
Abstract
Experimental and clinical data suggest that oxygen and/or glucose deprivation alters electrical transmission in the brain and generates free radicals, which may mediate neuronal death. We have analyzed the effects of oxygen and/or glucose deprivation on both excitatory transmission, by measuring field potential amplitude, and free radical production, by using electron spin resonance (ESR) spectroscopy, in a corticostriatal slice preparation. Combined oxygen and/or glucose deprivation (ischemia) lasting 10 to 20 minutes induced a long-term depression of field potential amplitude. The ascorbyl radical could only be detected in brain slices during the reperfusion-phase after 30 minutes of ischemia. It appeared in the early minutes after the washout of ischemic medium and remained stable throughout the reperfusion phase. This radical was never detected in the external medium. Ischemia induced only a slight, but progressive, release of lactate dehydrogenase (LDH) into the external medium during the reperfusion phase. In contrast, exposure of slices to hypoxia or hypoglycemia alone resulted in transient depression of field potential amplitude, and no generation of ascorbyl radicals was observed on reperfusion. We propose that the long-lasting loss of electrical signals is the early sign of neuronal damage during ischemia. On the other hand, ascorbyl radical formation may be considered an indicator of neuronal injury after prolonged energy deprivation.
Neuronal activity requires a continuous supply of oxygen and glucose. The impairment of oxidative metabolism causes a cascade of ionic and metabolic events leading to neuronal death (Siesjö, 1988; Choi, 1990; Beal, 1992; Haddad and Jiang, 1993; Martin et al., 1994). Experimental and clinical studies have shown that various brain areas express differential sensitivity to oxygen and/or glucose deprivation (Kass and Lipton, 1982; Hawker and Lang, 1990; Hara et al., 1993; Crepel et al., 1992). Although common mechanisms have been proposed to underlie neurologic damage after hypoglycemia, hypoxia, and ischemia (Martin et al., 1994), the relative contribution of each mechanism might depend on the experimental approach used. Inadequate synthesis of ATP has several consequences, such as cellular acidosis, through the stimulation of anaerobic glycolysis; disruption of ion distribution, caused by the loss of energy-dependent ion transport mechanisms; and loss of cytoskeletal integrity as a consequence of decreased synthesis of macromolecular assemblies required for maintaining cell structure (Hara et al., 1993; Martin et al., 1994). Cellular energy failure has also been hypothesized to result in calcium overload (Siesjö 1988; Choi, 1990), which is usually related to increased sensitivity to endogenous glutamate as well as to an increased release of excitatory amino acids (Rothman, 1983; Simon et al., 1984; Benveniste et al., 1984; Wieloch, 1985; Globus et al., 1988; Choi, 1990; Choi and Rothman, 1990).
Free radical production has been proposed to be involved in the pathogenesis of the ischemia-reperfusion neuronal damage (Demopoulos et al., 1982; Traystman et al., 1991; Schulz et al., 1995; Globus et al., 1995; Chan, 1996). The formation of free radicals has been detected by spin trapping techniques using electron spin resonance (ESR) spectroscopy during in vivo global ischemia (Zini et al., 1992; Sen and Phillis, 1993). Moreover, it has been demonstrated that pretreatment with free radical spin traps (Oliver et al., 1990; Schulz et al., 1995) or overexpression of the antioxidative enzyme superoxide dismutase in transgenic mice (Kinouchi et al., 1991; Chan, 1996) reduces ischemia-induced brain damage. In vitro experiments suggest that excitotoxicity and oxidative stress could be interrelated mechanisms in cerebral ischemia-reperfusion damage. It has been proposed that the release of excitatory amino acids may be triggered by the formation of oxygen free radicals, because both free radical scavengers and antioxidative enzymes were capable of reducing the excitatory amino acid output (Pellegrini-Giampietro et al., 1990).
The possible relationship between alterations of excitatory synaptic transmission observed during oxygen or glucose deprivation, or both, and the formation of free radicals has never been investigated. In the present study, we have used a new in vitro approach to study this relationship in the striatum, a brain region that shows a peculiar neuronal vulnerability to hypoxia (Calabresi et al., 1995 a, 1995b), hypoglycemia (Auer et al., 1984; Auer and Siesjö, 1988; Nakao et al., 1995; Kristian et al., 1995), and ischemia (Brierley, 1976; Pulsinelli, 1985; Xu, 1995). Free radical formation was measured directly in the same slices by monitoring the relatively stable monovalent oxidation product of endogenous ascorbate, which is considered a marker of oxygen radicals formation and can be detected without the use of spin trapping agents (Niki, 1991; Buettner and Jurkiewicz, 1993). In fact, it has already been reported that in vivo brain ischemia induces ascorbyl radical formation during the reperfusion phase (Matsuo et al., 1995). These authors suggested that neutrophils are the major source of ascorbyl radical. To further investigate this issue, we have used our in vitro approach to detect both ascorbyl radical formation and changes in cortically evoked field potentials. Thus, we have studied the formation of free radicals and the electrophysiologic alterations after hypoxia, hypoglycemia, and ischemia in a corticostriatal slice preparation. We show that brief periods of ischemia cause long-lasting depression of field potential amplitude whereas only longer periods lead to formation of ascorbyl radical on reperfusion and lactate dehydrogenase (LDH) release.
