
Abstract
The authors investigated the changes and the potential of cyclic nucleotide-dependent signal transduction, which induces smooth muscle relaxation, in the basilar artery with severe vasospasm in dogs with double experimental subarachnoid hemorrhage (SAH) to explore at which biochemical level the arterial dilative capability was impaired. The amount of cyclic adenosine and guanosine monophosphates (cAMP and cGMP) decreased significantly in the basilar artery after SAH. The activities of adenylate and guanylate cyclases also were decreased significantly in the smooth muscle cells of the basilar artery 4 days after SAH. In addition to the failure of the pathways to produce cyclic nucleotides, the activities of cAMP-and cGMP-dependent protein kinases, which are representative actual enzymes that amplify the signal for vascular dilation, also significantly decreased together with the almost total loss of activation by cyclic nucleotides in the same basilar artery after SAH. It was revealed that the system for smooth muscle relaxation was impaired severely in the cerebral arteries with severe vasospasm after SAH, on the biochemical basis of significantly less vasodilative capability and in several of the steps to produce the cyclic nucleotides of intracellular signal transduction.
Keywords
The impairment of endothelium-dependent smooth muscle relaxation, which is mediated by an endothelium relaxation factor identified as nitric oxide (NO), plays an important role in the pathogenesis of cerebral arterial narrowing after double experimental subarachnoid hemorrhage (SAH) (Kim, 1989; Kim et al., 1992a; Martin et al., 1986). Under these conditions, NO causes vascular relaxation by elevating the level of cyclic guanosine monophosphate (cGMP), which subsequently activates cGMP-dependent protein kinase (G-kinase) in the vascular smooth muscle cells. However, the mechanism underlying the impairment of endothelium-dependent smooth muscle relaxation in the vasospastic artery due to SAH remains controversial (Edwards et al., 1992; Kim, 1989). It was reported that the cerebral arterial response to NO-donor is not affected after SAH and that the release of NO is maintained even in severely vasospastic arteries, implying that the reduction of endothelium-dependent smooth muscle relaxation is derived from an increase in factors that capture NO, such as hemoglobin and superoxide (Macdonald and Wier, 1991). However, Kasuya et al. (1995) reported that the activities of nitric oxide synthase in the endothelium and guanylate cyclase in the smooth muscle cells are reduced primarily in the basilar artery with vasospasm in an experimental canine SAH model, indicating that the reduction of endothelium-dependent smooth muscle relaxation basically may be caused by disorders of the endothelium and smooth muscle cells in the vasospastic artery.
When applying β2-stimulants or prostaglandins to the cerebral artery, another intracellular signal transduction with a cyclic nucleotide, namely adenosine monophosphate (cAMP), also mediates vascular relaxation by modifying the intracellular free calcium (Ca++) level and the cGMP-dependent pathway (MacDaniel et al., 1994; Nakatsu and Diamond, 1989). In a clinical study on the treatment of vasospasm, an intra-arterial infusion of papaverine hydrochloride satisfactorily ameliorated vasospasm in some individuals with symptomatic cerebral vasospasm but not in others (Kaku et al., 1992; Kassell et al., 1992), where one of papaverine's actions to induce smooth muscle relaxation is the inhibition of the hydrolytic enzyme of cyclic nucleotides, phosphodiesterase, to elevate the amounts of cAMP and cGMP. Papaverine has other pharmaceutical effects, such as the inhibition of a voltage-dependent Ca++ current (Iguchi et al., 1992) and the inhibition of inositol-1, 4, 5-triphosphate cascade in releasing Ca++ from intracellular Ca++ store (Aoki et al., 1994). Therefore, to clarify the pathogenesis of and to explore strategies for treating cerebral vasospasm, understand that the activity of intracellular signal transduction with cyclic nucleotides might be impaired in cerebral arterial smooth muscle cells after SAH. In the present study, we first evaluated the amounts of cAMP and cGMP measured by a radioimmunoassay, adenylate and guanylate cyclase activities estimated by histochemistry, and the activities of cAMP- and cGMP-dependent kinases (A-kinase and G-kinase) in the basilar artery of a canine SAH model. This is because cyclic nucleotide-dependent arterial relaxation is mediated primarily by the subsequent activation of A- and G-kinases. We also assessed the responses of adenylate cyclase to isoproterenol (β2-stimulator) and of guanylate cyclase to sodium nitroprusside (NO-donor), and the effects of cyclic nucleotides on A- and G-kinases activities to examine the potential of intracellular signal transduction with cyclic nucleotides in smooth muscle cells with severely vasospastic arteries after SAH.
MATERIALS AND METHODS
Animals
This study was conducted in compliance with the guidelines for animal experimentation at Ehime University School of Medicine (Ehime, Japan). Adult mongrel dogs of both genders weighing 7 to 13 kg were anesthetized by an intramuscular injection of ketamine hydrochloride (5 mg/kg) and intubated after an intravenous injection of sodium thiamylal (10 mg/kg) and atropine sulfate (0.5 mg/kg). The right femoral artery was catheterized to continuously monitor the systemic blood pressure and pulse rate. Arterial blood gases and body temperature were monitored and maintained within the physiologic range using artificial ventilation and a heating blanket.
For a single-SAH—induced vasospasm, under sterile conditions, 0.8 mL/kg of autologous arterial blood obtained from the femoral arterial catheter was slowly injected intracisternally by puncturing the cisterna magna with a 22-gauge needle without drawing cerebrospinal fluid. For double-SAH—induced vasospasm, the intracisternal injection of arterial blood was repeated after the first 48 hours. The control group was injected with 0.8 mL/kg of saline in the same manner as those animals injected with arterial blood.
Isoproterenol (0.8 mg/kg) or sodium nitroprusside (1 mg/kg) was applied topically to the basilar arteries of the control group and 4 days after the initial SAH in the double-SAH group, after careful removal of the subarachnoid clot through a transclival exposure using an operating microscope.
Assessment of basilar arterial diameter
Vertebrobasilar angiography was performed serially before and after the intracisternal injection of autologous arterial blood or after the application of drugs. Five milliliters of iopamidol (Iopamiron, Japan Schering Co., Tokyo, Japan) was injected via a femoral catheter placed in the left vertebral artery. All angiograms were taken under a fixed magnification. The diameter of the basilar artery was measured with an MCID imaging analyzer (Imaging Research, St. Catherine, Ontario, Canada), and contractions were assessed by determining the mean percentage of the pretreatment diameter at a point 10 mm proximal from the basilar arterial head.
