
Abstract
White matter of the brain and spinal cord is susceptible to anoxia and ischemia. Irreversible injury to this tissue can have serious consequences for the overall function of the CNS through disruption of signal transmission. Myelinated axons of the CNS are critically dependent on a continuous supply of energy largely generated through oxidative phosphorylation. Anoxia and ischemia cause rapid energy depletion, failure of the Na+−K+-ATPase, and accumulation of axoplasmic Na+ through noninactivating Na+ channels, with concentrations approaching 100 mmol/L after 60 minutes of anoxia. Coupled with severe K+ depletion that results in large membrane depolarization, high [Na+]i stimulates reverse Na+–Ca2+ exchange and axonal Ca2+ overload. A component of Ca2+ entry occurs directly through Na+ channels. The excessive accumulation of Ca2+ in turn activates various Ca2+-dependent enzymes, such as calpain, phospholipases, and protein kinase C, resulting in irreversible injury. The latter enzyme may be involved in “autoprotection,” triggered by release of endogenous γ-aminobutyric acid and adenosine, by modulation of certain elements responsible for deregulation of ion homeostasis. Glycolytic block, in contrast to anoxia alone, appears to preferentially mobilize internal Ca2+ stores; as control of internal Ca2+ pools is lost, excessive release from this compartment may itself contribute to axonal damage. Reoxygenation paradoxically accelerates injury in many axons, possibly as a result of severe mitochondrial Ca2+ overload leading to a secondary failure of respiration. Although glia are relatively resistant to anoxia, oligodendrocytes and the myelin sheath may be damaged by glutamate released by reverse Na+–glutamate transport. Use-dependent Na+ channel blockers, particularly charged compounds such as QX-314, are highly neuroprotective in vitro, but only agents that exist partially in a neutral form, such as mexiletine and tocainide, are effective after systemic administration, because charged species cannot penetrate the blood–brain barrier easily.
These concepts may also apply to other white matter disorders, such as spinal cord injury or diffuse axonal injury in brain trauma. Moreover, whereas many events are unique to white matter injury, a number of steps are common to both gray and white matter anoxia and ischemia. Optimal protection of the CNS as a whole will therefore require combination therapy aimed at unique steps in gray and white matter regions, or intervention at common points in the injury cascades.
The mammalian central nervous system (CNS) has evolved into a marvelously complex machine controlling the key behavioral and homeostatic functions of all higher species. The unique role of the CNS is underscored by its intricate structure, protective enclosure, and continuous, tightly regulated blood supply; indeed, the human brain accounts for 20% of the total adult body oxygen utilization although representing only 2% of body weight (Clarke and Sokoloff, 1994). The innumerable neurons, axons, and supportive glial cells of the CNS display a high rate of metabolic activity, are fastidious with respect to their energy substrates (requiring continuous oxygen and glucose), and are consequently vulnerable to even brief disruptions of energy supply. A number of prevalent clinical conditions expose the CNS to anoxic and ischemic damage, of which stroke, cardiorespiratory arrest, and spinal cord injury (in which posttraumatic ischemia plays an important role in tissue injury [Sandler and Tator, 1976; Li and Taylor, 1997]) are but a few common examples. As a result, CNS ischemia represents a very significant clinical and socioeconomic problem.