METHODS
Preparation and treatment of coronal slices
Adult male Wistar rats (150 to 250 g) were used. The preparation and maintenance of coronal slices have been described previously (Calabresi et al., 1995 a, 1995b; 1996 a, 1996b). Briefly, corticostriatal coronal slices (200 to 300 μm) were prepared from tissue blocks of the brain with the use of a vibratome. All slices were preincubated 30 minutes at 35°C in Krebs solution (in mmol/L: 126 NaCl, 2.5 KCl, 1.2 MgCl2, 1.2 NaH2PO4, 2.4 CaCl2, 11 glucose, 25 NaHCO3; pH 7.4) gassed with 95% O2/5% CO2. A single slice was transferred to a recording chamber and submerged in the same continuously flowing Krebs solution (35°C, 2 to 3 mL/minute) gassed with 95% O2/5% CO2. Hypoglycemia was induced by switching to a glucose-free medium containing sucrose to maintain the osmolarity. In some experiments the osmolarity was balanced by increasing the NaCl concentration (Jiang and Haddad, 1992). Because experiments performed using these different procedures to replace glucose gave similar results, all the data were pooled together. Hypoxia was obtained by switching the perfusion from the oxygen-gassed solution to a Krebs solution (same composition) gassed with a mixture of 95% N2/5% CO2 (Calabresi et al., 1995 a, 1995b). Ischemia was obtained by combining glucose and oxygen deprivation. The solutions entered the recording chamber no later then 20 seconds after turning a 3-way valve. Complete replacement of the medium in the chamber took 90 seconds.
In some cases the slices used for electrophysiologic measurements were subsequently removed from the recording chamber and used for ESR studies. A different approach was applied in ESR time-course experiments, in which more slices were needed to ensure sufficient numbers of data points. Typically, 10 to 12 slices from the same preparation were incubated at 35°C in holding chambers containing 5 mL of the Krebs solution per slice. Hypoxic, hypoglycemic, and ischemic conditions were obtained by gassing the medium with the gas mixtures described above, in the presence or absence of glucose. For reperfusion, the slices were rapidly transferred to other holding chambers corresponding to the preincubation conditions. The latter experimental approach was necessary to measure the possible formation of radicals not only in the tissue, but also in the external medium, as well as the release of LDH.
Electrophysiologic methods
Extracellular recording electrodes were filled with 2 mol/L NaCl (1 to 10 MΩ). An Axoclamp 2A amplifier (Axon Instruments, Foster City, CA, U.S.A.) was used for voltage recordings. Traces were displayed on an oscilloscope and stored digitally. For synaptic stimulation, bipolar electrodes were used. These stimulating electrodes were located either in the cortical areas close to the recording electrode or in the white matter between the cortex and the striatum to activate corticostriatal fibers. The frequency of the stimulation was 0.05 Hz. The field potential amplitude was defined as the average of the amplitude from the peak of the early positivity to the peak negativity, and the amplitude from the negativity to the peak late positivity (Calabresi et al. 1996 a, 1996b).
Electron spin resonance measurements
For detection of ascorbyl radical formation, single slices, or alternatively 40 μL incubation medium, were drawn into glass capillaries at the indicated time points; the capillaries were sealed at one end and immediately frozen in liquid N2. At the end of each experiment ESR measurements were made at room temperature with an ESP 300 instrument (Bruker, Rheinstetten, Germany) set at 10 mW power at 9.83 GHz, 1 G modulation, 100 G scanning in 5 seconds, 16 scans accumulated.
Lactate dehydrogenase activity determination
In some experiments in which the activity of the cytosolic enzyme LDH was determined during reperfusion in the medium to assess cellular necrosis, 0.2 mL of medium was collected at the indicated times, and the amount of LDH, expressed as percent of total activity of the tissue, was calculated. The activity of LDH was determined spectrophotometrically from the change in absorbance at 340 nm, using as substrate 0.18 mmol/L NADH and 0.72 mmol/L pyruvate in 50 mmol/L phosphate buffer, pH 7.4 at 25°C.
Statistical analysis
Values given in the text and in the figures are mean ± SEM of changes. Student's t-test (for paired and unpaired observations) was used to compare the means.
RESULTS
Electrophysiologic effects of hypoglycemia
The effect of different periods of hypoglycemia on corticostriatal field potential amplitude are shown in Fig. 1. A decrease of the potential induced by hypoglycemia was observed 10 minutes after the onset of glucose deprivation. The amplitude of this reduction was dependent on the duration of the hypoglycemic period; after 40 minutes of hypoglycemia the potential was completely suppressed. On reperfusion the potential returned completely to the baseline when hypoglycemia had been applied for only 15 minutes. After longer periods (25, 30, or 40 minutes) the recovery was slower and gradually became less complete.
