
Abstract
In anesthetized piglets, endothelial and neuronal nitric oxide synthase (eNOS and nNOS, respectively) levels were investigated after global cerebral ischemia. Increased intracranial pressure was used to produce 5 or 10 minutes of global ischemia, which was verified visually by observing pial arteriolar blood flow and by a microsphere technique. At 4 to 6 hours of reperfusion, parietal cortex, hippocampus, and cerebellum were collected for immunohistochemical or immunoblot analysis. Immunohistochemical examination localized eNOS only to blood vessels and nNOS only to nonvascular cells, which were primarily neurons in all regions examined. Analysis of immunoblot data revealed significant increases in eNOS levels from 47 ± 22 pixels/μg protein for time controls to 77 ± 36 pixels/μg protein (75% increase) for ischemia in parietal cortex (n = 9 to 10) and 22 ± 10 for control to 40 ± 16 pixels/μg protein (40% increase) for ischemia in hippocampus (n = 7 to 8). Levels of eNOS in cerebellum also tended to be higher but were variable and not significant (n = 5 to 6). In contrast, changes in nNOS levels were not detected at 4 or 6 hours. The increase in eNOS levels detected on immunoblots also was apparent on tissue sections as an increase in intensity of staining. Cyclooxygenase-dependent mechanisms were investigated with respect to the ischemia-induced increase in eNOS levels. Pretreatment with the cyclooxygenase inhibitor indomethacin (5 mg/kg intravenously) abolished the ischemia-induced eNOS increase in parietal cortex and hippocampus (n = 7). Thus, we conclude that the eNOS response is rapid, specific to vessels, and involves an indomethacin-sensitive mechanism.
Hypoxic/ischemic stress occurs often in the perinatal period and usually is followed by neurologic sequelae (Rice et al., 1981; Vannucci et al., 1993; Volpe, 1994). The neonatal cerebral cortex, hippocampus, and cerebellum are particularly susceptible to anoxic stress (Vannucci et al., 1993). Limited evidence from neonatal animals suggests that nitric oxide (NO) derived from the constitutive nitric oxide synthase (NOS) isoforms (neuronal nitric oxide synthase [nNOS] and endothelial nitric oxide synthase [eNOS]) can effect the evolution of hypoxic/ischemic injury (Hamada et al., 1994; Ferriero et al., 1995; Ashwal et al., 1995). However, little is known about brain levels of NOS after recovery from anoxic stress in the neonate. Altered levels of NOS might influence the ability of an already compromised brain to withstand secondary insults such as seizure activity and intracranial bleeding, which are known to occur after successful reoxygenation of asphyxiated babies.
Measurements of NO and NOS activity indicate that NO levels change during and immediately after focal cerebral ischemia in adult rats (Malinski et al., 1993; Yoshida et al., 1995; Zhang et al., 1995). In addition to immediate effects on cellular function after ischemia, NO may continue to have important effects, which are dependent on NOS levels, substrate availability in brain, or both. Changes in regional distribution and timing of NOS expression have not been fully characterized, especially during the first 12 hours after cerebral ischemia/reperfusion. Using immunohistochemical study, which is a semiquantitative technique, eNOS and nNOS levels have been reported to increase rapidly in cerebral cortex and striatum after focal cerebral ischemia in rodents (Zhang et al., 1993, 1994). Unlike the constitutive enzymes, inducible NOS mRNA is not present until 12 hours after cerebral ischemia in adult rats (Iadecola et al., 1995, 1996). Mechanisms involved in immediate elevation of nNOS or eNOS levels remain to be examined.
This study examines the effects of total, global ischemia on NOS levels in brains of newborn pigs in the immediate (4- to 6-hour) postischemic period. We tested the hypothesis that levels of nNOS and eNOS would increase under these circumstances. A global ischemia model was used because it more closely matches the clinical situation in babies. We used complimentary immunohistochemical and immunoblot approaches to characterize the effect of ischemia on parietal cortex, hippocampus, and cerebellum. Thus, immunohistochemical examination was used to localize NOS, and immunoblot analysis was used to quantify changes in NOS levels. We also examined whether indomethacin pretreatment affects changes in NOS levels after ischemia. Indomethacin, which is a widely used cyclooxygenase inhibitor, is commonly given to stressed newborn babies to reduce the risk of intracranial hemorrhage and aid in closure of the ductus arteriosus (Volpe, 1994). Indomethacin also has been reported to prevent or attenuate cerebrovascular and neuronal dysfunction after anoxic stress in newborn pigs (Bari et al., 1996a; Busija et al., 1996).
MATERIALS AND METHODS
Global cerebral ischemia
All procedures were approved by the Animal Care and Use Committee at the Bowman Gray School of Medicine. Global cerebral ischemia was produced as described previously (Bari et al., 1996a; Busija et al., 1996). Newborn piglets (1 to 4 days old) of either gender were anesthetized initially with 30 mg/kg of sodium thiopental given intraperitoneally. Intravenous administration of 5 to 10 mL α-chloralose (75 mg/kg) was used to maintain anesthesia. The animals were intubated by tracheotomy and ventilated with room air throughout the entire procedure. A catheter was placed in a femoral artery to record blood pressure, determine blood gases, and measure pH. To administer fluid and drugs, a catheter also was placed in a femoral vein. Body temperature was maintained at 37° to 38°C using a heating pad.
Using cyanoacrylate ester (SuperGlue, Atlanta, GA) and dental acrylic, a stainless steel ring with a glass window was cemented into a craniotomy over the left parietal cortex. A microscope (Wild M36, Heerbrugg, Switzerland) equipped with a video camera (Panasonic, Secaucus, NJ, U.S.A.) and a video monitor was used to observe pial blood flow during ischemia/reperfusion. Anterior to the steel ring, a hollow bolt was cemented into the parietal bone without disrupting the dura mater. Using the bolt, artificial cerebrospinal fluid was delivered into the epidural space to raise intracranial pressure, thus producing global cerebral ischemia. During increased intracranial pressure, blood flow through pial vessels ceased. The artificial cerebrospinal fluid consisted of the following: 2.9 mmol/L KCl, 1.4 mmol/L MgCl2, 1.2 mmol/L CaCl2, 132 mmol/L NaCl, 24.6 mmol/L NaHCO3, 6.7 mmol/L urea, and 3.7 mmol/L glucose. During ischemia, blood was withdrawn through the femoral vein using a heparinized syringe to counteract increases in arterial blood pressure.