Cyclic nucleotide measurements
The level of cyclic nucleotides in the basilar artery was measured in the control group and as follows in the others: at 1 and 48 hours, 4, 7, and 14 days after the arterial blood injection in the single-SAH group; 1 hour after the second injection (49 hours after the first SAH) and 4, 7, and 14 days after the first arterial blood injection in the double-SAH group, and 15 minutes after the application of the drugs to the normal basilar artery and to the basilar artery 4 days after the initial SAH in the double-SAH group because a significant increase of cyclic nucleotides was observed 15 minutes after the application of drugs and was not seen at 30 minutes in the normal basilar arteries in our preliminary study. Under the same anesthetic, the animals were killed by an intracardiac injection of 1 L of ice cold saline during exsanguination via a femoral venous catheter, and the brain quickly was removed. After the careful removal of periarterial subarachnoid clots in an ice-cold bath using an operative microscope, the basilar artery was weighed (wet weight), frozen in liquid nitrogen, and stored at −80°C until measurement. The basilar arteries were homogenized in 1 mL of 6% trichloroacetic acid and centrifuged at 3,000 rpm for 10 minutes at 4°C. The supernatant was removed and stored at 4°C. The precipitate was extracted again with trichloroacetic acid, and the supernatant was removed by centrifugation at 3,000 rpm for 10 minutes at 4°C. Water-saturated ether (6 mL) was added to a mixture of both supernatants, and then the ether was removed by centrifugation at 5,000 rpm for 10 minutes at 4°C. This procedure was repeated three times, and 20 mL of 500 mmol ethylenediaminetetraacetic acid was added. After a 10-minute incubation of 0.1 mL of sample with 0.1 mL of 0.1 mol succinic trimethylamine, samples in 0.8 mL of 0.3 mol imidazole buffer (pH 6.5) were processed for the radioimmunoassays of cAMP and cGMP using monoclonal antibodies (Yamasa Shoyu Co., Chiba, Japan) (Broadus, 1977; Honma et al., 1977). Samples were incubated with 0.1 mL of 125I-succynil cAMP or cGMP tyrosine methyl ester (59.2 kBq) and 0.1 mL of 4.4% anti-cAMP or 2.3% anti-cGMP antibody for 12 hours at 4°C. After a 30-minute incubation with 0.5 mL of dextran-coated charcoal at 0°C, the sample was centrifuged at 3,000 rpm for 5 minutes at 4°C. The radioactivity level in 0.5 mL of supernatant was measured using an Aloka-600 system (Aloka, Tokyo, Japan). Standard curves for cAMP and cGMP were in the range of 0 to 10 nmol/mL and 0 to 150 pmol/mL, respectively.
Electron microscopic histochemistry of adenylate and guanylate cyclases
Adenylate and guanylate cyclase activities were examined histochemically in the normal basilar artery (n = 4, each) and the basilar artery 4 days after the first arterial blood injection in the double-SAH group (n = 4, each) with and without the topical application of isoproterenol or sodium nitroprusside. Under the same anesthetic, the animals were killed by intracardiac perfusion fixation with 1 L of ice-cold saline followed by 1 L of 2% paraformaldehyde and 0.5% glutaraldehyde in 0.1 mol cacodylate buffer (pH 7.4) containing 5% dimethyl sulfoxide and 8% sucrose at a constant pressure of 120 mm Hg via a femoral venous catheter. The basilar artery was removed and frozen with Tissue-Tek O. C. T. compound on dry ice (Miles Inc., Elkhart IN, U.S.A.). Sections (60 mm thick) were made and postfixed with 2% glutaraldehyde in 0.1 mol cacodylate buffer (pH 7.4) containing 8% sucrose at 4°C for 30 minutes. Specimens were washed with 0.1 mol cacodylate buffer (pH 7.4) containing 5% dimethyl sulfoxide and 8% sucrose five times and incubated in the following reaction media at 37°C for 60 minutes: for adenylate cyclase histochemistry, 80 mmol Tris-malate buffer (pH 7.4), 0.5 mmol adenylate imidodiphosphate (AMP-PNP) (Boehringer Mannheim, Tokyo, Japan), 10 mmol NaF, 4 mmol MgSO4, 2 mmol theophylline, 2 mmol lead nitrate, 0.25 mol sucrose, 5% (v/v) dimethyl sulfoxide, 2.5 mmol levamisole (Sigma Chemical Co., St. Louis, MO, U.S.A.); for guanylate cyclase, 80 mmol Tris-malate buffer (pH 7.4), 0.5 mmol guanylate imidodiphosphate (GMP-PNP) (Boehringer Mannheim), 3 mmol MnCl2, 2 mmol theophylline, 2 mmol lead nitrate, 0.25 mol sucrose, 5% (v/v) dimethyl sulfoxide, and 2.5 mmol levamisole. Thereafter, the specimens were fixed by 2% glutaraldehyde in 0.1 mol cacodylate buffer (pH 7.4) containing 8% sucrose at 4°C for 30 minutes and washed with 0.1 mol cacodylate buffer (pH 7.4) containing 8% sucrose 5 times at 4°C. The specimens were postfixed with 1% OsO4 in 0.1 mol cacodylate buffer (pH 7.4) for 30 minutes at 4°C, stained with uranyl acetate and dehydrated through a graded series of ethanol baths. The specimens were embedded in Quetol 812 (Nishin E. M. Co., Tokyo, Japan), and then 70 nm thick sections were cut with an ultramicrotome and stained with uranyl acetate and lead citrate. Sections were examined under an electron microscope (H-800, Hitachi, Tokyo, Japan). For the histochemical control, AMP-PNP or GMP-PNP was removed from the reaction medium (Ogawa et al., 1986).
Measurement of cyclic adenosine monophosphate- and cyclic guanosine monophosphate-dependent protein kinase activities
The activities of the compartmentalized cAMP-dependent protein kinase (A-kinase) and cGMP-dependent protein kinase (G-kinase) were measured in the normal basilar artery and those 4 days after the first arterial blood injection in the double-SAH group in the absence or presence of cAMP or cGMP in the samples. Each basilar artery was suspended at 4°C in 10 volumes of homogenizing buffer (pH 6.8) containing 10 mmol potassium phosphate, 10 mmol ethylenediaminetetraacetic acid, 0.5 mmol methyl isobutylxanthine and 0.5 mmol 1,4-dithiothreitol. For homogenization, 2 mmol cAMP or 0.2 mmol cGMP was added to half of the samples. Approximately 50 to 70 mg of the basilar artery was disrupted with a Polytron homogenizer, and the homogenate was centrifuged immediately at 30,000 revolutions per minute for 15 minutes at 4°C to separate the soluble (supernatant) and particulate (pellet) fractions. The supernatant fraction was immediately assayed for both protein kinase activities. The pellet containing the particulate fraction was washed gently twice with homogenizing buffer to remove adhering material. The washed pellet was dispersed with a hand-held homogenizer in 1 mL of homogenizing buffer containing 0.1% Triton X-100. Thereafter, the samples were placed on ice for 10 minutes, then centrifuged at 30,000 revolutions per minute for 15 minutes. Particulate protein kinase activity was estimated in the supernatant. The A- or G-kinase activity was determined by measuring the transfer of 32P from [32P]-adenosine triphosphate (ATP) to histone or from [32P]-guanosine triphosphate (GTP) to G-kinase—specific peptide (Sigma). The reaction was started by adding 20 mL of the supernatant to 50 mL of the reaction mixture containing 20 mmol potassium phosphate (pH 6.8), 10 mmol NaF, 10 mmol MgCl2, 100 mmol [32P]ATP or 20 mmol [32P]GTP and 100 mg histone II-A or 0.2 mg G-kinase—specific peptide. The incubation proceeded at 37°C for 10 minutes. The reaction was terminated by placing 60 mL aliquots of the reaction mixture onto filter paper discs (Whatman International, Maidstone, England) using pipettes, and dropping the discs immediately into 10% ice-cold trichloroacetic acid. The filters were washed four times in trichloroacetic acid, followed by one 5-minute wash in 95% ethanol and one wash in ether. Then the filters were dried and counted in ACS II scintillant (Amersham, Arlington Heights, IL, U.S.A.) using a liquid scintillation counter. Protein kinase activities are expressed as picomoles of phosphate transferred per milligram of protein. The concentration of protein in the homogenate was determined according to the Lowry method, using bovine serum albumin as the standard (Vegesna and Diamond, 1984; Landgraf et al., 1986).