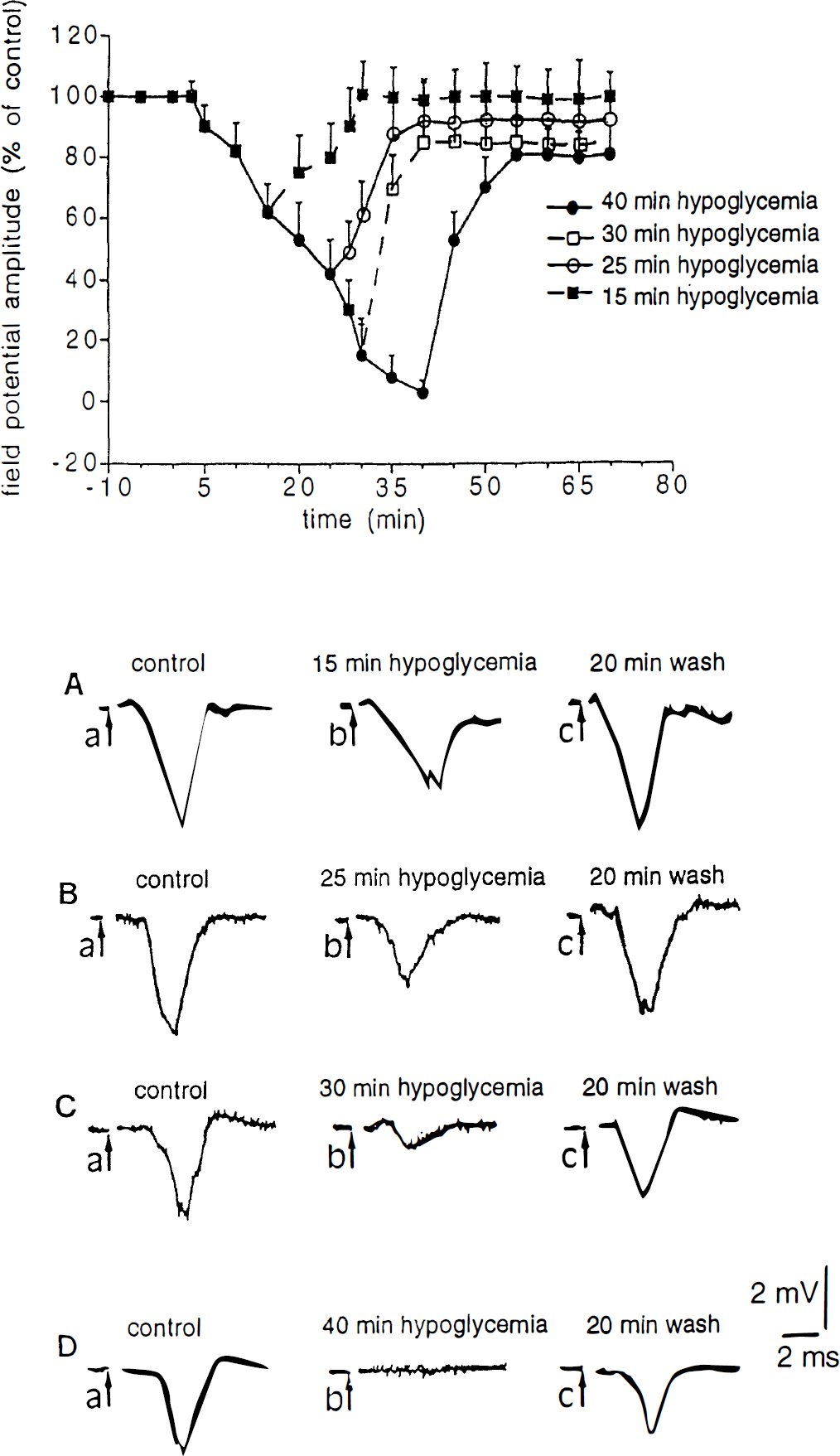
Effects of glucose deprivation on corticostriatal field potentials. The upper part shows the effects of different periods of hypoglycemia on the field potential amplitude. In all experiments hypoglycemia started at time 0 and lasted 15 minutes (▪), 25 minutes (○), 30 minutes (□), or 40 minutes (•). In this graph and in the two following figures each data point represents the average of at least four single experiments. In the lower part
Electrophysiologic effects of hypoxia
Figure 2 shows the action of hypoxia on field potential amplitude. The hypoxia-induced reduction of the potentials was rapid and time-dependent: at 5 minutes a decrease of about 80% of control values was obtained, and full suppression was observed after 20 minutes. Reperfusion restored the control amplitude for periods of hypoxia lasting 10, 20, and 30 minutes. Only 40 minutes of hypoxia induced a significant (P < 0.001) long-lasting decrease of the electrophysiologic signal.