Lack of blood flow to the various regions of the brain was verified using a microsphere technique. Briefly, colored microspheres were injected into the left ventricle, and a reference blood sample was collected from the femoral artery. Analysis of the number of microspheres detected in each region of the brain indicated that ischemia was widespread and total. Before ischemia, cerebral blood flow to the parietal cortex, hippocampus, and cerebellum was 39 ± 15 mL/min/100 g (n = 5), 31 ± 19 mL/min/100 g (n = 5), and 33 ± 26 mL/min/100 g (n = 3), respectively. During ischemia, cerebral blood flow ceased in all three regions, as indicated by the lack of spheres detected (<1 mL/min/100 g).
Experimental protocol
The animals remained anesthetized during 4 to 6 hours of reperfusion and were carefully monitored the entire time. The following three treatments were examined: ischemia, indomethacin pretreatment (indomethacin), and sham-operated time controls (time control). Baseline information was obtained from a fourth group of untreated piglets. Indomethacin (5 mg/kg intravenously) was administered 20 minutes before ischemia. At the conclusion of the experiments, the brains were removed and dissected on ice. Parietal cortex, hippocampus, and cerebellum from each animal were collected for immunoblot analysis, immunohistochemical study, or both. The number of tissues used for each analysis is indicated in the results section.
Immunoblot analysis
Tissue was immediately frozen in 2-methyl butane on dry ice and stored at −60°C. Protein was extracted from about 1 mm of tissue in boiling lysis buffer (1% 1 mol/L Tris and 1% sodium dodecyl sulfate). The samples were sonicated, heated at 95°C for 5 minutes, and centrifuged for 5 minutes at 12,000 rpm at 4°C. An aliquot of the supernatant was removed for protein concentration determination (Lowry et al., 1951).
An equal volume of sample buffer (42% 0.5 mol/L Tris, pH 6.8; 42% glycerol; 5% bromophenol blue; 1% sodium dodecyl sulfate) was added to each sample. Equal protein was separated on a 4% to 20% gradient mini gel (Bio Rad, New York, U.S.A.) and transferred to nitrocellulose. After blocking in 5% milk, primary antibody was applied, followed by horseradish peroxidase–conjugated secondary antibody. Both of the nNOS and eNOS antibodies are monoclonal, developed against the human isoforms (Transduction Laboratory, Kentucky, U.S.A.). The nNOS antibody was used at 1:2000, whereas the eNOS antibody was used at 1:1000. Electrochemiluminescence was used to visualize the bands. Standards for each protein (Transduction Laboratory) and molecular weight markers (Bio Rad) were included on each blot.
Immunohistochemistry
Tissue was removed and immersion fixed in 4% paraformaldehyde in 0.1 mol/L phosphate buffer, pH 7.4 for 15 hours. After washing in 10 mmol/L phosphate buffer containing 0.9% saline, pH 7.4 (PBS), 50-μm vibratome sections were cut and collected in 10 mmol/L PBS containing 0.1 mg/L of thimerosal. All washes and solutions were made in PBS unless otherwise stated. The sections were incubated in 50 mmol/L ammonium chloride for 1 hour, washed, and incubated in 0.3% Triton X-100 for an hour. After washing, the sections were blocked in 10% normal goat serum/0.1% tween 20 (NGS-T) for 4 hours. Antibodies specific for nNOS and eNOS were diluted 1:500 in NGS-T and incubated with sections overnight (15 hours) at room temperature. After rinsing sections in PBS, endogenous peroxidase was blocked by incubating the sections in 3% H2O2/10% methanol for 30 minutes. After washing, sections were incubated in biotinylated goat anti-mouse IgG (Vector Laboratories, Burlingame, CA, U.S.A.), diluted 1:1000 in 2% normal goat serum for 2 hours. The sections were incubated with Vector ABC reagent for 30 minutes, washed, and reacted with diaminobenzidine. After washing, the sections were mounted on slides and dried, and cover slips were added.
Photomicrographs were created with Adobe Photoshop software (Adobe Systems Inc., San Jose, CA, U.S.A.) from original 35-mm color slides, and negatives and were printed on a Fujix Pictrography 3000 digital printer (Digital Image Lab, Encino, CA, U.S.A.). Contrast and intensity of scanned images were altered only to replicate the originals. The images were converted to gray scale using filters as necessary, formatted to plate form, and labeled.
Statistical analysis
Immunoblots were scanned using an Hewlett Packard DeskScan II (Roseville, CA, U.S.A.). The density of individual bands then were determined using the NIH Image Analysis software (version 1.55). Values are reported as pixels per microgram of protein within a given series of immunoblots. Data were analyzed with analysis of variance and Student-Newman-Keuls test. In addition, the treatment values were converted to percent change from averaged time control values in some cases. All values are reported as mean ± SD, and P < 0.05 was regarded as statistically significant.
RESULTS
Immunolocalization of nitric oxide synthase
Immunolocalization of nNOS and eNOS in parietal cortex and cerebellum from untreated piglets is shown in Fig. 1. No apparent differences in nNOS immunostaining were observed between tissue obtained from untreated and control animals (not shown). Based on cellular morphologic makeup and location within the parenchyma, most of nNOS immunoreactive cells probably are neurons. Vascular nNOS immunostaining was never observed.
