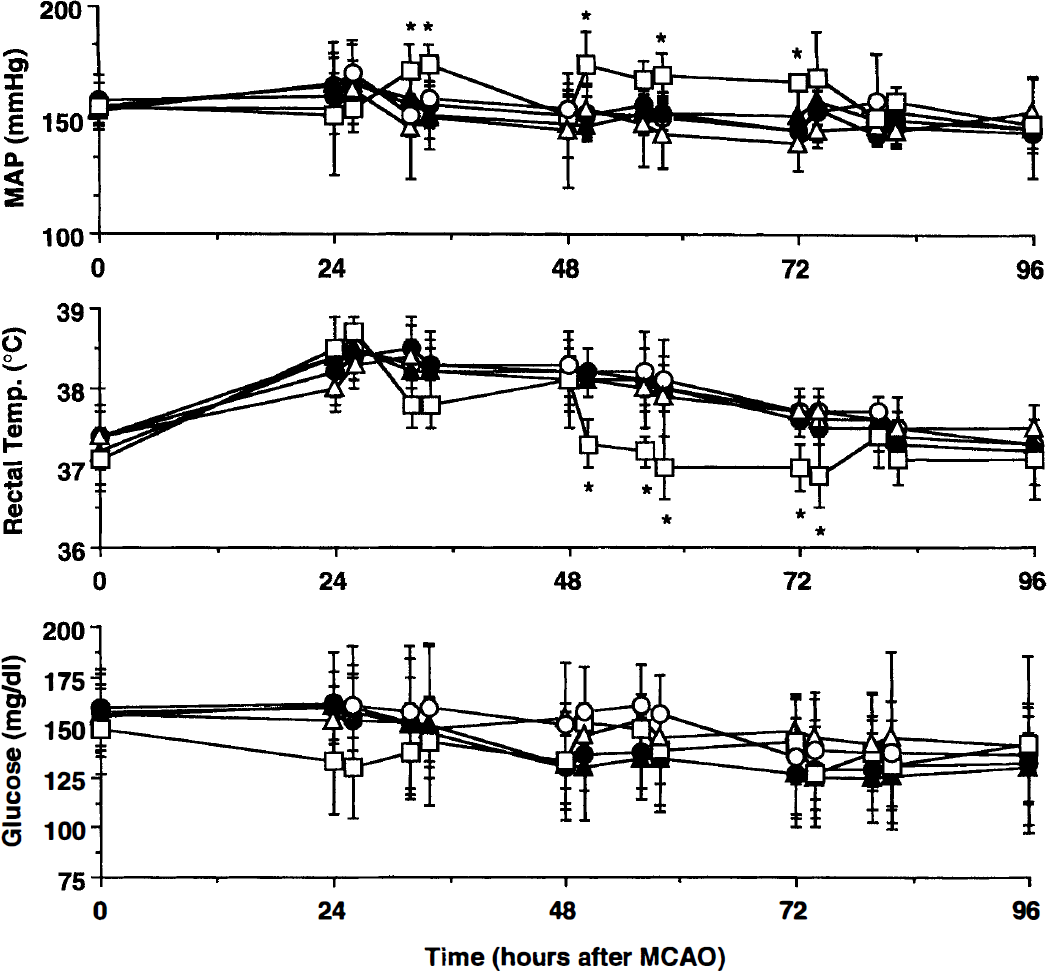
Effect of administration of saline (vehicle), or aminoguanidine (AG; 50 to 400 mg/kg) on mean arterial pressure, rectal temperature, and serum glucose. Arterial pressure did not differ among groups (P > .05, analysis of variance and Tukey-Kramer honestly significant difference test), except for the 400-mg/kg group, in which treatment produced a sustained but mild increase in arterial pressure (P < .05). Rectal temperature was increased by 1°C to 1.5°C after ischemia and did not differ among groups (P > .05), except for the 400-mg/kg group in which the temperature was decreased at some time points, in comparison to vehicle-treated rats (P < .05). No differences in serum glucose were observed (P > .05). AG, aminoguanidine; MAP, mean arterial pressure; MCAO, middle cerebral artery occlusion. *P < 0.05 from vehicle. Mean ± SD. (○) Vehicle (n = 9). (•) AG 50 mg/kg (n = 8). (▵) AG 100 mg/kg (n = 7). (▴) AG 200 mg/kg (n = 7). (□) AG 400 mg/kg (n = 5).