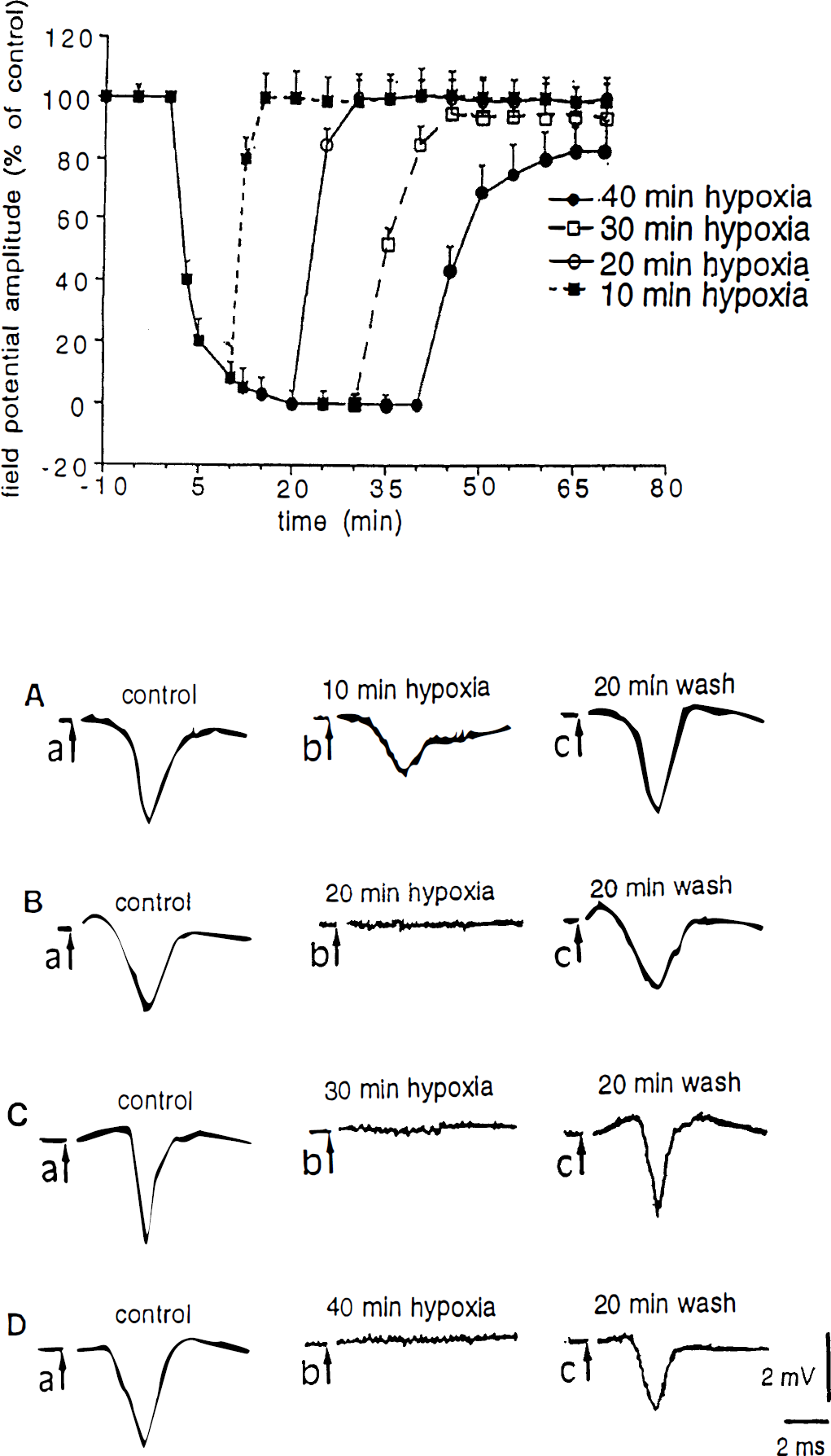
Effects of oxygen deprivation on corticostriatal field potentials. The upper part shows the effects of different periods of hypoxia on the field potential amplitude. In all experiments hypoxia started at time 0 and lasted 10 minutes (▪), 20 minutes (○), 30 minutes (□), or 40 minutes (•). In the lower part
Electrophysiologic effects of ischemia
The effects of ischemia were more dramatic. Only 5 minutes of both glucose and oxygen deprivation were sufficient to abolish the field potential (Fig. 3). However, after reperfusion, the signal returned to the control values. When ischemia lasted 10 minutes or longer, the potential was fully suppressed and only partial recovery (less than 30%) was achieved after a 40-minute wash. After 20 minutes of ischemia no sign of recovery was observed.