Representative photomicrographs of parietal cortex and cerebellum demonstrating localization of neuronal nitric oxide synthase (nNOS) and endothelial nitric oxide synthase (eNOS) immunoreactivity (IR) in normal piglet brain. 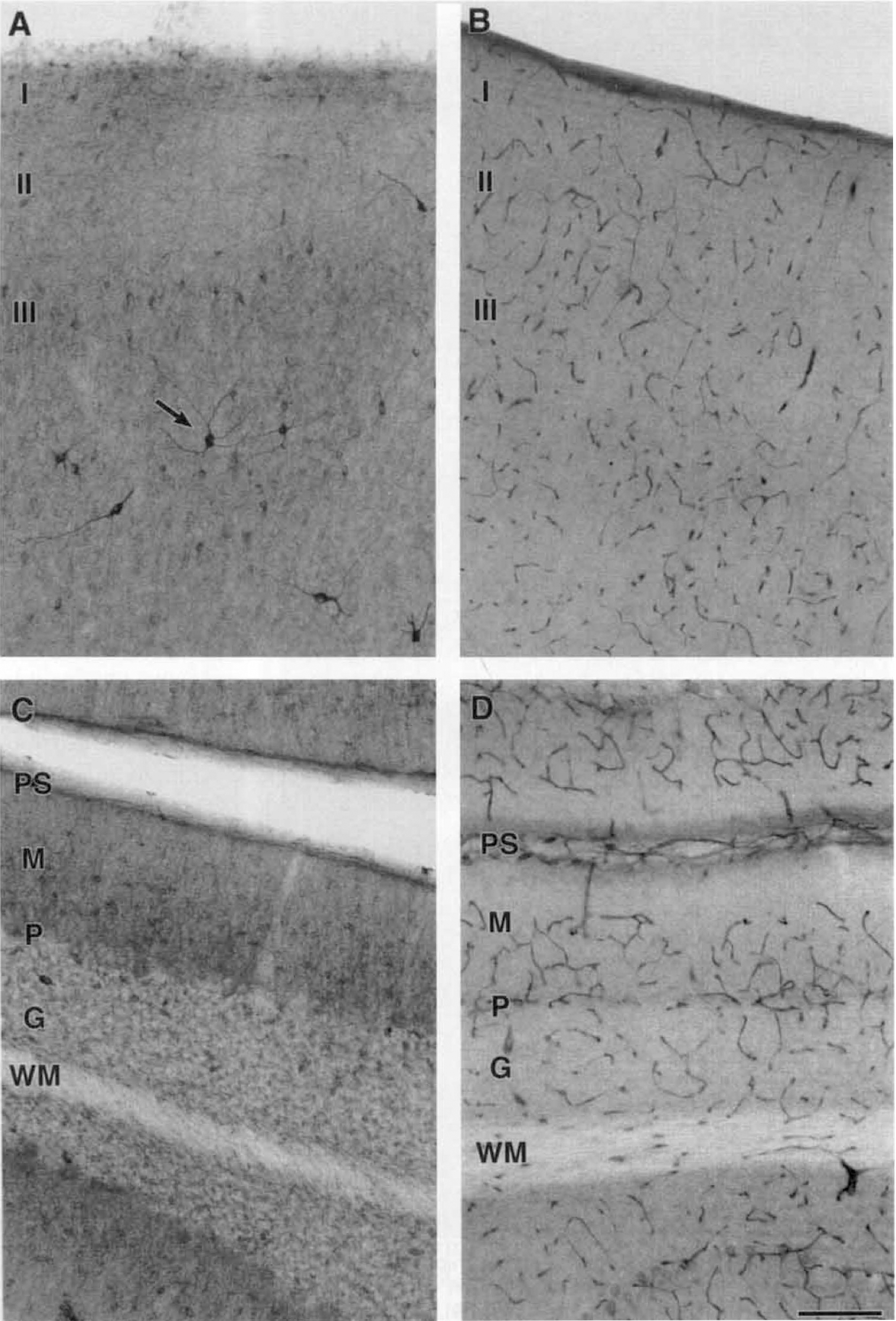
In parietal cortex, nNOS immunoreactivity (IR) was obvious but sparse (Fig. 1A). The most consistent site of nNOS IR was in cortical layer III. Neuronal NOS IR usually occurred in the soma and was uniform in intensity. However, bipolar and multipolar neurons in adjacent layers occasionally displayed nNOS IR, which was more intense than normally observed (Fig. 1 A, arrow). In addition to greater intensity in the cell bodies, the processes of these neurons also were stained.
Neuronal NOS IR in the cerebellum was localized to small neurons of the molecular and granular layers (Fig. 1C). Only the cell bodies appeared to be nNOS-positive in each cerebellar layer. Neuronal NOS IR was not observed in the immature cells located in the external granular layer. As indicated by the unstained areas at the junction of the molecular and granular layers, Purkinje cells were not immunoreactive for nNOS (Fig. 1C).
As in the other regions, untreated and control piglets did not differ in hippocampal nNOS immunostaining (not shown). Most striking was the immunostaining of the granular cells of the dentate gyrus and the mossy fiber layer (Fig. 2). The nNOS IR within the granular layer was localized to the cell bodies. The mossy fiber layer, which contains the axons from the granular cells, was consistently nNOS-positive. Higher magnification of the CA3 region demonstrated nNOS IR in nearby interneurons and occasional pyramidal cells (Fig. 2B). In general, the staining intensity was uniform throughout the hippocampus. However, multipolar neurons located in the hippocampal molecular layer often displayed darker nNOS IR than most of the immunopositive cells (Fig. 2C, arrow). Also evident on these multipolar neurons were processes with varicosities (Fig. 2C, arrowheads).