The time course of the effect of AG on the neurologic deficits resulting from MCA occlusion is shown in Figure 2 Scores for the postural reflex test and the beam balance test did not differ between treated and untreated rats at 24 hours after MCA occlusion (P > .05, Kruskal-Wallis and Tukey-Kramer HSD tests). In rats receiving vehicle, neurologic deficits remained stable throughout the monitoring period (24 to 96 hours after MCA occlusion; Fig. 2). However, in rats treated with AG, the deficits improved between 24 and 96 hours after MCA occlusion. The effect was related to the dose of AG and was maximal at 200 to 400 mg/kg (P < .05). The data suggest that AG treatment decreases infarct volume and improves the neurologic deficits resulting from MCA occlusion.
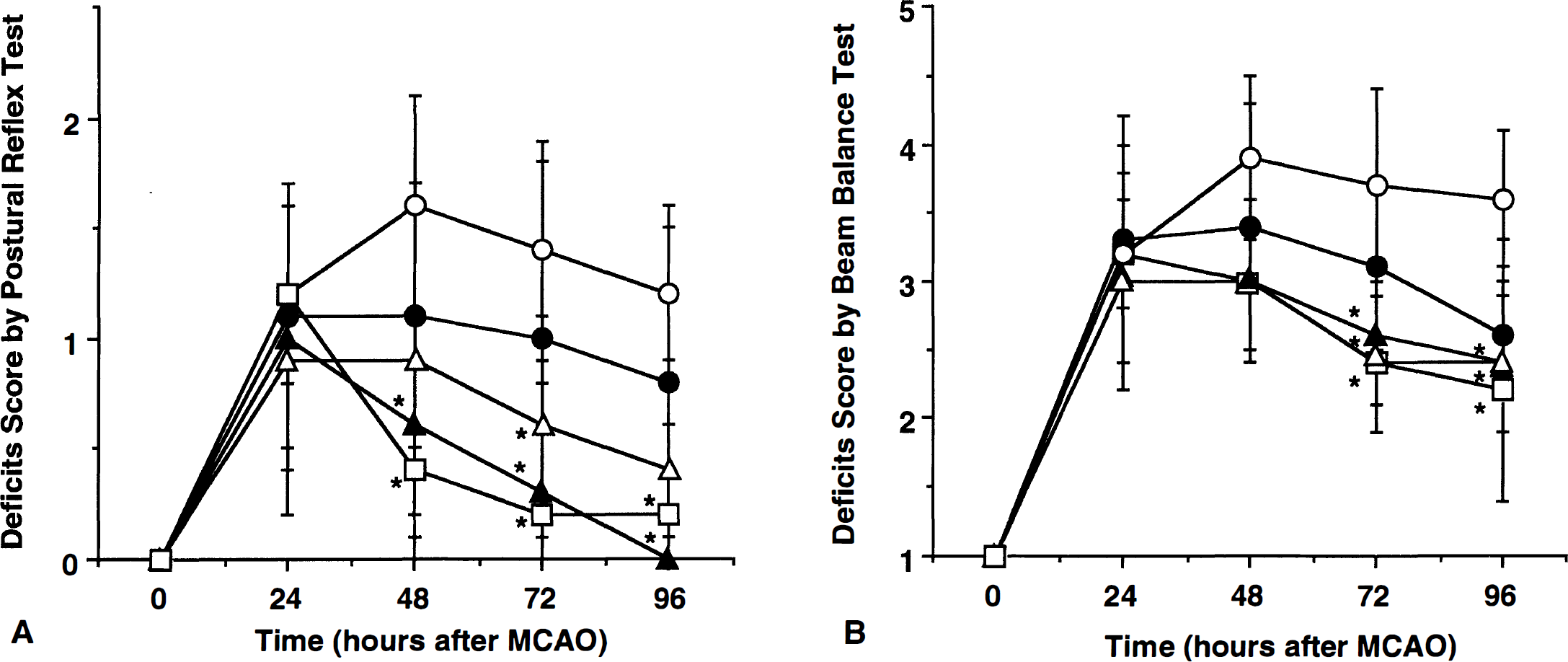
Time-course of the effect of aminoguanidine on the neurologic deficits resulting from middle cerebral artery occlusion. Neurologic deficits were evaluated by the postural reflex test
Effect of the timing of administration of AG relative to MCA occlusion
In these studies we investigated the relationship between the time interval between MCA occlusion and administration of AG (100 mg/kg) and the observed decrease in infarct size. As shown in Fig. 3, arterial pressure, serum glucose, and rectal