Statistical analysis
All values are given as the mean ± standard deviation. Statistical comparisons were made by a two-factor analysis of variance (Scheffe's test for multiple comparisons).
RESULTS
Changes in basilar arterial diameter
In the single-SAH group, the basilar artery was contracted by 74 ± 6% and 71 ± 5% of the pretreatment diameter 24 and 48 hours after SAH induction, respectively, and almost completely returned to the pretreatment value 7 days after SAH. Conversely, in the double-SAH group, the basilar artery narrowed by approximately 60%, a value that was much more persistent than in the single-SAH group after the second arterial blood injection. An approximate 40% decrease in the diameter persisted from 4 to 14 days after the initial SAH (Fig. 1).
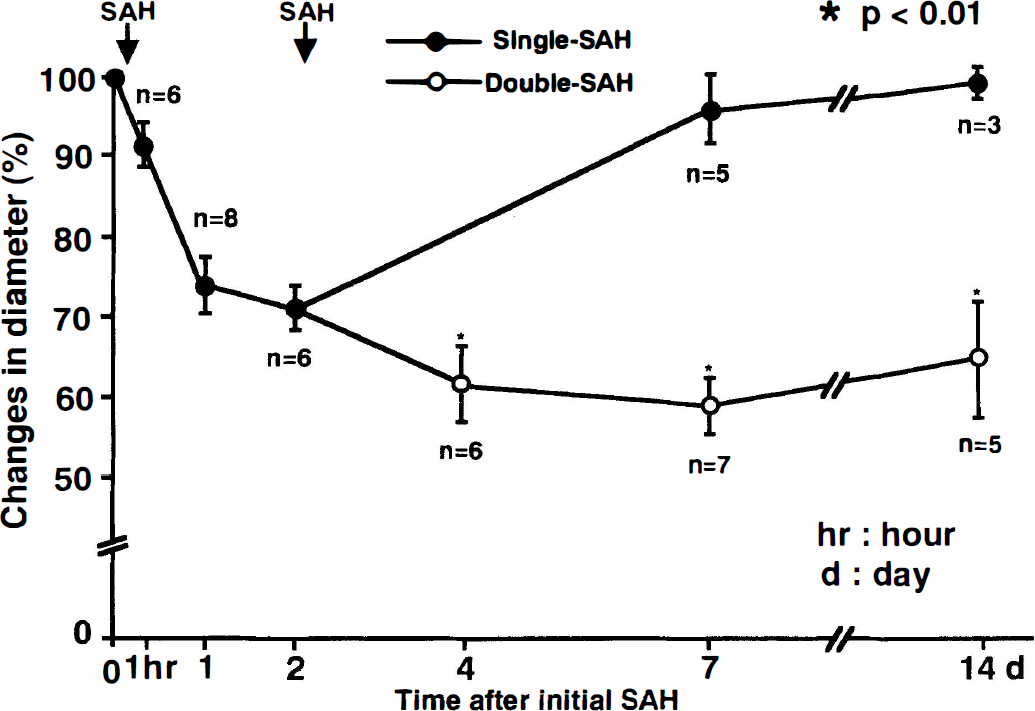
Changes in the diameter of the basilar artery after subarachnoid hemorrhage (SAH) (the control diameter was taken as 100%). Each value represents the mean ± standard deviation. In the single-SAH group, the diameter of the basilar artery was significantly decreased 24 hours after the initial SAH and gradually recovered by day 7. In the double-SAH group, significant basilar arterial narrowing persisted for 4 to 14 days after the initial SAH. (* P < 0.01 compared with the single-SAH group.)
Isoproterenol and sodium nitroprusside dilated maximally the normal basilar artery, to 123 ± 1% and 150 ± 1% of the pretreatment value 15 minutes after the drug application. However, 4 days after SAH, the dilation of the basilar artery in response to these drugs was suppressed to increases of only 8 ± 1% and 10 ± 2%, respectively, 15 minutes after the drug application. Thus, the arteries remained more narrow than the controls (65 ± 7% to 70 ± 9% of the pre-SAH diameter with isoproterenol and 67 ± 8% to 74 ± 1% with sodium nitroprusside), and no further dilation was observed until 2 hours later.
Cyclic nucleotides in the basilar artery
Cyclic adenosine monophosphate level. In the normal basilar artery, the cAMP level was 1.47 ± 0.18% (nmol/mg wet weight: n = 4). In the single-SAH group, the cAMP level was decreased significantly to 0.92 ± 0.07 nmol/mg (62.7 nmol/mg of control value: n = 4) 2 days after SAH, and it recovered close to the control value 4 days after SAH, whereas a significant decrease in the amount of cAMP (around 30% of the control value: n = 4) persisted from 4 to 14 days after the initial injection in the double-SAH group (Fig. 2). Although the cAMP content in the normal basilar artery after the topical application of isoproterenol significantly increased to 4.52 ± 0.61 nmol/mg (306% elevation from the control value: n = 4), this evidently was suppressed to only 34% elevation (1.35 ± 0.32 nmol/mg: n = 4) from the preadministration value (1.01 ± 0.25 nmol/mg: n = 4) in the basilar artery 4 days after the initial SAH in the double-SAH group.