Representative photomicrographs of hippocampal nNOS IR in normal piglet brain. 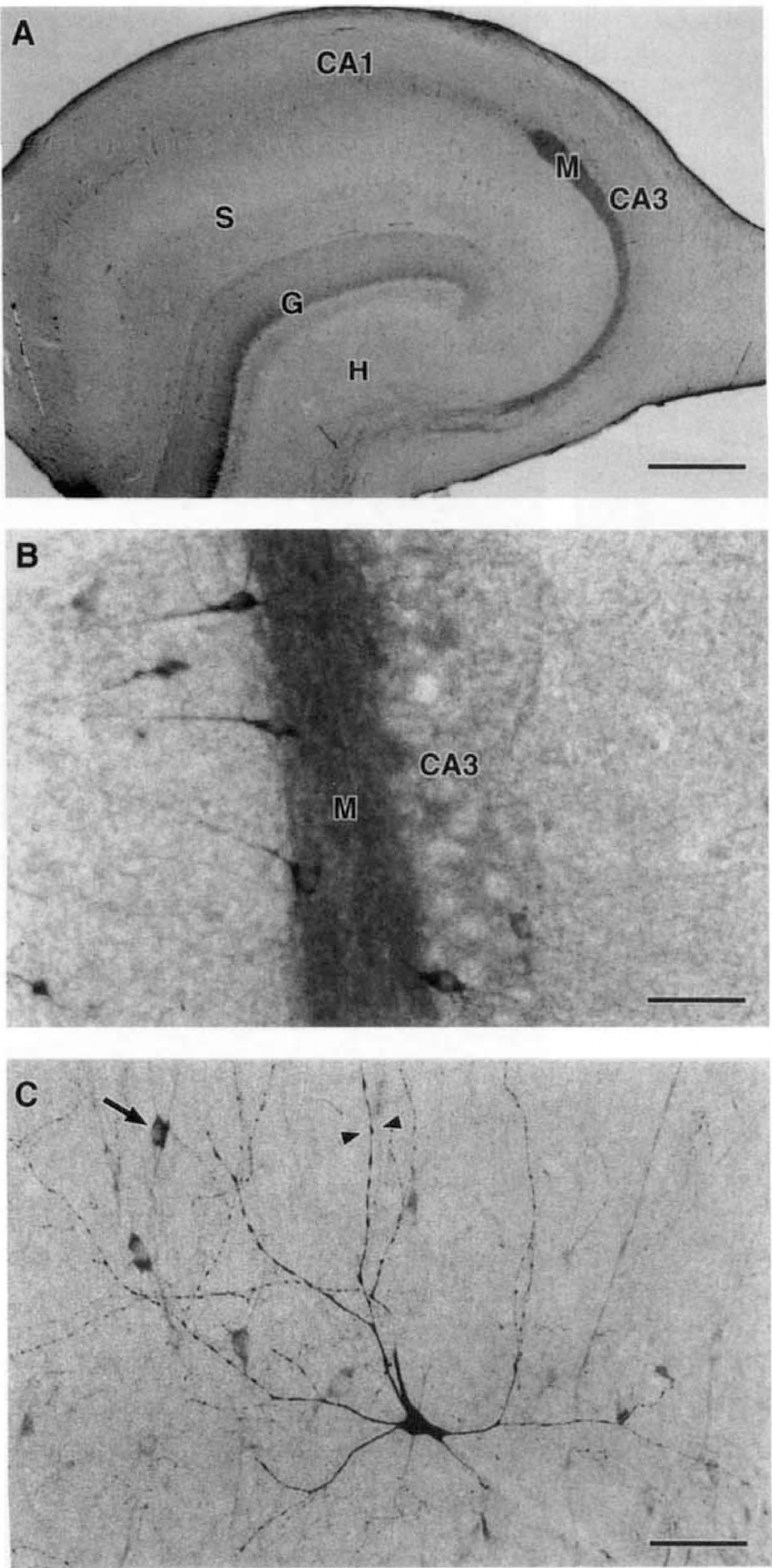
Endothelial NOS IR was localized to blood vessels within the parietal cortex, cerebellum (Fig. 1B and D), and hippocampus (Fig. 3). Cresyl violet staining provided morphologic evidence that the stained structures were blood vessels (not shown). The lumen and vascular walls were evident in vessels that lacked significant eNOS IR. The homogeneous eNOS staining of the vessels indicated that the IR probably was localized to the vessel wall and not to cells within the lumen. Immunoreactive vessels were similar in size and uniformly distributed throughout each region (Figs. 1B, 1D, and 3). Immunoreactivity for eNOS was not seen in cell types other than vascular cells.
Representative photomicrographs show changes in hippocampal eNOS IR 6 hours after ischemia. 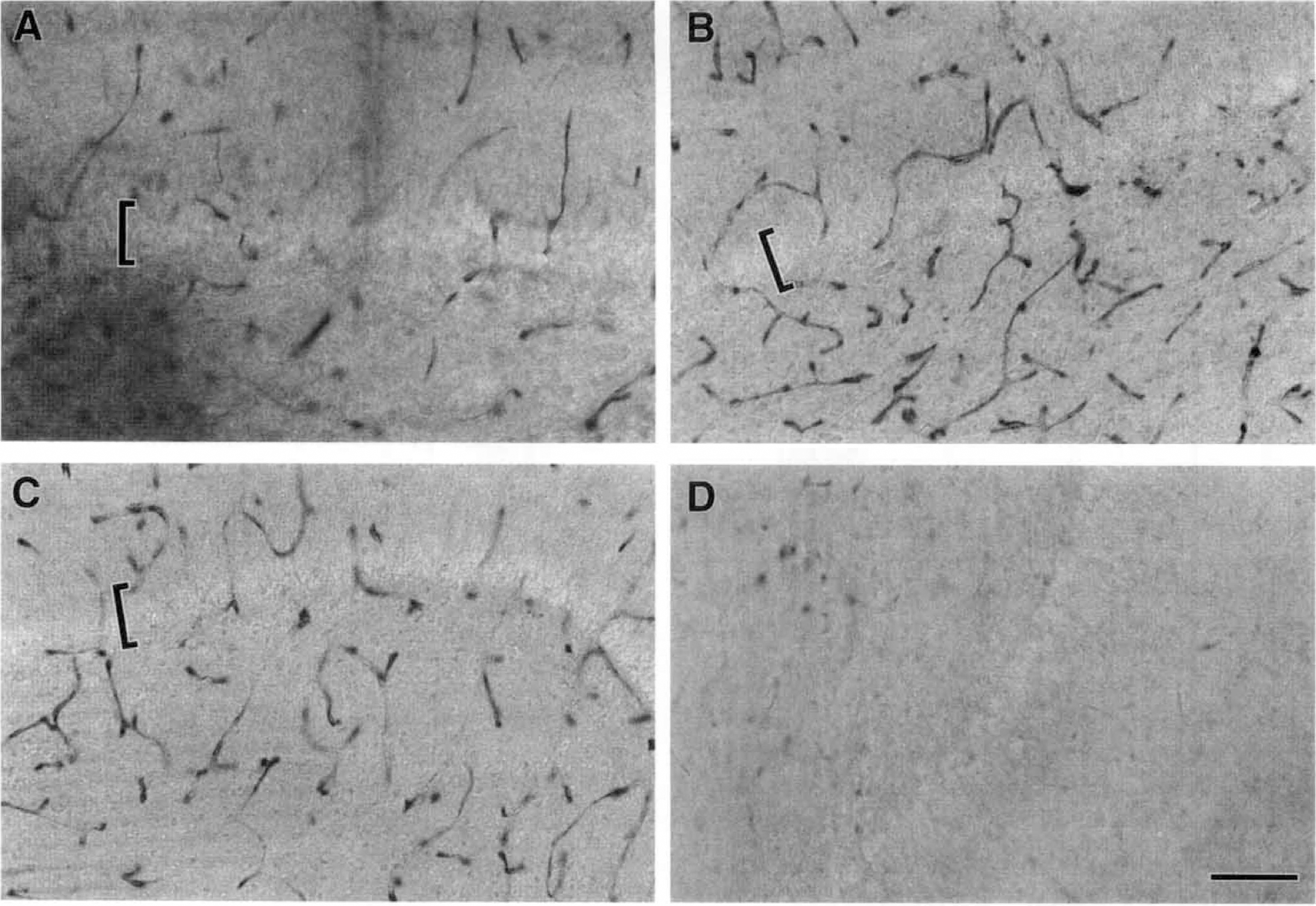
Immunolocalization of nitric oxide synthase after ischemia
The intensity and pattern of nNOS IR did not change in any region 4 to 6 hours after ischemia. In addition, no detectable morphologic alterations in nNOS-positive neurons were observed in any region (not shown). In contrast, increased eNOS IR was observed in parietal cortex and hippocampus within 6 hours after