temperature did not differ among the groups studied (P > .05). Baseline pCO2, pO2, pH, and hematocrit in the vehicle group were, respectively, 42 ± 4 mm Hg, 79 ± 5 mm Hg, 7.41 ± 0.08 and 50.9 ± 1.6%. No differences in these variables were noted among the different groups during the course of the study (P > .05). Administration of AG starting 6 hours after MCA occlusion decreased infarct size by 13 ± 23% (corrected for swelling), a change that did not reach statistical significance (P > .05; n = 6; Table 2). Treatments started 12 hours (n = 9) after MCA occlusion decreased neocortical infarct volume by 26 ± 17% (P < .05) (Table 2). The decrease in infarct volume observed when AG was administered 12 hours after MCA occlusion was identical to that observed when AG was administered 24 hours after MCA occlusion (see 100 mg/kg-group of the dose-response study). The volume of tissue swelling did not differ among the groups studied (P > .05). Therefore, the protection exerted by AG is greater when the drug is administered 12 or 24 hours than 6 hours after MCA occlusion.
Effect of the time of onset of aminoguanidine treatment on infarct volume after middle cerebral artery occlusion
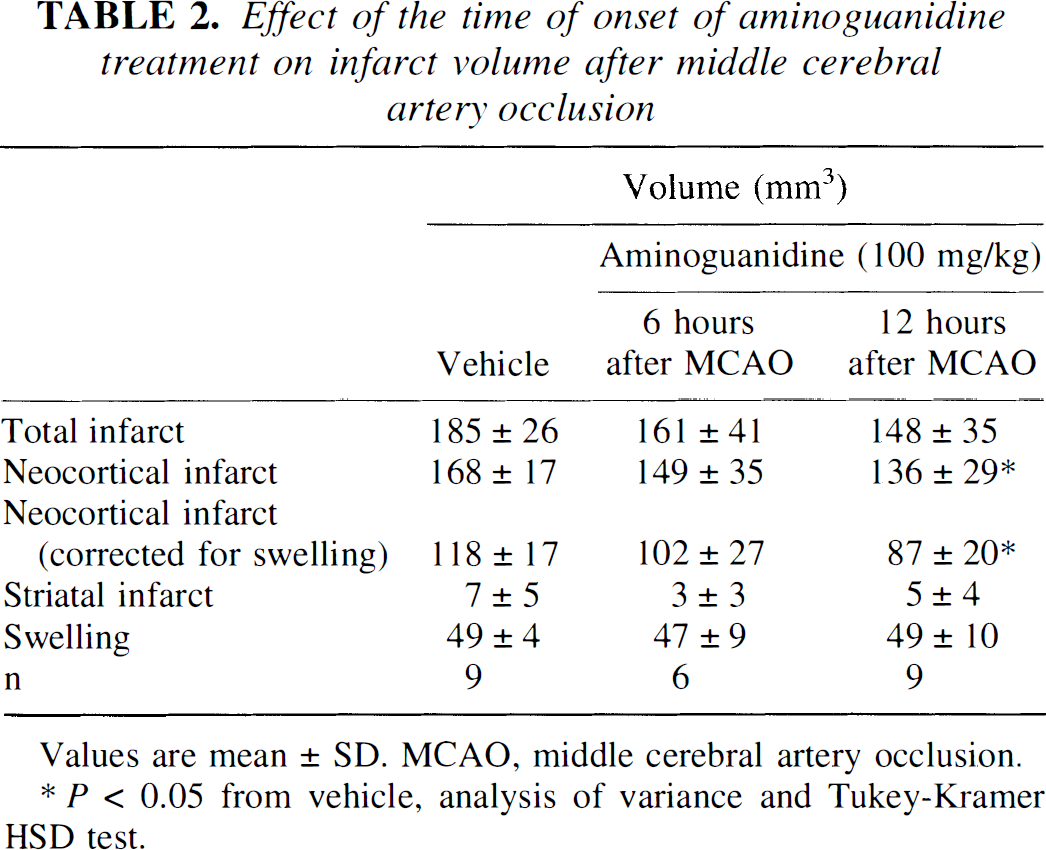
P < 0.05 from vehicle, analysis of variance and Tukey-Kramer HSD test.