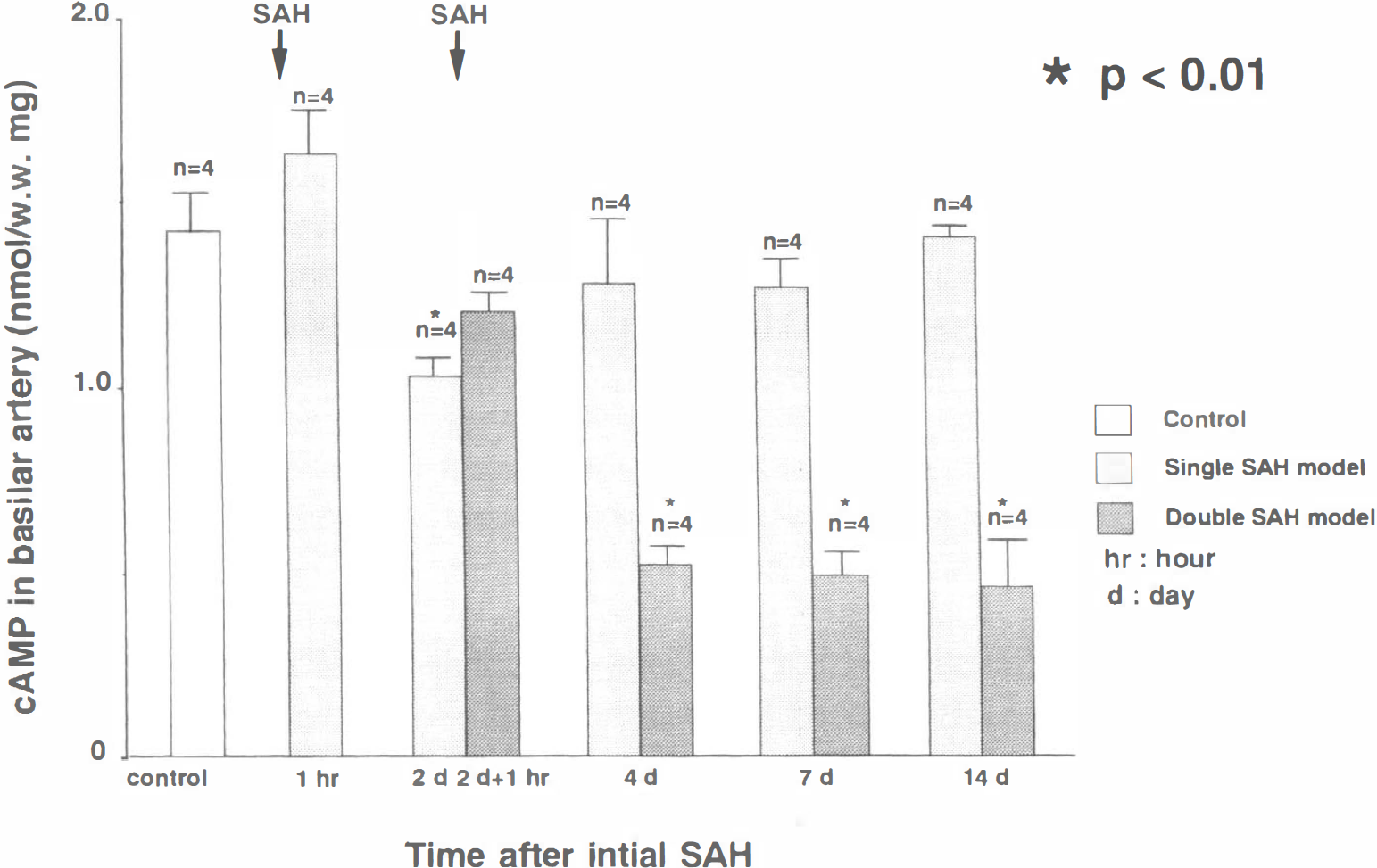
Bar graph showing the time-dependent changes in the amount of cyclic adenosine monophosphate (cAMP) in the basilar arteries of canine single- and double-subarachnoid hemorrhage (SAH) models. Each value represents the mean ± standard deviation. In the single-SAH group, the amount of cAMP significantly decreased 2 days after the initial SAH and recovered close to the control value 4 days after SAH. In the double-SAH group, the significant decrease persisted from 4 to 14 days after the initial SAH. (*P < 0.01 compared with controls.)
Cyclic guanosine monophosphate level. In the normal basilar artery, the cGMP level was 121 ± 9 pmol/mg (pmol/mg wet weight: n = 4). The cGMP level in the single-SAH group tended to decrease 2 days after SAH and did not significantly alter until 14 days. In the double-SAH group, the cGMP significantly decreased to 87 ± 14% (72% of control value: n = 4) 4 days after the initial SAH, and this decrease persisted for 14 days (Fig. 3). Although the cGMP content in the normal basilar artery after the topical application of sodium nitroprusside significantly increased to 270 ± 57 pmol/mg (123% elevation from the control value: n = 4), this was suppressed an only to 28% elevation (111 ± 21 pmol/mg: n = 4) of the preadministration value (87 ± 14 pmol/mg: n = 4) in the basilar artery 4 days after the initial SAH in the double-SAH group. The cGMP levels and the low response to the stimulator were similar to those of cAMP.
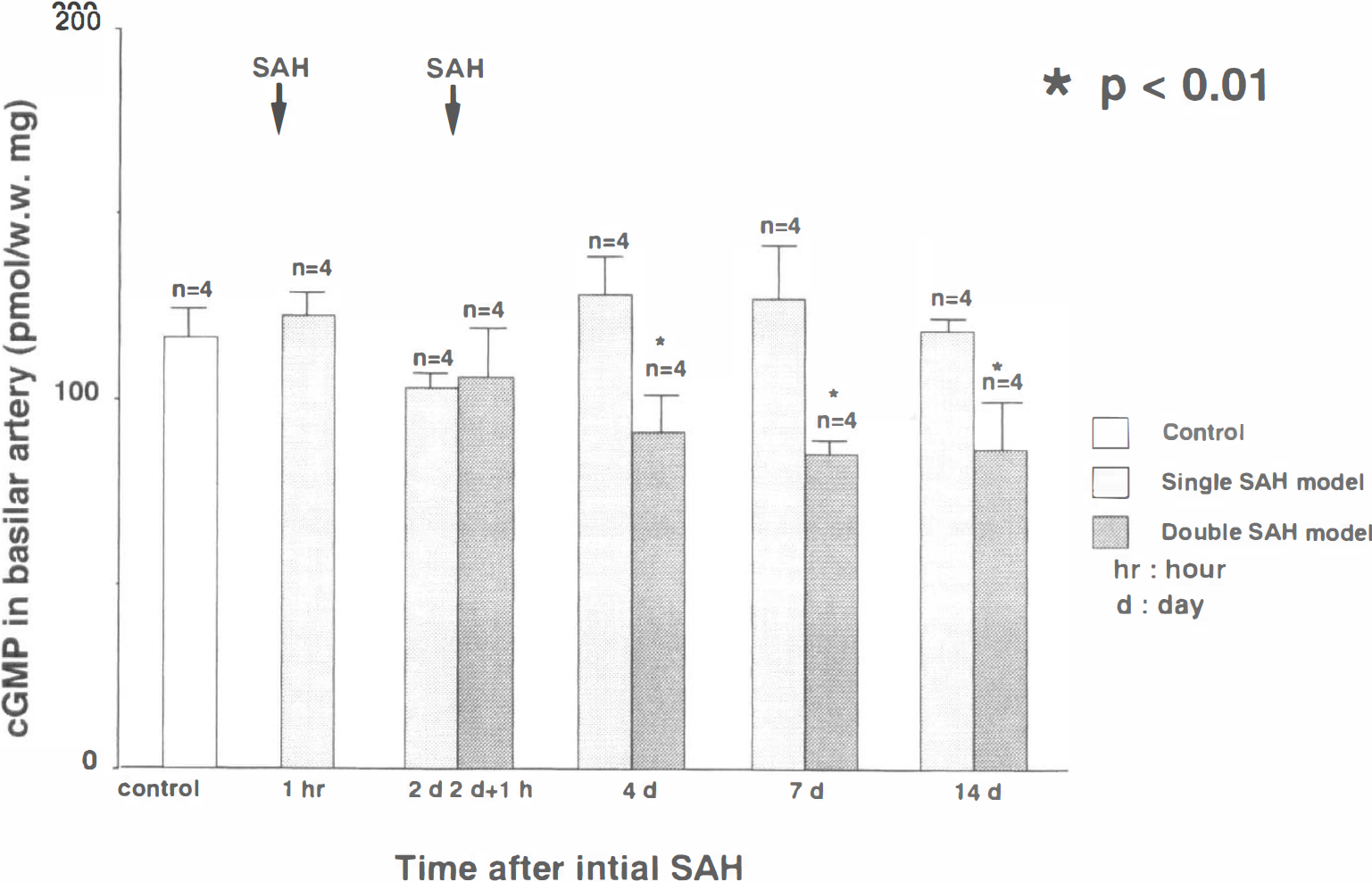
Bar graph showing the time-dependent changes in the amount of cyclic guanosine monophosphate (cGMP) in the basilar arteries of the single- and double-subarachnoid hemorrhage (SAH) groups. Each value represents the mean ± standard deviation. In the single-SAH group, the amount of cGMP was not significantly altered throughout the duration of the experiment. In the double-SAH group, it significantly decreased from 4 to 14 days after the initial SAH. (*P < 0.01 compared with controls.)