ischemia. Representative changes in hippocampal eNOS IR at 6 hours after ischemia are shown in Fig. 3. The most obvious change detected was an increase in intensity of eNOS IR after ischemia (Fig. 3A and B). Whereas all identified vessels appeared to be immunopositive for eNOS after ischemia (Fig. 3B), unstained vessels were observed in both control and indomethacin-pretreated sections (Fig. 3A and C).
Indomethacin pretreatment resulted in a decrease in eNOS IR intensity compared with ischemia alone (Fig. 3B and C). Although only the hippocampus is shown in Fig. 3, indomethacin modulation of eNOS IR also was observed in the parietal cortex. The eNOS immunopattern in control tissue was similar to that of the untreated tissue, suggesting that observable changes in eNOS IR could not be attributed to surgical manipulation. Omission of the primary antibody revealed only background IR (Fig. 3D).
Immunoblot analysis
Both the nNOS and eNOS antibodies reacted with the corresponding piglet isoform, as indicated by the appearance of a single immunoreactive band migrating at the appropriate molecular weight and with the protein standard (Figs. 4 and 5). Omission of the primary antibody resulted in the lack of an immunoreactive band (2° lane in Figs. 4 and 5). Data obtained from immunoblots were used to demonstrate the lack of change in nNOS and to quantify ischemia-induced changes in eNOS levels.