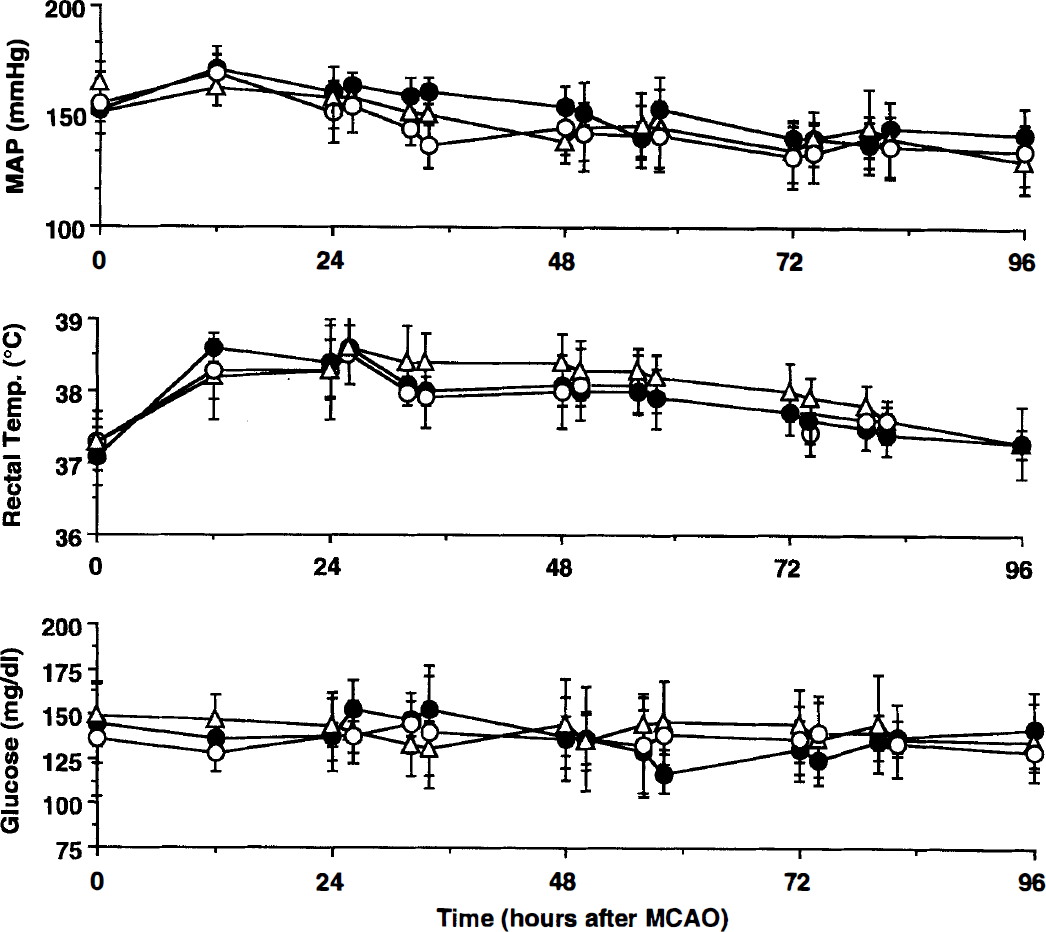
Mean arterial pressure, rectal temperature, and serum glucose in rats in which the effect of the timing of aminoguanidine administration on infarct volume was studied. These variables did not differ among the groups of rats. AG, aminoguanidine; MAP, mean arterial pressure; MCAO, middle cerebral artery occlusion. Mean ± SD. (○) Vehicle (n = 9). (•) AG from 6 hours after MCAO (n = 6). (▵) AG from 12 hours after MCAO (n = 9).