Electron microscopic histochemistry of adenylate and guanylate cyclases
In the control basilar artery, electron-dense deposits representing reaction products of adenylate cyclase accumulated mainly beneath the plasma membrane of smooth muscle cells, and several reaction products of guanylate cyclase were scattered over the cytoplasm of smooth muscle cells in the control basilar artery (Figs. 4A and 5A). Although a few reaction products were located in the extracellular space in the histochemical control sections incubated in medium without AMP-PNP and GMP-PNP, no black deposits were detected inside the smooth muscle cells. The number and size of both the adenylate and guanylate cyclase reaction products were smaller in the smooth muscle cells of the basilar artery 4 days after the initial SAH in the double-SAH group, without any apparent histologic degeneration, in comparison with those in the normal basilar artery (Figs. 4B and 5B).
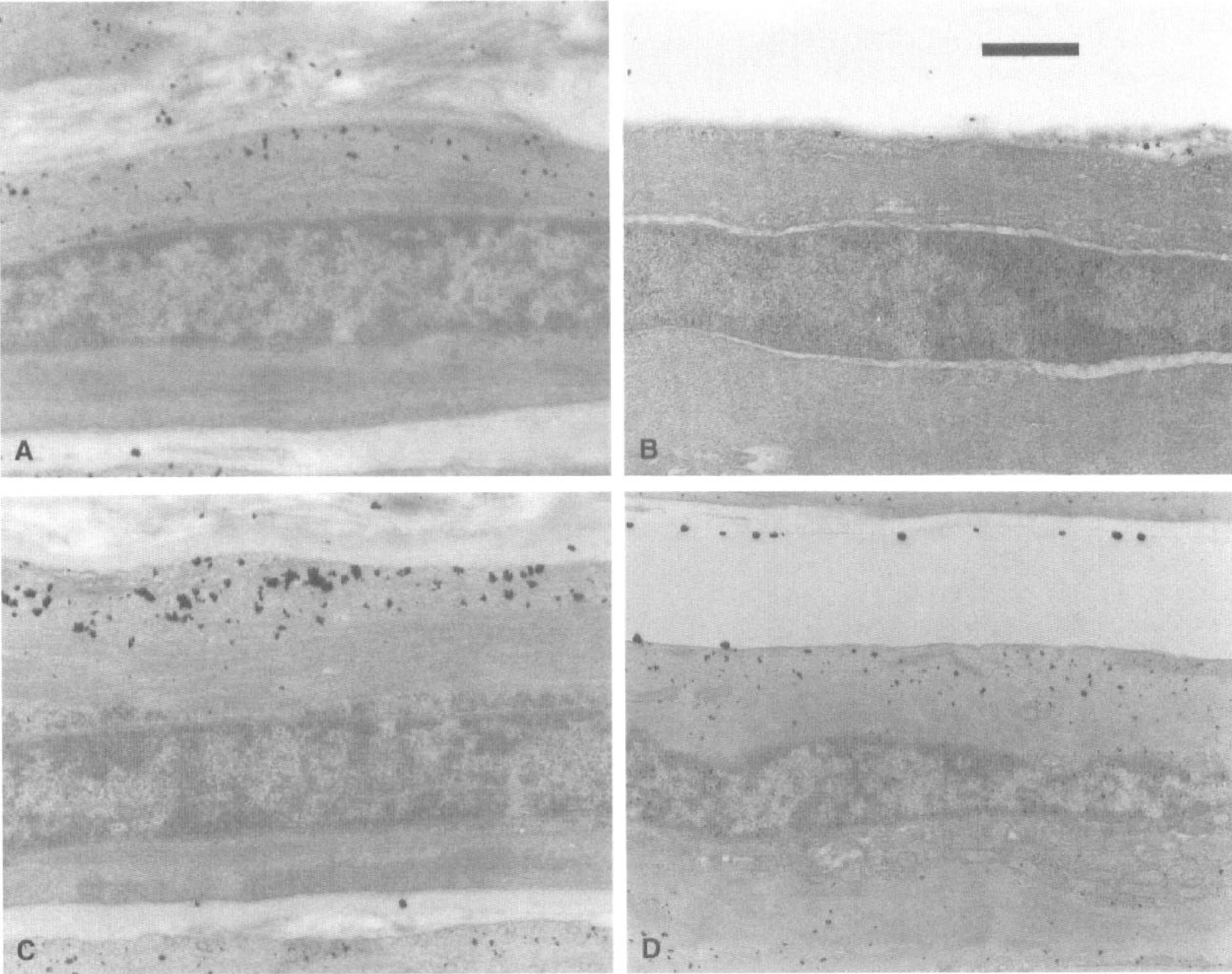
Electron microphotographs showing adenylate cyclase histochemistry in the smooth muscle cells of the basilar artery. In the normal basilar artery, electron-dense black deposits representing reaction products were located mainly beneath the plasma membrane in smooth muscle cells (
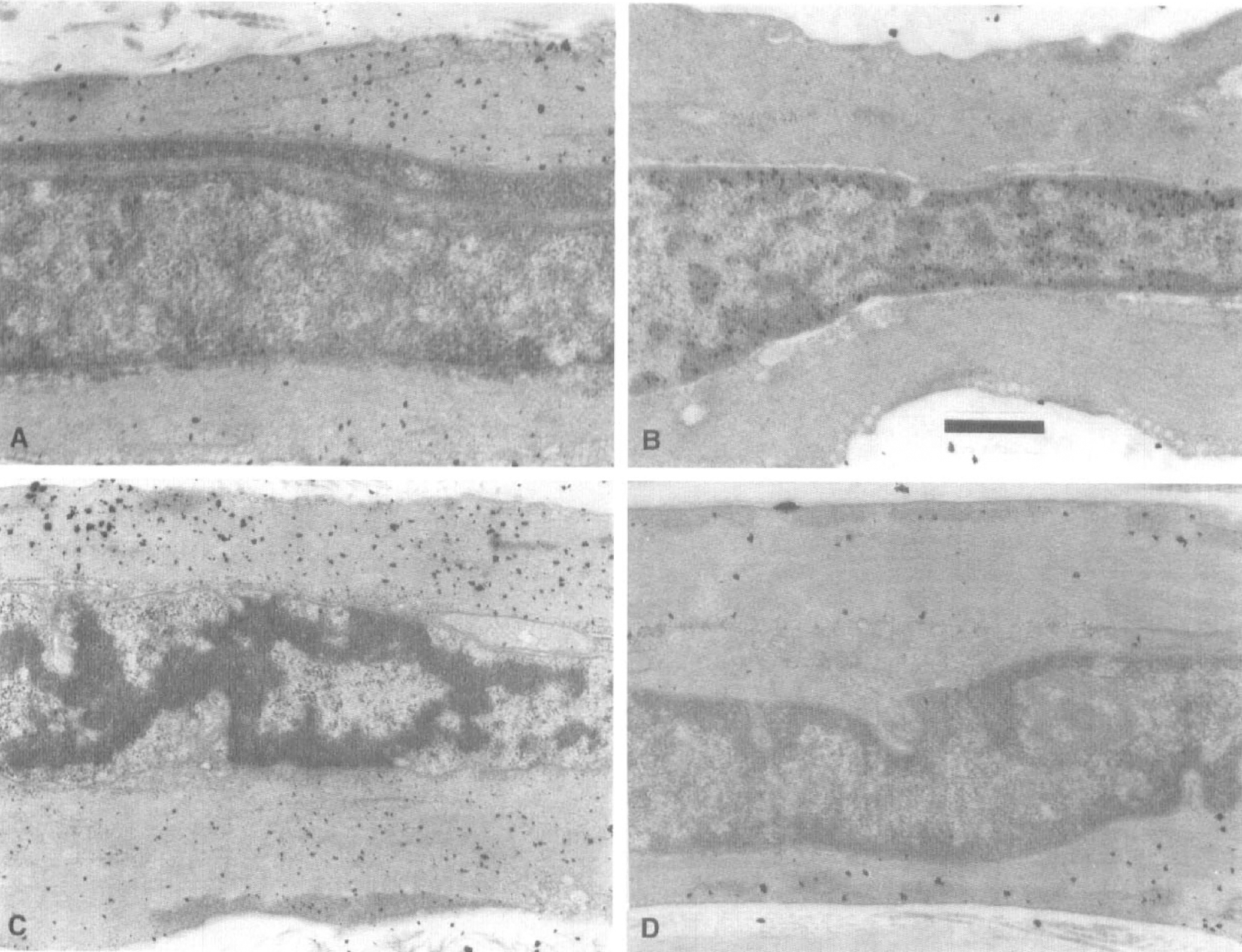
Electron microphotographs showing guanylate cyclase histochemistry in a smooth muscle cell of the basilar artery. In the normal basilar artery, electron-dense black deposits representing reaction products were located mainly over the cytosol in the smooth muscle cells (
After the application of isoproterenol or sodium nitroprusside to the normal basilar artery, many more and larger reaction products of adenylate and guanylate cyclase were found beneath the plasma membrane or over the cytosol of smooth muscle cells compared with the controls (Figs. 4C and 5C). Conversely, the smooth muscle cells of the basilar artery 4 days after the initial SAH in the double-SAH group responded to drugs by producing only a few more reaction products (Figs. 4D and 5D).