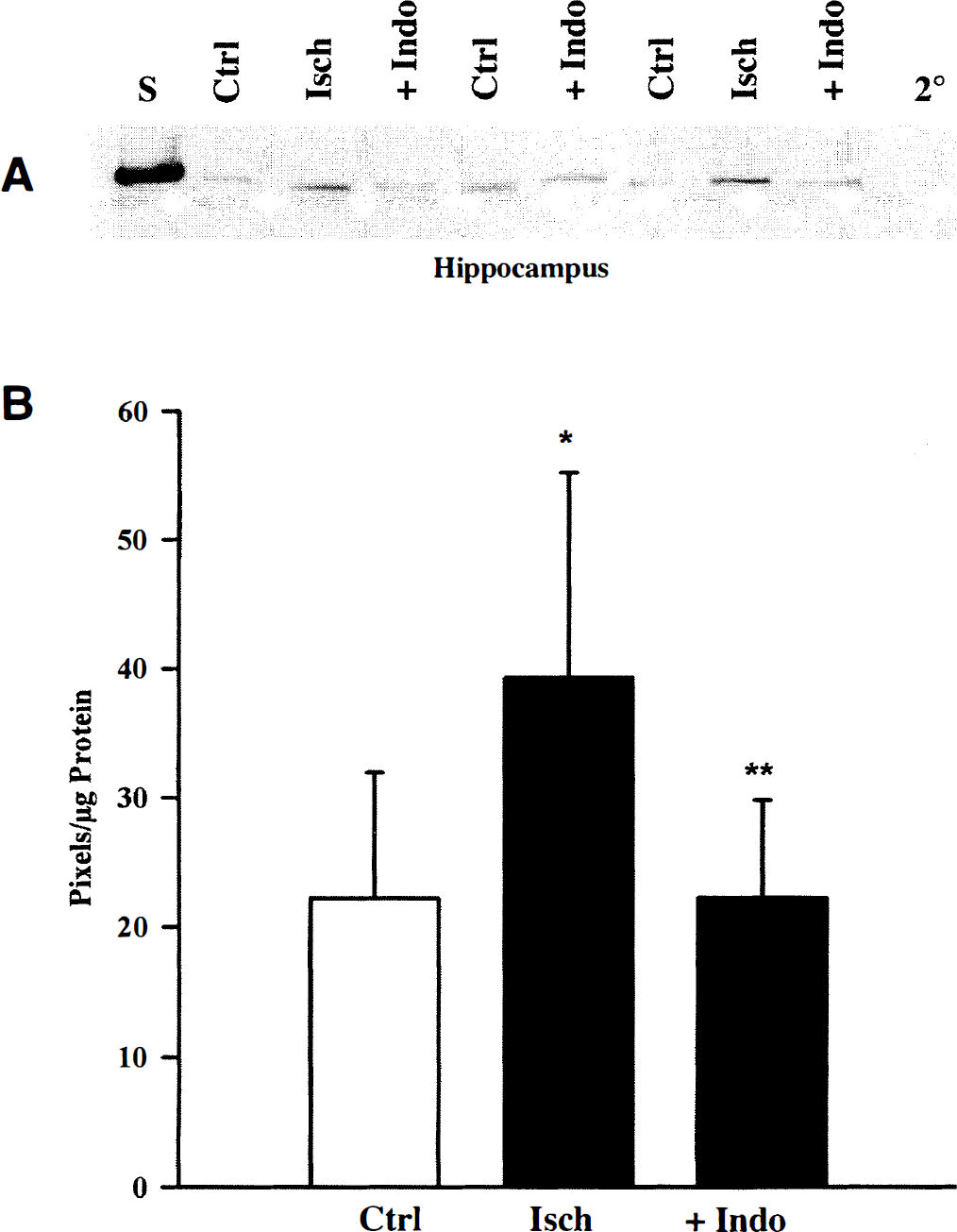
Hippocampal nNOS levels 6 hours after ischemia. Endothelial NOS levels 6 hours after global cerebral ischemia. 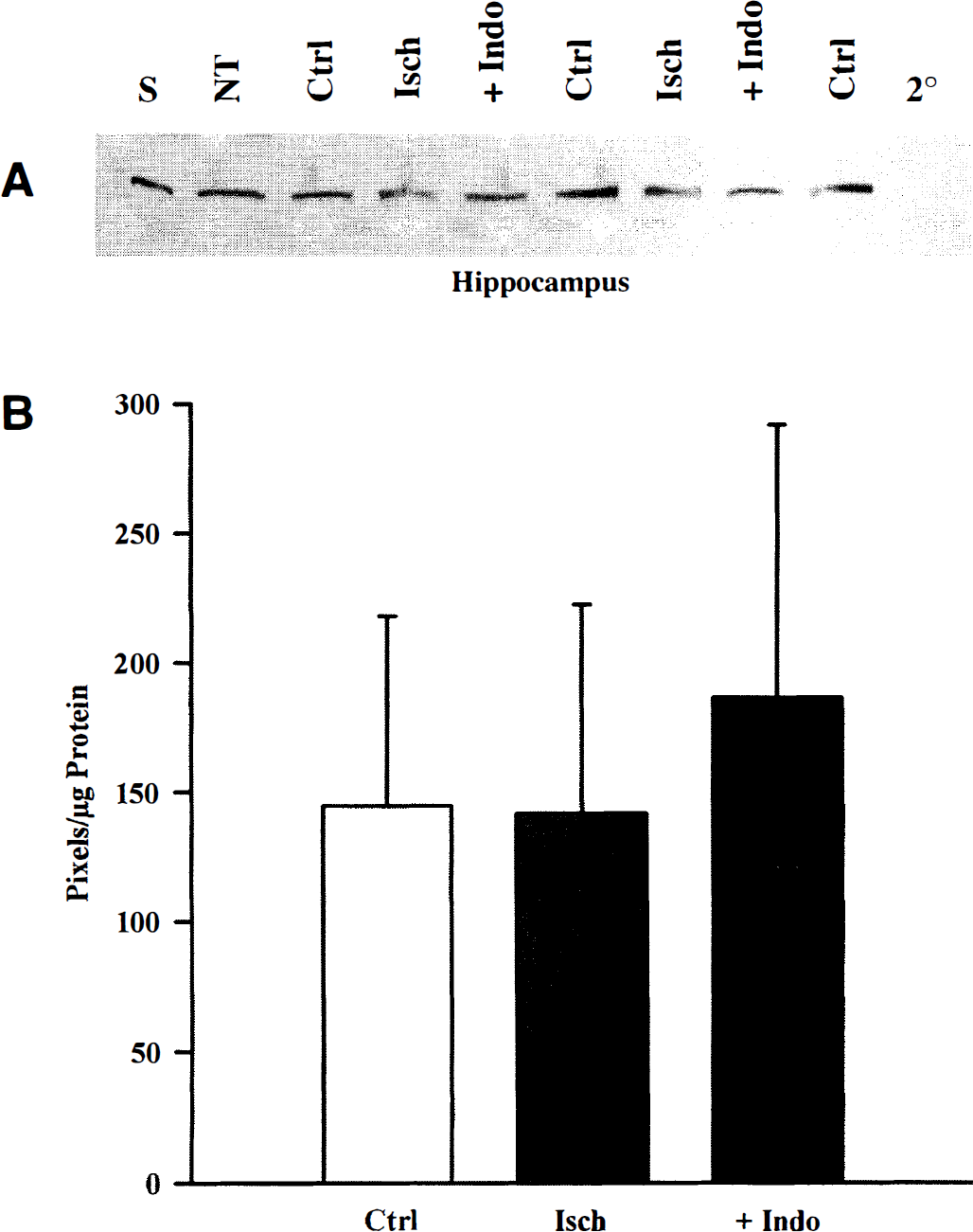
Lack of changes in levels of neuronal nitric oxide synthase
Global cerebral ischemia had no detectable effect on nNOS levels within 6 hours of reperfusion in any region examined. Immunoblots for nNOS revealed bands of equal intensity, regardless of treatment. A representative hippocampal blot at 6 hours is shown in Fig. 4A. Comparison of the band densities (pixels per microgram of protein) confirmed the lack of change in nNOS after ischemia and indomethacin pretreatment in hippocampus and parietal cortex (Figs. 4B and 6A). Further, no changes in nNOS levels were detected in cerebellum at 4 to 6 hours after global cerebral ischemia (not shown). Comparisons for cerebellum data at 4 and 6 hours were based on n = 8,9, and 7 for time control, ischemia, and indomethacin pretreatment, respectively. Administration of indomethacin before ischemia did not appear to significantly alter levels of nNOS in hippocampus (Fig. 4), parietal cortex (Fig. 6A), or cerebellum (not shown).