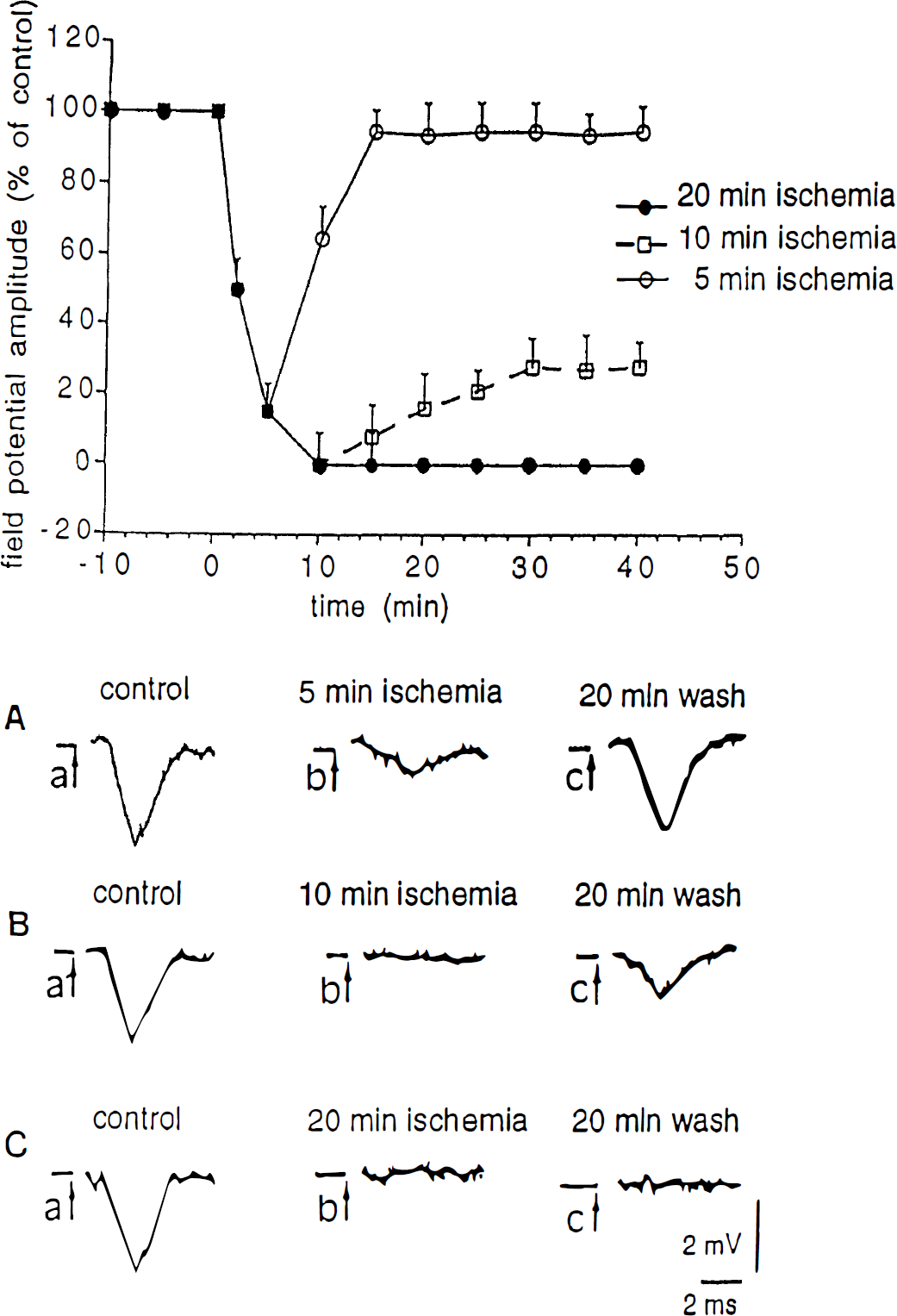
Effects of ischemia on corticostriatal field potentials. The upper part shows the effects of different periods of ischemia on the field potential amplitude. In all the experiments ischemia started at time 0 and lasted 5 minutes (○), 10 minutes (□), or 20 minutes (•). In the lower part
Ascorbyl radical formation
The corticostriatal slices prepared for electrophysiologic measurements could also be used directly for ESR studies of free radical production. Figure 4A shows the resulting ESR spectra of intact slices measured during ischemia and after reperfusion. In the reperfusion phase a large two-line signal appeared in the spectrum. From its characteristic shape and position this signal was identified as the spectrum of the ascorbyl radical, the monovalent oxidation product of the intracellular antioxidant ascorbic acid. No significant amounts of ascorbyl radical were detected in control experiments with sham-treated slices or during different periods of ischemia. The ascorbyl signal appeared rapidly after the onset of reperfusion, and the intensity remained stable throughout the whole reperfusion phase for up to 60 minutes (Fig. 4B). High levels of ascorbyl were only seen after prolonged ischemia (30 minutes or more), whereas little radical production was found after 10 or 20 minutes of ischemia. Slices exposed to hypoxia or hypoglycemia showed only insignificant ascorbyl radical formation, even after prolonged treatment (Fig. 4B and C).