Neurons of the brain and spinal cord concern themselves predominantly with signal processing, relying on a vast array of axonal connections for reliable signal transmission. Both functions are critical for the normal operation of the CNS, and understanding the basic mechanisms of injury for the rational design of therapeutic intervention in both gray and white matter is of utmost importance to provide optimal protection of this organ. A large body of work exists dealing with the pathophysiology of gray matter anoxic and ischemic injury (for reviews see Siesjö, 1986; Choi, 1990; Haddad and Jiang, 1993; Siesjö and Wieloch, 1996). An emerging theme implicates cellular overload of Ca2+, occurring largely through glutamate-gated receptors and possibly voltage-gated Ca2+ channels, cell swelling as a result of excessive Na+ and Cl− influx, free radical production, and delayed apoptotic neuronal death. In contrast, much less is known about the fundamental mechanisms of anoxic and ischemic injury to CNS myelinated axons, despite the fact that white matter has been shown to be very vulnerable to this type of injury (Follis et al., 1993; Pantoni et al., 1996). A number of reports have been published on ischemic spinal cord in vivo (e.g., Madden et al., 1990; Kochhar et al., 1991a; Kochhar et al., 1991b; von Euler et al., 1994), in which gray matter, white matter, and vascular effects are easily confounded. Although informative and clinically relevant, it is inherently difficult to formulate mechanistic inferences from such whole animal studies. This article will focus on studies that emphasize fundamental cellular mechanisms of white matter injury, a thorough understanding of which is essential for efficient and logical design of neuroprotective intervention. The results of these basic studies have led to the successful development of therapeutic strategies that will in turn guide future in vivo experiments aimed at developing effective clinical treatments.
MYELINATED AXONS—STRUCTURE AND FUNCTION
Myelinated axons have a unique architecture allowing them to transmit action potentials reliably and in an energy-efficient manner (Waxman and Ritchie, 1993; Salzer, 1997). There is a highly segregated distribution of ion channels and transporters on the axon membrane that is designed to carry the ionic currents required for action potential generation and propagation (Fig. 1). The most striking is the high density of voltage-gated Na+ channels at the node of Ranvier (≈1,000 to 2,000 channels per μm2; Waxman, 1995). Internodal K+ channels are thought to contribute largely to the maintenance of a hyperpolarized resting membrane potential of approximately −80 mV (Chiu and Ritchie, 1982; Chiu and Ritchie, 1984; Bostock and Grafe, 1985; Morita et al., 1993; Stys et al., 1991a). The Na+–K+-ATPase is the main active ion transporter designed to maintain adequate transmembrane gradients of Na+ and K+. A number of other transport mechanisms also contribute to overall ionic homeostasis. Together with electrically tight paranodal myelin seals, axonal ion channels generate transmembrane currents that are conducted from node to node in a saltatory fashion, thus allowing rapid and efficient signal conduction. Important supportive roles of adjacent astrocytes and their processes have also been proposed (Black et al., 1995; Newman, 1995).