Histogram of mean pixels per microgram of protein ± SD from cortical nNOS 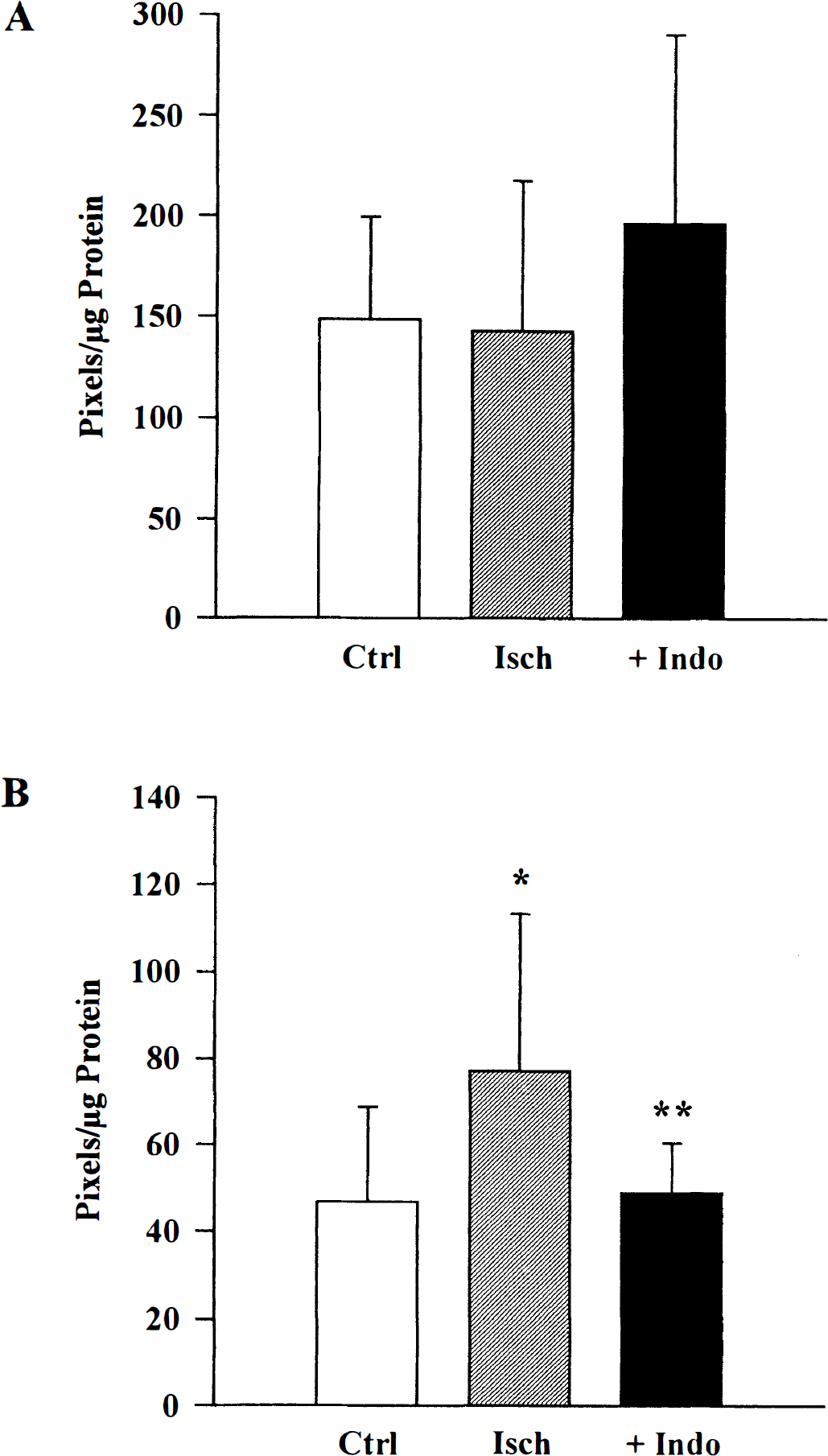
Ischemia-induced changes in endothelial nitric oxide synthase
Within 6 hours after global cerebral ischemia in newborn pigs, eNOS levels were greater than time control levels in hippocampus and parietal cortex (Figs. 5A and B, and 6). Further, indomethacin pretreatment practically abolished the ischemia-induced increase in eNOS in both regions (Figs. 5A and B, and 6B). Analysis of actual band densities (pixels per microgram of protein) revealed that in hippocampus and parietal cortex, global cerebral ischemia induced a significant increase in eNOS levels compared with the time controls (Figs. 5B and 6B). Calculation of percent change from averaged time control values revealed that ischemia induced a 40% increase in hippocampus and a 75% increase in parietal cortex. In contrast, indomethacin pretreatment band densities were not different from time control values in either hippocampus or parietal cortex (Figs. 5 and 6B). Thus, pretreatment with indomethacin results in a significant attenuation in eNOS levels compared with ischemia alone (Figs. 5B and 6B).
Unlike the hippocampus and parietal cortex, global cerebral ischemia had an inconsistent effect on eNOS levels in cerebellum. Analysis of the raw data indicated values of 45 ± 20 pixels/μg protein for time controls, 82 ± 48 pixels/μg protein for ischemia, and 46 ± 13 for indomethacin pretreatment. Whereas levels of eNOS after ischemia (n = 6) tended to be higher than in time controls (n = 5), the difference was not significant (P > 0.05). Likewise, indomethacin pretreatment (n = 5) resulted in levels of eNOS that were not statistically different from time control levels (P > 0.05). Thus, global cerebral ischemia appeared to selectively increase eNOS through an indomethacin-sensitive mechanism.
DISCUSSION
Characterization of eNOS and nNOS in newborn piglets reveals three novel findings. First, eNOS and nNOS are localized to vessels and neurons, respectively, in parietal cortex, hippocampus, and cerebellum. Second, eNOS levels are increased within 6 hours after global cerebral ischemia, whereas nNOS levels have not changed at this time. And third, indomethacin pretreatment significantly attenuates the ischemia-induced increase in eNOS. These results indicate that the eNOS response is rapid, specific to blood vessels, and subject to pharmacologic intervention.