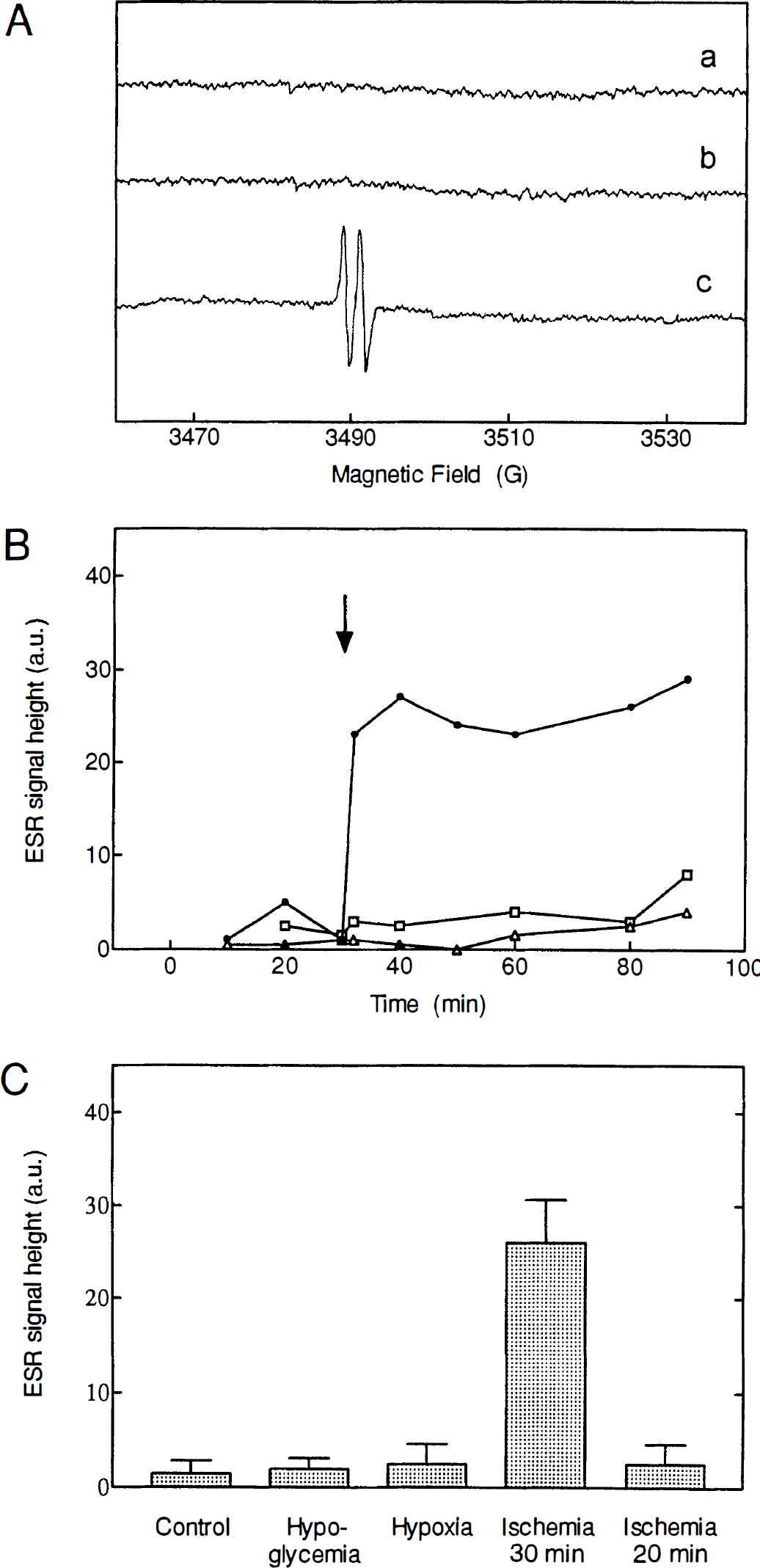
Formation of ascorbyl radicals in the reperfusion phase after ischemia, but not after hypoglycemia or hypoxia.
Apart from the ascorbyl radical production no other radical signals could be detected in the ESR spectra for any of the treatments studied. No ascorbyl radical was found in samples of the solution surrounding the slices measured at different times of reperfusion. Thus, the entire radical signal should be attributed to ascorbyl trapped within the tissue slice.
Lactate dehydrogenase release
As shown in Fig. 5, a small but significant increase in the release of LDH from the slices was observed in the reperfusion phase after prolonged ischemic conditions (30 minutes). This release was time-dependent, reaching a maximal value at 50 to 60 minutes of reperfusion. Shorter periods of ischemia did not produce significant changes in LDH release when compared with control experiments. In the same way, neither hypoxia nor hypoglycemia resulted in additional LDH release into the reperfusion medium, at least for periods of treatment up to 30 minutes.