Diagram of ion channel and transporter distributions in myelinated axons. A high density of nodal Na+ channels carries the inward depolarizing current and initiates the action potential. A variety of K+ channel subtypes with fast (KF), intermediate (K,), or slow (KS) kinetics, chemically modulated K+ channels (by axoplasmic ATP, Ca2+, or Na+ ions), and voltage-independent “flicker” channels serve to set the resting membrane potential, modulate firing patterns, and in some fibers aid in action potential repolarization. Anion channels and transporters have also been identified. Ionic homeostasis is maintained by several ATP-dependent pumps, such as the Na+–K+- and Ca2+-ATPases, as well as the Na+–Ca2+ antiporter. The precise distribution of these channels varies between species and also differs in myelinated axons of the peripheral nervous system and CNS. The tight electrical seal at the paranode is critical for isolating the various currents, allowing fast and reliable signal propagation. Modified and reprinted with permission from Vogel and Schwarz, 1995. Copyright© 1995 by Oxford University Press, Inc.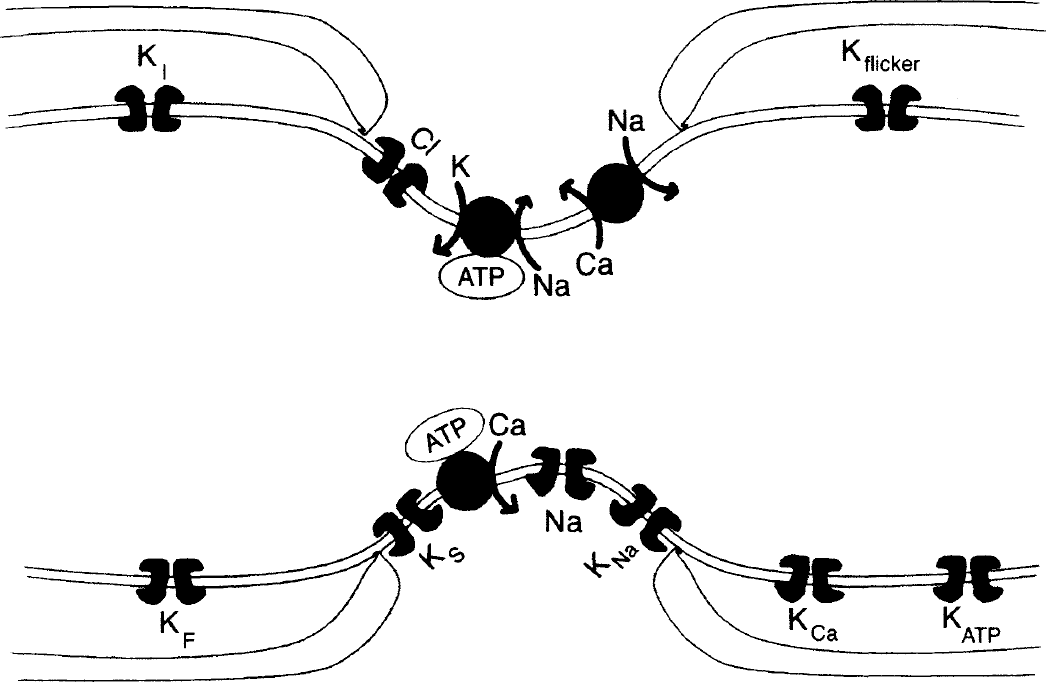
The highly specialized architecture of myelinated axons renders them vulnerable to injury. Disruption of any of the previously mentioned ion transporters, the cytoskeleton (which imparts structure to the axon cylinder and serves to maintain the required distribution of channels [Salzer, 1997]), or glia (including the overlying myelin sheath) may result in impairment or complete failure of impulse transmission. Many of these structural and functional elements are irreversibly disrupted by anoxia and ischemia.
ANOXIA AND ISCHEMIA: ACUTE LOSS OF AXONAL EXCITABILITY
Many studies have been performed over the years on the effects of anoxia and ischemia in peripheral axons (for a review see Stys et al., 1995). Despite the strong similarity in structure and function between central and peripheral fibers, their responses to energy failure are quite different, and therefore findings in the peripheral nervous system cannot be extrapolated to central axons. An elegant study by Utzschneider et al. (1991) emphasized the dramatically different effects of hypoxia on central (dorsal root ganglion cell processes in spinal dorsal columns) versus peripheral (dorsal roots) axons. The central component is very sensitive to hypoxia, whereas the peripheral projections originating from the same cell are completely unaffected by a 30-minute exposure (Fig. 2). Even more striking is the observation that although longer hypoxic exposures result in significant deregulation of elemental content (including large accumulations of Ca2+) in the axoplasm of peripheral myelinated fibers, reoxygenation promotes complete recovery (Lehning et al., 1996b); this is in stark contrast to central axons, in that not only does reoxygenation fail to correct the pathologic translocation of ions caused by the hypoxic insult, but many axons continue to deteriorate, accumulating more Ca2+ and presumably suffering more damage (Stys and LoPachin, 1996) (see Reoxygenation, below).