Immunolocalization of nNOS and eNOS in piglet parietal cortex, hippocampus, and cerebellum is consistent with the pattern of staining observed in postnatal day 7 rats, adult rats, and humans (nNOS, Bredt and Snyder, 1994; Dinerman et al., 1994; Egberongbe et al., 1994; eNOS, Miyawaki et al., 1995; Tomimoto et al., 1994; Pollock et al., 1993; Zhang et al., 1993). The staining for piglet nNOS in cerebellum and eNOS in hippocampus reveals additional information regarding localization of these two enzymes. First, although the pattern of nNOS IR in the piglet cerebellum is similar to that reported for postnatal day 7 rats (Bredt and Snyder, 1994), the positive signal does not appear to be as intense or widespread as in the adult (Dinerman et al., 1994). This difference in nNOS staining may be related to developmental age, since the piglet and postnatal day 7 rats are more similar than the piglet and adult rodent. Second, although vascular eNOS IR has been observed in the adult rat cerebral cortex, bovine cerebellum, and human cerebral cortex (Miyawaki et al., 1995; Pollock et al., 1993; Zhang et al., 1993), a single study of the adult rat brain localized eNOS to neuronal cell populations in hippocampus (Dinerman, et al., 1994). Interestingly, the pattern of eNOS IR reported by Dinerman, et al. (1994) more closely resembles the nNOS staining observed in piglet hippocampus. Thus, nNOS and eNOS localization in newborn piglet cerebral cortex, hippocampus, and cerebellum revealed immunostaining of distinct cellular populations for each isoform of NOS.
No detectable changes in either nNOS IR or levels were observed within 6 hours after global cerebral ischemia in piglets. Although some studies suggest that nNOS-derived NO is correlated with ischemia-induced cellular injury (Huang et al., 1994; Zhang et al. 1996), the lack of detectable changes in nNOS levels in piglets is consistent with the previous finding that nNOS levels remained unchanged after 4 hours of reperfusion (Busija et al., 1996). However, altered NO production also can be achieved by increasing enzyme activity. Whereas activity was not measured in the current experiments, no change in cortical NOS activity was observed within 4 hours after anoxic stress (Busija et al., 1996; Groenendaal et al., 1996). Thus, the current data suggest that nNOS levels remain unaltered during the immediate recovery period after cerebral anoxia in piglets.
Unlike nNOS, a significant increase in eNOS levels was observed within 6 hours after global cerebral ischemia in piglet parietal cortex and hippocampus. However, the physiologic significance of this selective increase in eNOS is unclear. A NO-dependent delayed vasodilation has been observed 12 to 24 hours after cerebral hypoxic ischemia in fetal sheep, and blockade of this response is correlated with increased cellular injury (Marks et al., 1996). Further, in adult models of focal cerebral ischemia, early exposure to NO donors appears to attenuate injury (Zhang and Iadecola, 1993; Zhang et al., 1994), whereas infarct size is enlarged in eNOS-deficient mice (Huang et al., 1996). A rapid increase in eNOS levels is consistent with the potential beneficial role of this enzyme during the initial recovery period after global cerebral ischemia.
The indomethacin attenuation of the ischemia-induced increase in eNOS suggests that cyclooxygenase-dependent mechanisms are involved in regulating this enzyme. Cyclooxygenase catalyzes the rate limiting reaction of arachidonic acid metabolism, resulting in prostanoid and superoxide anion production. Cerebral ischemia/reperfusion has been reported to increase cortical superoxide and prostaglandin production, which is prevented by indomethacin pretreatment (Armstead et al., 1988; Pourcyrous et al., 1993). Interestingly, both superoxide anions and prostanoids have been implicated in regulating the synthesis of various proteins. For example, ischemia-induced increases in inducible heat shock protein 70 in swine heart and liver are dependent on superoxide anion production (Schoeniger et al., 1994a, 1994b). Further, the eukaryotic transcription factor NF-κB, which is involved in regulating NOS synthesis (Remacle et al., 1995; Feinstein et al., 1996), appears to be dependent on reactive oxygen species (Pahl and Baeuerle, 1996). Alternatively, cyclooxygenase-derived prostaglandins may regulate protein synthesis. Using murine 3T3-L1 cell cultures, indomethacin appears to preserve arachidonic acid suppression of GLUT4 gene expression (Long and Pekala, 1996). Indomethacin also has been reported to suppress prostaglandin E2–induced transcription of the human corticotropin-releasing hormone (Makrigiannakis et al., 1996). Thus, whereas the indomethacin attenuation of ischemia-induced increases in eNOS levels probably is related to inhibition of cyclooxygenase activity and/or reduced superoxide anion production, precise mechanisms are unclear.
Whether the attenuation of eNOS levels is beneficial or detrimental is unknown. In piglets, indomethacin diminishes cerebral blood flow, which returns to baseline within 4 hours (Pourcyrous et al., 1994). Despite the transient attenuation of cerebral blood flow, indomethacin has been reported to have beneficial effects when administered before anoxic stress. For example, preservation of cerebrovascular response to vasoactive substances supports the beneficial effects of indomethacin (Bari et al., 1996b; Busija et al., 1996). As mentioned earlier, indomethacin is given clinically to premature babies to reduce the risk of intracranial hemorrhage and aid in closure of the ductus arteriosus (Volpe, 1994). Perhaps the indomethacin reduction of injurious superoxide anion production eliminates the need or signal for increased eNOS production. However, the current study reveals that indomethacin-sensitive mechanisms appear to be involved in the ischemia-induced increase in eNOS levels and does not address whether this attenuation will ultimately be beneficial or detrimental.
In summary, global cerebral ischemia results in a rapid increase in eNOS levels without any detectable effects on nNOS in newborn pigs. The increase in eNOS occurs in parietal cortex and hippocampus regions known to be particularly sensitive to anoxic stress in newborn babies. Indomethacin pretreatment attenuates the increase in eNOS, suggesting that ischemia induction of eNOS involves mechanisms that can be pharmacologically altered. Thus, indomethacin-sensitive mechanisms appear to be involved in ischemia-induced increases in eNOS levels after global cerebral ischemia in neonates.