Changes in activities of A- and G-kinases
The soluble and particulate A-kinase activities were 455 ± 109 (n = 4) and 320 ± 55 (n = 4) pmol-transferred phosphate/mg protein, respectively, in the normal basilar artery. When 2 mmol cAMP was added to the sample, the A-kinase activities in soluble and particulate fractions were increased significantly to 5298 ± 743 (n = 4) and 3772 ± 336 pmol-transferred phosphate/mg protein (n = 4), respectively. In the basilar artery 4 days after the initial SAH in the double-SAH group, both the soluble and particulate A-kinase activities basically were significantly lower (254 ± 30 and 192 ± 45 pmol-transferred phosphate/mg protein; n = 4, each) than those in the normal basilar artery, and the activation of A-kinase in both fractions by cAMP also was significantly suppressed to below the control values (Fig. 6).
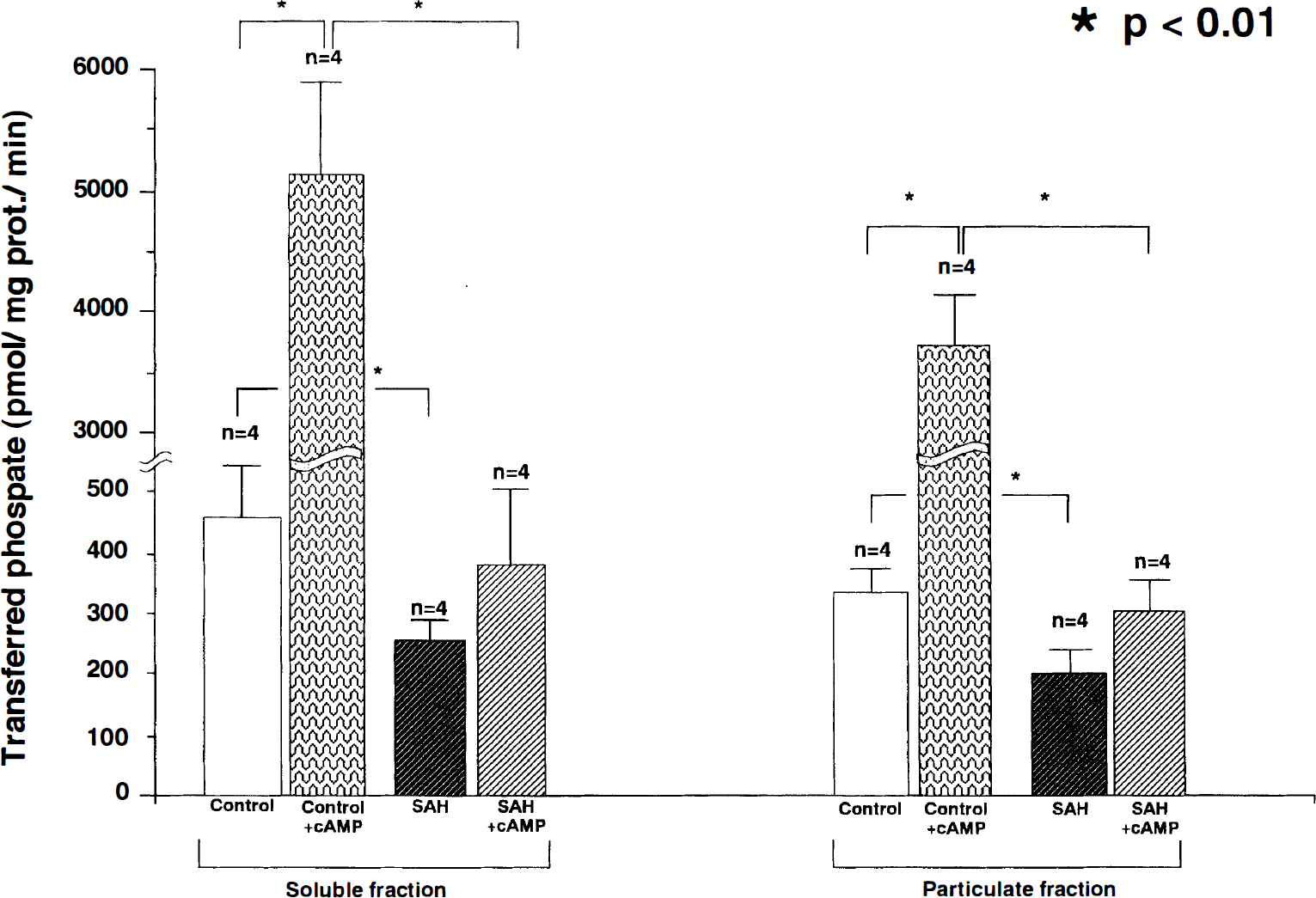
Bar graph showing cyclic adenosine monophosphate (cAMP)-dependent protein kinase activities (A-kinase) in the soluble and particulate fractions in the normal basilar artery and that 4 days after the initial subarachnoid hemorrhage (SAH) in the double-SAH group with or without 2 mmol of cyclic adenosine phosphate (cAMP). Each value represents the mean ± standard deviation. There were significant differences in the basic activities of A-kinase in the soluble and particulate fractions between the normal basilar artery and that 4 days after the initial SAH in the double-SAH group. In the presence of cAMP, although the A-kinase activities in both the soluble and particulate fractions were increased significantly in the normal basilar artery, the increase in A-kinase activities in both fractions was suppressed significantly in the basilar artery 4 days after the initial SAH in the double-SAH group. (* P < 0.01.)