Diagram illustrating the different susceptibility of central and peripheral axons to hypoxia. Despite the strong similarity in structure and function between peripheral and central fibers, the former are quite resistant to hypoxia. 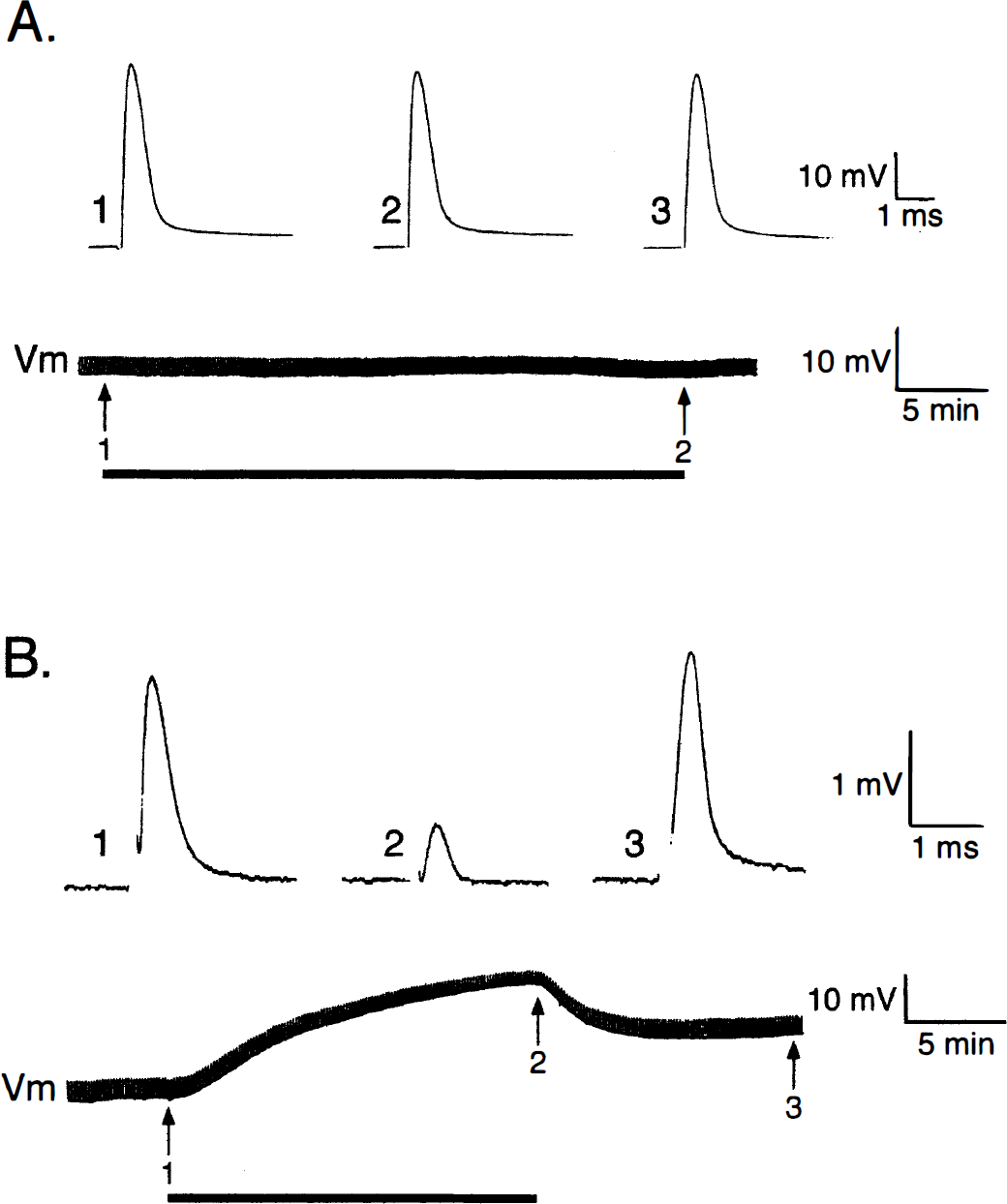
A series of studies begun in the early 1990s using the in vitro rat optic nerve as a model of central white matter injury has yielded a number of important mechanistic insights into the acute phase of axonal injury in the CNS. The optic nerve is a central axonal tract consisting of myelinated axons and glia that is devoid of synaptic machinery (Foster et al., 1982; Hildebrand and Waxman, 1984); this simplifies the interpretation of data because the complexities of the synapse, dendrite, and neuronal soma are eliminated. Figure 3 illustrates the early response of optic nerves to in vitro anoxia or glycolytic block. Propagated compound action potentials are shown in the upper panels, and corresponding trajectories of compound resting membrane potentials appear below. Anoxia causes a rapid decline in the magnitude of the compound action potential in central white matter tracts (Stys, 1996; Imaizumi et al., 1997) with a concomitant large membrane depolarization (Leppanen and Stys, 1997b) (Fig. 3A and B). This indicates that CNS white matter is critically dependent on a continuous supply of energy substrates and is heavily dependent on oxidative metabolism for maintenance of excitability, although a minor component of ion pumping is fueled by glycolysis (Leppanen and Stys, 1997b) (Fig. 3B). Glycolytic block reveals interesting differences from pure anoxia (Fig. 3C and D). First, the onset of massive depolarization is delayed, in contrast to anoxia in which function begins to fail within minutes. This is likely caused by temporary utilization of alternate substrates such as amino acids or fatty acids fed directly into the tricarboxylic acid cycle (Stryer, 1988; Silver