DISCUSSION
We have shown that administration of AG decreases the size of the infarct resulting from MCA occlusion in a dose-dependent fashion and that such a decrease is associated with an improvement in neurologic deficits, as assessed by the postural reflex and beam balance tests. The effect of AG is more marked when the drug is administered 12 to 24 hours rather than 6 hours after MCA occlusion. The observation that AG improves the neurologic deficits of the animals is of interest because it suggests that the brain tissue spared from infarction by AG is functional and results in a better neurologic outcome. Although it had previously been shown that AG is beneficial to the postischemic brain (Iadecola, et al., 1996; Iadecola, et al., 1995a), the present study extends these observations by identifying the most effective dose (200 to 400 mg/kg) and timing of administration (12 to 24 hours after ischemia). Most importantly, the present study shows that the decrease in histologic damage is reflected in improvement of some of the neurologic deficits associated with MCA occlusion. These new observations provide additional evidence supporting the validity of AG as a potential treatment for the late stages of cerebral ischemia.
The effect of AG cannot be a consequence of changes in arterial pressure, blood gases, hematocrit, rectal temperature, or serum glucose because these variables were carefully monitored and did not differ among the groups studied. In rats treated with the highest dose of AG (400 mg/kg) a slight increase in arterial pressure and a decrease in rectal temperature were observed. However, these small changes are unlikely to be responsible for the protective effect because smaller doses of AG, which did not alter arterial pressure or rectal temperature, were also effective. Similarly, the decrease in infarct volume is not a consequence of effects of AG on ischemic edema because the volume of swelling of the ischemic brain was not influenced by treatment with AG.
It was found that AG was more effective when it was administered 12 or 24 hours after induction of ischemia. This observation suggests that AG counteracts the delayed progression of cerebral ischemic damage that occurs after MCA occlusion. Recent evidence suggests that the evolution of ischemic brain injury is more protracted in time than previously believed. Histologic data indicate that neuronal damage occurs rapidly in the center of the ischemic territory, whereas at the periphery the damage develops more slowly over days (Garcia et al., 1995; Garcia et al., 1993). Studies by positron emission tomography suggest that brain potentially rescuable from infarction is still present many hours after the onset of ischemia (Furlan et al., 1996; Marchal et al., 1996; Touzani et al., 1995). Similarly, studies by magnetic resonance imaging have shown that the area of irretrievable damage grows over several days after ischemia in patients (Baird et al., 1997). The mechanisms of the delayed progression of the damage have not been firmly established. Experimental studies indicate that postischemic expression of iNOS, an enzyme that produces cytotoxic amounts of nitric oxide, contributes to the damage (Iadecola et al., 1997; Iadecola, et al., 1995a,1995b). Similarly, expression of cyclooxygenase-2, an enzyme that produces toxic prostanoids and free radicals, may also contribute to the postischemic evolution of the damage (Nogawa et al., 1997). In addition, programmed cell death, a process that develops over days after ischemia, may also participate in the delayed evolution of the damage (MacManus and Linnik, 1997). The finding that AG is effective even its administration is delayed by 24 hours after ischemia suggests that AG counteracts some of the factors contributing to the delayed extension of the damage. However, the time-course of iNOS expression in transient ischemia, produced by intraluminal occlusion of the MCA, is shifted to earlier time points (Iadecola, et al., 1996). It is, therefore, likely that the therapeutic window will be shorter in transient ischemia than in permanent ischemia.