The soluble and particulate G-kinase activities were 668 ± 91 (n = 4) and 427 ± 60 (n = 4) pmol-transferred phosphate/mg protein, respectively, in the normal basilar artery. When 0.2 mmol cGMP was added to the sample, G-kinase activities in soluble and particulate fractions were increased significantly to 6124 ± 735 (n = 4) and 3513 ± 621 (n = 4) pmol-transferred phosphate/mg protein. In the basilar artery 4 days after the initial SAH in the double-SAH group, both the soluble and particulate G-kinase activities were significantly lower (405 ± 33 and 274 ± 32 pmol-transferred phosphate/mg protein) (n = 4, each) than those in the normal basilar artery, and the activation by cGMP also was suppressed significantly (Fig. 7).
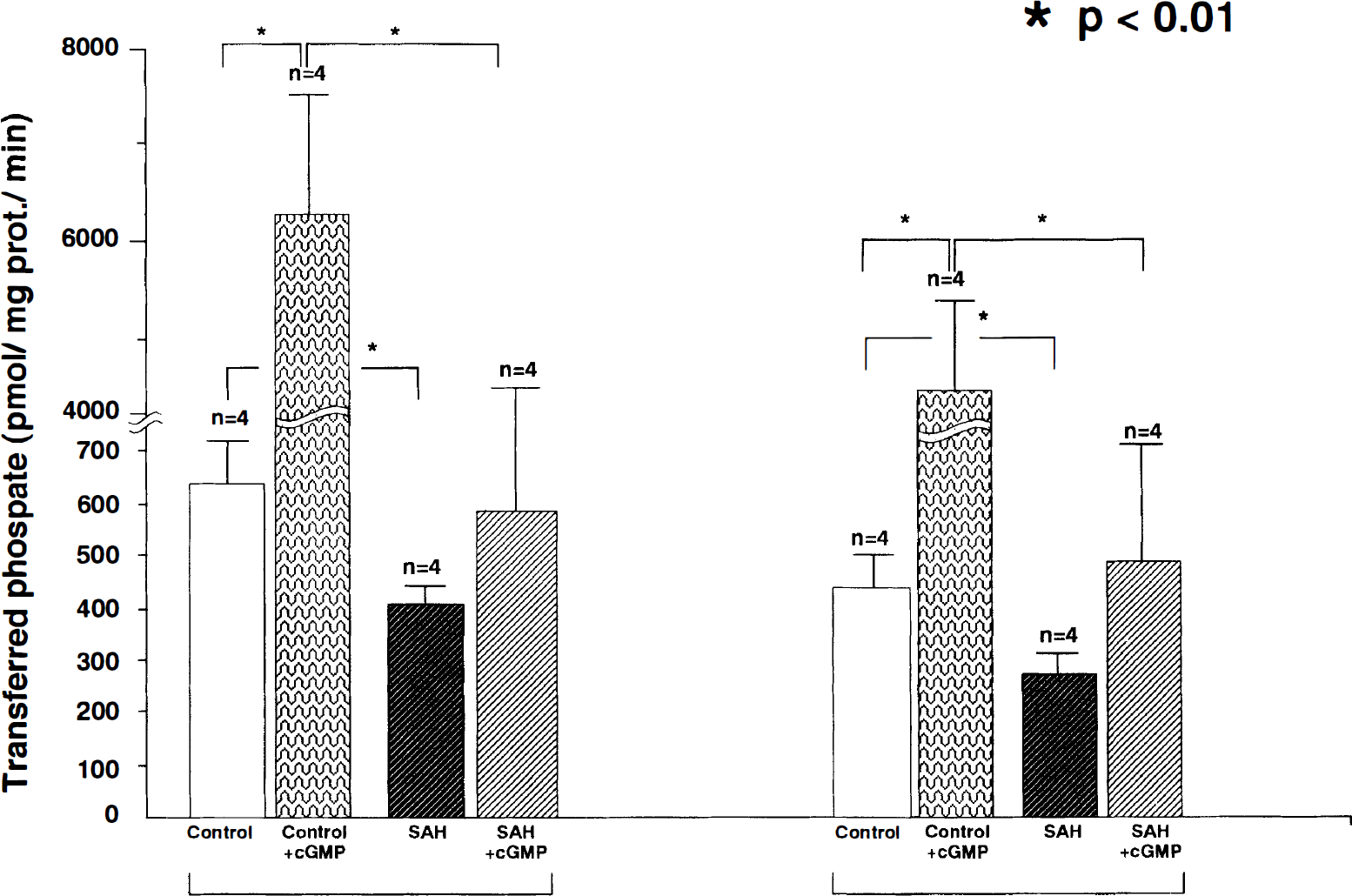
Bar graph showing cyclic guanosine monophosphate (cGMP)-dependent protein kinase activities (G-kinase) in the soluble and particulate fractions in the normal basilar artery and the basilar artery 4 days after the initial subarachnoid hemorrhage (SAH) in the double-SAH group with or without 0.2 mmol of cGMP. Each value represents the mean ± standard deviation. There were significant differences in the basic activities of G-kinase in the soluble and particulate fractions between the normal basilar artery and that 4 days after the initial SAH in the double-SAH group. In the presence of cGMP, the G-kinase activities in both the soluble and particulate fractions were increased significantly in the normal basilar artery, but in neither fraction in the basilar artery 4 days after the initial SAH in the double-SAH group. (* P < 0.01.)
DISCUSSION
In the present study, we demonstrated that significant decreases in the amounts of cAMP and cGMP persisted from 4 to 14 days in the basilar arteries, with severe vasospasm after the second SAH insult in the double-SAH group. Both of these compounds are well known as the first modulators in the intracellular signal transduction that initiates smooth muscle cell relaxation (MacDaniel et al., 1994; Nakatsu and Diamond, 1989). The increases of cAMP and cGMP content in response to the stimulation of adenylate and guanylate cyclases, such as isoproterenol (β2-stimulator) and sodium nitroprusside (NO-donor), were suppressed significantly, being lower than the basal levels in the basilar artery 4 days after SAH in the double-SAH group. This result was in good agreement with the decreased dilation that demonstrated resistance to these drugs. Several mechanisms that induce the reduction of these cyclic nucleotides in the early phase of cerebral vasospasm after SAH could be considered. One is a lack of substances that activate generative enzymes, such as NO and prostaglandin I2. These substances were reported to be reduced significantly in the cerebral arteries after SAH (MacDonald and Wier, 1991; Toda, 1990). Another is a deficiency of ATP and GTP, which are precursors of cAMP and cGMP and were significantly decreased in the basilar arteries of the same double-SAH model (Kim et al., 1992a; Yoshimoto et al., 1993). Phosphodiesterase, which hydrolyzes both cAMP and cGMP, might be activated in the vasospastic cerebral arteries because several authors have reported that the intra-arterial administration of papaverine or a phosphodiesterase inhibitor clinically and experimentally prevents or ameliorates cerebral vasospasm (Alksne and Branson, 1979; Norwood et al., 1976). Another possibility is a reduction of adenylate and guanylate cyclase activities in the cerebral arterial smooth muscle cells after SAH.