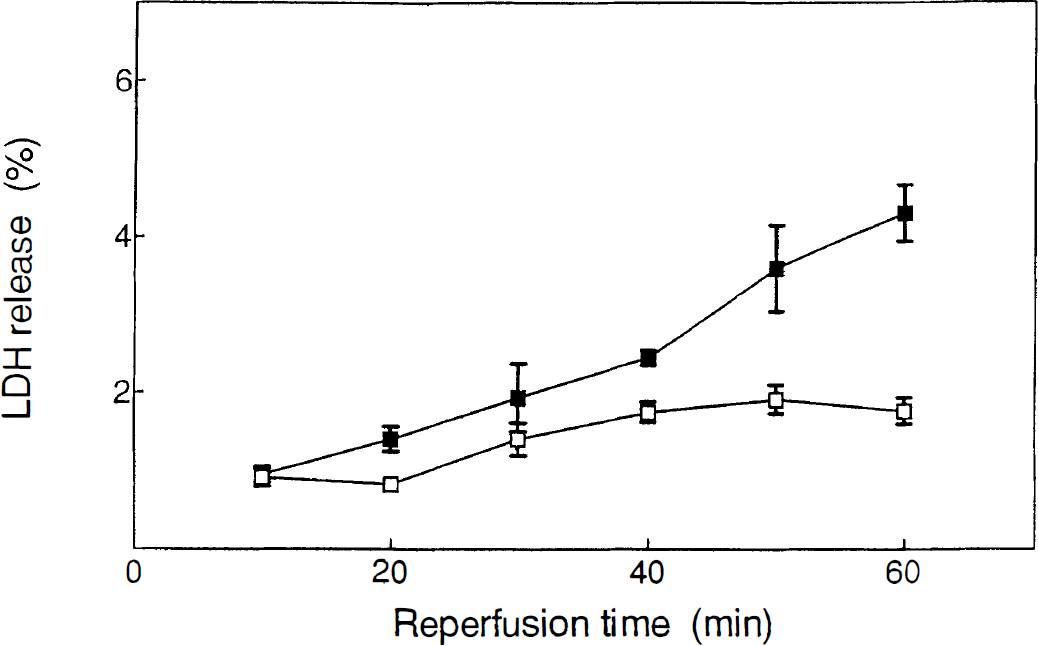
Release of lactate dehydrogenase from corticostriatal slices during postischemic reperfusion. Filled squares represent release of LDH into the medium during the reperfusion phase after 30 minutes of ischemia. The open squares indicate the LDH activity present in the medium after 30 minutes of incubation of the slices in control conditions. Data are expressed as percentage of the total LDH activity present in the tissue. Each data point is the mean of at least four single experiments. Experiments were significantly different (P < 0.05) from control at times 40, 50, and 60 minutes. LDH, lactate dehydrogenase.
DISCUSSION
The present study represents the first attempt to correlate electrophysiologic parameters with free radical formation during induction of neuronal damage through separate or combined glucose and/or oxygen deprivation. We have used a corticostriatal brain slice preparation that offers several advantages in comparison with in vivo models of ischemia. First, it allows us to induce hypoxia, hypoglycemia, and ischemia in a specific brain region that has been reported to be particularly vulnerable to these pathologic events (Auer et al., 1984; Auer and Siesjö, 1988; Pulsinelli, 1985; Xu, 1995). Second, the use of an in vitro system allows us to investigate the effects of known concentrations of selective glutamate receptor antagonists during energy metabolism failure. Third, the brain slice preparation can be used to dissect the differential role between brain cells and circulating cells in free radical formation and brain damage. Furthermore, in this study, the application of noninvasive ESR spectroscopy allows us to analyze the formation of free radicals directly in the tissue and in the perfusion medium. Thus, possible artifacts deriving from chemical or mechanical disruption of the tissue before the measurements of free radicals with techniques other than ESR can be avoided (Traystman et al., 1991).
The main finding of the present study is that 10 to 20 minutes' ischemia caused a long-lasting decrease of the field potential amplitude whereas significant formation of ascorbyl radical in the reperfusion phase appeared only after 30 minutes of ischemia. Accordingly, a significant, but small, increase in LDH release from the tissue, which is a biochemical marker of tissue necrosis, was only observed in the reperfusion phase after 30 minutes of ischemia. These findings suggest that the long-lasting loss of electrical signals is the early sign of neuronal damage during ischemia whereas ascorbyl radical formation may be considered an indicator of neuronal injury after prolonged energy deprivation.
In our experimental model prolonged hypoxia and hypoglycemia (up to 40 minutes) induced a depression of field potential amplitude that was almost completely reversible, suggesting that these pathophysiologic conditions induce a less severe damage to the tissue. It is likely that periods of oxygen or glucose deprivation alone longer than those used in the present study could cause a permanent loss of the electrical signals and formation of free radicals. However, this hypothesis cannot be easily tested because the loss of viability of the tissue maintained in vitro might affect the results obtained in long-term experiments.