et al., 1997). Second, the depolarization was often preceded by a brief hyperpolarization, which was Ca2+-dependent (Leppanen and Stys, 1997b). The latter may be related to activation of a Ca2+-dependent K+ conductance, since it is reduced by tetraethylammonium, a broad spectrum K+ channel blocker (Leppanen and Stys, 1997b). Preliminary data also suggest that the source of this early axoplasmic [Ca2+] rise could be from intracellular stores, rather than entry from outside the axon (Leppanen and Stys, 1997b). Even more interesting is the fact that this hyperpolarizing phenomenon was never observed with anoxia, raising the intriguing notion of selective energy supply of intracellular Ca2+ stores in axons by glycolytic, rather than mitochondrial, ATP (Xu et al., 1995).
Effect of anoxia or glycolytic block on optic nerve propagated compound action potentials (CAP) or resting membrane potentials (Vm). 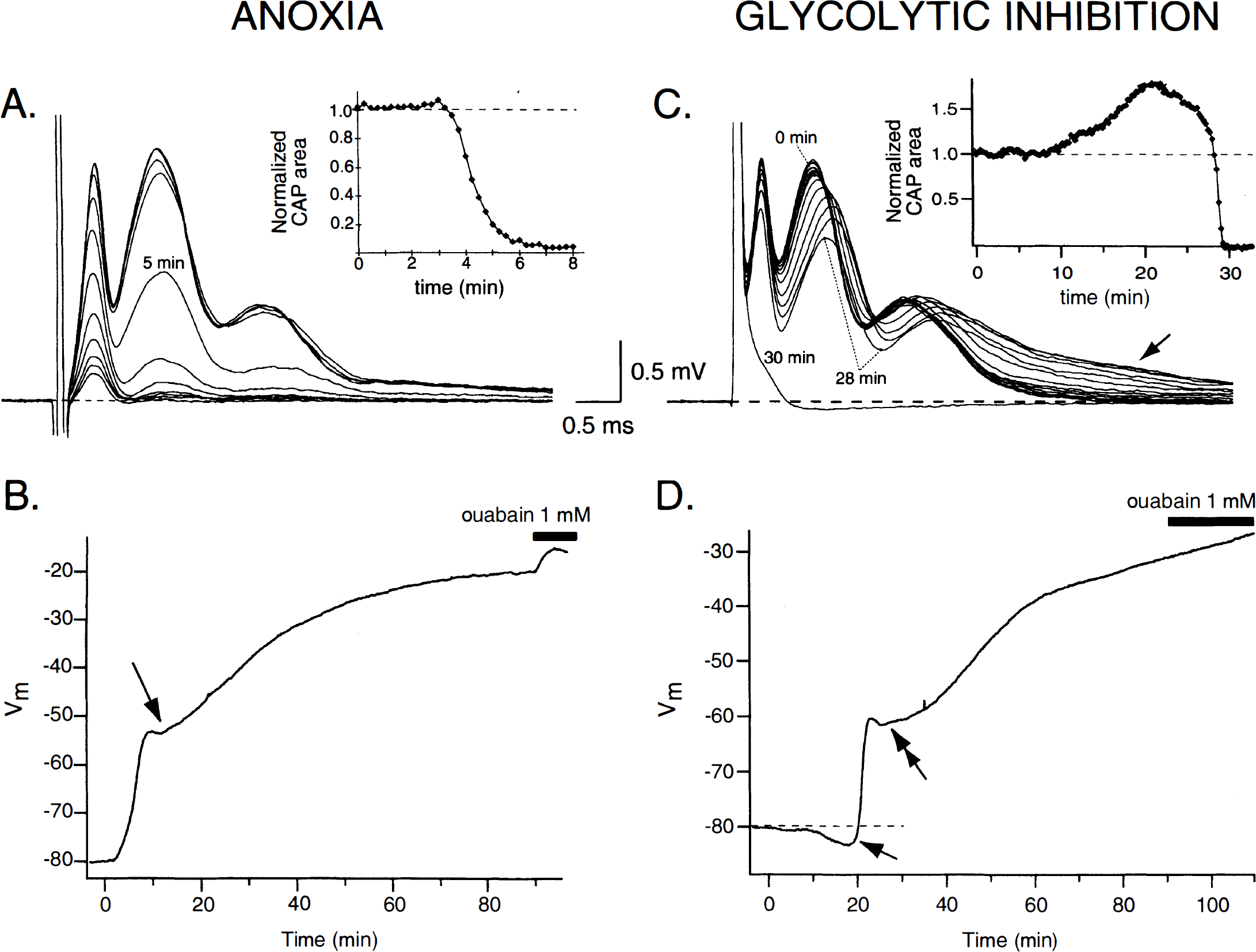
A common phenomenon observed with both anoxia and glycolytic block is a sudden change in the rate of depolarization within minutes of the rapid phase of membrane potential collapse (Fig. 3B and D). This is not observed during pure Na+–K+-ATPase inhibition with ouabain, in which the depolarization is always monotonic (Leppanen and Stys, 1997a). Given that depolarization promotes deleterious Ca2+ influx into axons (see Ca2+ overload, below), perhaps this rate change represents an attempt to limit the degree of depolarization, reflecting an “autoprotective” mechanism (see Ca2+ overload, below, for further discussion). The precise mechanisms of the abrupt change in dVm/dt, which occurs only during energy depletion, but not during Na+–K+-ATPase inhibition alone, are currently under investigation in our laboratory.