The cAMP and cGMP contents and their elevation in response to the drugs were decreased significantly and the maximum basilar arterial contraction was initiated 4 days after SAH in the double-SAH group. To clarify whether these findings reflected the reduced activities of adenylate and guanylate cyclases activities exactly in the smooth muscle cells of the basilar artery after SAH, we employed a histochemical technique to estimate the adenylate and guanylate cyclase activities in the smooth muscle cells instead of biochemical assay because the contamination of endothelium and inflammatory cells infiltrating the adventitia after SAH could not be completely excluded in the biochemical analysis; only the histochemical method can identify these enzyme activities in smooth muscle cells. The electron-dense black deposits representing the reaction products—the size and number of which indicated adenylate or guanylate cyclase activities—were located predominantly beneath the plasma membrane and over the cytosol of smooth muscle cells in the normal basilar arteries, respectively. These observations corresponded to the localization of these enzymes because high adenylate cyclase activity was found in the purified particulate fraction, including the plasma membrane, and greatly increased guanylate cyclase activity was found in the soluble fraction. In the normal basilar arteries, isoproterenol eventually increased the number and enlarged the size of the deposits of adenylate cyclase beneath the plasma membrane and sodium nitroprusside brought out the same effect on the cytosolic reaction products of guanylate cyclase. However, in the basilar artery of the double-SAH group 4 days after SAH, there was little of either reaction product compared with those in the normal basilar artery. Furthermore, the stimulators did not increase significantly either reaction product, even when the maximum arterial dilation was attained, although the arterial dilative responses also were fairly restricted. These results indicated that decreased activities of adenylate and guanylate cyclases were likely to be the primary cause of the reduced cAMP and cGMP contents in the basilar artery with severe vasospasm because the substrates for these enzymes (AMP-PNP and GMP-PNP) were replenished sufficiently instead of ATP, and GTP or theophylline, which inhibit phosphodiesterase, also were included in the reaction media. Although the possibility exists that shifts in the dynamics of the responses to the stimulators result in the apparent differences in the increase of cyclic nucleotides and reaction products in the basilar arteries after SAH, although the times of the peak responses in the vasodilation were identical between the normal basilar arteries and the basilar arteries 4 days after SAH in the double-SAH group, we considered that also the smooth muscle cells with severe vasospasm hardly activated the adenylate and guanylate cyclase activities, as demonstrated by the too-low responses to the drugs along with significant suppression of vasodilation.
Generally, cyclic nucleotides modulate several cellular functions (including smooth muscle relaxation) through the activation of A- and G-kinases (Lincoln and Cornwell, 1991). The activation of these enzymes relaxes vascular smooth muscle cells through several pathways. Both enzymes lower the intracellular Ca++ concentration by suppressing the intracellular Ca++ influx by phosphorylating the Ca++ channel (Felbel et al., 1988; Ishikawa et al., 1993; Xiong et al., 1994) and by enhancing the Ca++-adenosine triphosphatase (ATPase) activity (Furukawa and Nakamura, 1987; Yoshida et al., 1991) and the Na+–Ca++ exchanger on the plasma membrane to export Ca++ (Furukawa et al., 1991) and on the smooth endoplasmic reticulum to sequester free Ca++ from the cytosol (Twort and van Breemen, 1988). A- and G-kinases directly phosphorylate myosin light chain kinase to prohibit the interaction of myosin and actin through reducing the affinity of myosin light chain kinase for Ca++-calmodulin (de Lanerolle et al., 1984). G-kinase also may phosphorylate other contractile thin filaments (Marston and Smith, 1985). The intracellular compartmentalization of A-kinase and G-kinase in particulate fraction may play a more pivotal role in arterial smooth muscle relaxation rather than total protein kinase activity; the higher activity in the particulate fraction may promote more relaxant capability, even when the total kinase activity is identical (Vegesna and Diamond, 1984). We separately measured A- and G-kinase activities in the soluble and particulate fractions and found that both enzyme activities in both fractions were decreased significantly in the basilar artery 4 days after the initial SAH in the double-SAH group. Furthermore, we measured the potential of these enzymes to be activated by cyclic nucleotides in vitro. The addition of cyclic nucleotides to the homogenate increased the particulate activities of A- and G-kinases to levels tenfold higher than those before the addition in the normal basilar artery. Conversely, their increase by the cyclic nucleotides was limited to less than 1.5-fold in the basilar arteries 4 days after the initial SAH in the double-SAH group. It was revealed in the present study that smooth muscle cells with severe vasospasm also lose the potential to activate A- and G-kinases as well as adenylate and guanylate cyclases, even during the early phase of chronic vasospasm.
It is not clear why several steps of the cyclic nucleotide-dependent signal transduction in the basilar artery with severe vasospasm were impaired significantly during the early phase of chronic vasospasm. It might be induced by the free radical reaction initiated in the subarachnoid clots because several authors suggested that lipid peroxidation after the free radical reaction causes a loss of membrane fluidity and alters its components in the cellular membrane, leading to changes in the plasma membrane potential, conformational changes of membrane-bound proteins and the inactivation of enzymes in the cerebral arteries after SAH (Hubschmann and Nathanson, 1985; Waters and Harder, 1985; Wang et al., 1994). However, the activities of guanylate cyclase and A- and G-kinases in the soluble fraction also were decreased in the present study, although they were not membrane-bound enzymes. Free radicals and lipid peroxidase directly attack proteins, degenerating them. Proteolysis by protease is enhanced and protein synthesis is suppressed in the cerebral arterial smooth muscle cells after SAH (Gardner, 1979; Matsushita, 1975; Oka et al., 1996, Yamaura et al., 1993).
The cause of attenuated vasorelaxant capacity in the cerebral vasospasm after SAH has been speculated to be an impairment of the system of Ca++ efflux from the smooth muscle cells (Wang et al. 1994), continuous smooth muscle membrane depolarization (Waters and Harder 1985), and less generation of the substances that induce vasorelaxation (Romero et al., 1984). In addition to these mechanisms, the results of the present study suggest that defective intracellular signal transduction with cyclic nucleotide in the smooth muscle cells after SAH is one of the cause of impaired cerebral arterial relaxation. The amounts of cAMP and cGMP, the activities of their generative enzymes (adenylate and guanylate cyclases) and of subsequent enzymes that are activated by cyclic nucleotides (A- and G-kinases) were decreased significantly during the early phase of prolonged chronic severe cerebral vasospasm after SAH, even if less NO and prostaglandin I2 were generated in the endothelium of the vasospastic artery. In addition, severe vasospasm could not be ameliorated completely by the administration of papaverine, an NO donor and β2-stimulator, because the responses were impaired severely in all steps examined, indicating that smooth muscle cell viability itself might be damaged. On the contrary, the diminished capacity of cyclic nucleotide-dependent signal transduction might disturb smooth muscle cellular integrity, leading to delayed histologic degeneration, such as myonecrosis, because it also plays a crucial role in maintaining cellular viability as well as in smooth muscle relaxation. How cyclic nucleotide-dependent signal transduction is damaged in vasospastic arteries and whether its disturbance is a cause or an effect of the loss of smooth muscle viability, as well as how that viability can be restored, needs continued investigation.
Footnotes
Abbreviations used
Acknowledgments
The authors thank Morishige Tanaka of the Central Research Laboratory and Kansei Wada and Kazushige Ohno of the Laboratory Animal Center at the Ehime University School of Medicine for their technical assistance.