The apparent discrepancy between the present data and our previous report showing that hypoxia is able to produce irreversible membrane potential changes in spiny striatal neurons (Calabresi et al., 1995 a, 1995b) can be explained by considering the different electrophysiologic techniques applied to detect the effects of energy deprivation. In our previous study intracellular recordings were used to measure the effect of hypoxia on a single neuronal subtype. In fact, that study only included spiny neurons, which represent the majority of the striatal neuronal population, whereas striatal interneurons were not included. Differently, the extracellular field potential measurements used in the present investigation reflect the synchronous activity of a heterogeneous neuronal population, which may have differential sensitivity to energy metabolism failure. Accordingly, most of the striatal interneurons have been reported to be less vulnerable than spiny neurons to hypoxic and ischemic insults (Pulsinelli et al., 1982; Pulsinelli, 1985).
The formation of partially reduced oxygen species, namely superoxide anion, hydroxyl radical, and hydrogen peroxide, has been reported to occur during reperfusion of different ischemic organs (McCord, 1985; Flaherty and Weisfeldt, 1988; Werns and Lucchesi, 1990; Traystman et al., 1991; Flitter, 1993). Damage to lipids, proteins, and nucleic acids has been observed concomitantly with their production, ultimately resulting in cell function impairment and death (Halliwell and Gutteridge, 1990). The high reactivity of oxygen free radicals means that their lifetime is too short to allow detection by ESR spectroscopy. Instead these radicals may react with suitable compounds called spin traps to produce more stable types of radicals that can be observed. By this application of ESR spectroscopy and the aid of spin traps, the formation of free radicals during cerebral ischemia-reperfusion has been demonstrated (Sen and Phillis, 1993; Zini et al., 1992). In the present study, rather than using exogenous spin trapping molecules, the measurements rely on endogenous ascorbic acid, which can be considered a physiologic spin trap. In fact, ascorbyl radical, the stable monovalent oxidation product of ascorbate, can be used as an indicator of oxygen radical formation (Niki, 1991; Buettner and Jurkiewicz, 1993). Interestingly, a remarkably high concentration of ascorbate is present in the central nervous system (Demopoulos et al. 1982; Grünewald, 1993).
It should be noticed that by avoiding the use of exogenous spin trapping agents we could overcome two experimental limitations: spin traps lack specificity and the radical adducts are generally not stable in contact with cells (Rice-Evans et al., 1991). Therefore their use often depends on experimental systems involving perfusion and microdialysis (Zini et al., 1992). Moreover, because their free radical-scavenging activity has been reported to prevent ischemia-induced damage (Oliver et al., 1990; Schulz et al., 1995), their presence may actually obscure the relationship between free radical formation and the neuronal injury.
Several mechanisms have been considered as responsible for the formation of oxygen radicals during reperfusion after organ ischemia (Halliwell and Gutteridge, 1990; Traystman et al., 1991). Reoxygenation during reperfusion provides oxygen for numerous enzymatic oxidation reactions, producing reactive oxidants. McCord (1985) originally proposed that during ischemia the catabolism of ATP to ADP and AMP causes accumulation of xanthine. On reoxygenation the oxidation of xanthine to uric acid by xanthine oxidase generates O − [over] 2 and H2O2 (McCord, 1985). In our experiments, ascorbyl radical formation only occurred when the lack of oxygen was concomitant with the absence of glucose (ischemia). The radical did not accumulate when hypoxia or hypoglycemia were applied separately. This phenomenon can be related to McCord's theory of free radical generation on reoxygenation. In the presence of glucose, ATP is provided by anaerobic glycolysis, despite the failure of oxidative metabolism, and thus accumulation of AMP does not occur and free radical production is prevented.
Another possible source of partially reduced oxygen species during ischemia-reperfusion events is activated neutrophils (Lucchesi et al., 1989). In particular, they were suggested to be the major source of oxygen radicals during reperfusion after focal cerebral ischemia because the administration of an antineutrophil monoclonal antibody before occlusion of the cerebral artery in the rat decreased the formation of extracellular ascorbyl radical (Matsuo et al., 1995). Our in vitro experimental protocol seems to rule out a major role of circulating neutrophils in free radical formation.
It is known that formation of ascorbyl from ascorbate can depend very much on the presence of small amounts of metal ions (Buettner, 1990), and it has been suggested that release of iron from damaged cells is responsible for the generation of ascorbyl in the central nervous system (Halliwell, 1992). However, our data on LDH release during reperfusion clearly show that intracellular components are released slowly and continuously. The ascorbyl radical, in contrast, appears immediately after the reintroduction of oxygen, and its concentration does not increase much further on. There is therefore little reason to believe that the mechanism of radical generation depends on the release of intracellular iron. Interestingly, ascorbate is released from brain cells in association with the activity of glutamatergic neurons by a mechanism of glutamate-ascorbate heteroexchange (Grünewald, 1993). Moreover, ascorbic acid-mediated protection against excitotoxin has been demonstrated in cultures of rat cerebral cortex (Majewska and Bell, 1990), probably by alteration of glutamate binding to N-methyl-D-aspartate receptors (Majewska et al., 1990). Thus, further studies are required to investigate the possible interaction between ascorbyl radical and glutamate in the generation of ischemia-induced damage.
Footnotes
Acknowledgements
The authors thank M. Tolu, E. Marchese, and G. Gattoni for their excellent technical assistance.