There are several mechanisms by which AG could protect the brain from the delayed evolution of the damage. One possibility is that AG decreases infarct size by inhibiting iNOS activity in the postischemic brain. This hypothesis is supported by several observations. First, AG, at doses comparable to those used in the present study, inhibits calcium-independent NOS activity selectively in the postischemic brain (Iadecola, et al., 1995a). Second, the effect of AG is counteracted by coadministration of the nitric oxide precursor L-arginine, but not by D-arginine (Iadecola, et al., 1995a). Third, the protection exerted by AG is observed during the period when iNOS is expressed in the postischemic brain (Zhang and Iadecola, 1998). The magnitude of the decrease in infarct size afforded by AG is virtually identical to that observed in null mice lacking the iNOS gene (Iadecola, et al., 1997). These observations support the hypothesis that the protective effect exerted by AG is related to iNOS inhibition. However, AG has also other pharmacologic actions that might contribute to its neuroprotective effects. For example, AG is a potent inhibitor of diamine oxidase (Beaven et al., 1969), an enzyme that participates in the degradation of histamine and polyamines (Sessa and Perin, 1994). The terminal oxidative deamination of polyamines by diamine oxidase leads to the production of toxic aldehydes (Sessa and Perin, 1994), compounds that could potentially contribute to cerebral ischemic damage. Because increased polyamine synthesis and degradation begins early after an ischemic insult (Paschen et al., 1993), it is unlikely that, under the experimental conditions of the present study, inhibition of diamine oxidase contributes to the protective effect of AG. Another potential mechanism by which AG could decrease tissues damage is by inhibition of advanced glycation end-products (AGE) production or cross-linking (Brownlee et al., 1986). Systemic administration of AGE before, but not after ischemia, exacerbates cerebral ischemic damage, an effect that is blocked by AG (Zimmerman et al., 1995). However, it has not been shown whether ischemia leads to AGE formation. Therefore, further studies will be required to clarify the role of AGE in cerebral ischemic damage. Irrespective of the mechanisms of the effect, the present results provide additional evidence that delayed administration of AG decreases cerebral ischemic damage. Therefore, this drug could be useful to specifically target the damage that occurs in the late stages of cerebral ischemia.
We also found that AG was less effective when its administration was started 6 hours after ischemia. One possible explanation for this finding is that AG also inhibits endothelial NOS (eNOS) (Laszlo et al., 1995). The observation that AG at 400 mg/kg increases arterial pressure is consistent with the hypothesis that, in our preparation, AG inhibits eNOS. Nitric oxide produced by eNOS is beneficial in the early stages of cerebral ischemia, probably, by producing vasodilation and by inhibiting platelet aggregation and leukocyte adhesion (Huang et al., 1996). Therefore, eNOS inhibition by AG could result in further flow decrease in regions at risk for infarction and could facilitate postischemic microvascular occlusions by platelets and leukocytes. The worsening of the initial damage resulting from eNOS inhibition could offset the protective effect observed in the late stages of cerebral ischemia. In apparent contradiction with this hypothesis, however, is the finding that pretreatment or early post-treatment with AG (160 mg/kg) has been reported to decrease infarct size in a model in which the right common carotid artery is ligated, the right MCA severed, and then the left common carotid artery transiently occluded in Lewis rats (Cockroft et al., 1996; Zimmerman, et al., 1995). Although the reasons for this discrepancy are unclear at the present time, differences in the stroke models, dose of AG, and/or survival times are likely to play a role. Further studies in which the effect of AG on CBF in the early postischemic period is determined would help define the potential hemodynamic effect of AG after induction of ischemia.
In conclusion, we have shown that AG decreases ischemic cerebral damage and enhances neurologic recovery in a dose-dependent fashion. Delayed treatment is more beneficial than early treatment. The data suggest that AG counteracts the delayed progression of cerebral ischemic damage that occurs in the postischemic period. The fact that AG is effective when administered 12 to 24 hours after induction of cerebral ischemia indicates that this drug could be very valuable in the treatment of human stroke by specifically targeting the late stage of the damage.
Footnotes
Acknowledgements
The authors thank Ms. Karen MacEwan for excellent editorial assistance.