Optic nerve axons are capable of resisting glycolytic failure when supplied with exogenous lactate or pyruvate (Leppanen and Stys, 1991b; Ransom and Fern, 1997). This suggests that these axons have the necessary transporters to take up lactate and convert it to pyruvate through lactate dehydrogenase (or to take up and use pyruvate directly), thus supplying substrate for continuous operation of the tricarboxylic acid cycle and oxidative phosphorylation provided that sufficient oxygen is available. Ransom and Fern (1997) have recently proposed that lactate generated by optic nerve astrocytes may constitute an exogenous source of this substrate, exported by glia for the purpose of supplying energy to the axons under normal conditions. The rate of compound action potential failure (Ransom and Fern, 1997) and axonal membrane depolarization (Leppanen and Stys, 1997b) is much slower with simple aglycemia compared with aglycemia with the addition of glycolytic inhibitors, such as 2-deoxyglucose (Ransom and Fern, 1997) or iodoacetate (Leppanen and Stys, 1997b). With aglycemia alone astrocytes could continue to produce lactate from their glycogen stores, thereby supplying axons with substrate until these stores themselves become depleted. Addition of either of the two pharmacologic blockers of glycolytic metabolism would additionally impair astrocytic lactate production because breakdown and metabolism of glial glycogen stores would be impaired under these conditions. Taken together, these observations indirectly but convincingly point to white matter glia as playing a role in supporting the energy requirements of myelinated axons, probably in the form of lactate export. This would be analogous to other parts of the CNS in which glial-to-neuronal transfer of energy substrates has been shown to occur (Tsacopoulos and Magistretti, 1996).
DISRUPTION OF AXONAL ION HOMEOSTASIS
The large membrane depolarization and action potential failure induced by glycolytic or mitochondrial block are caused by a massive translocation across the axolemma of Na+ and K+ ions down their electrochemical gradients. Experiments with K+-sensitive microelectrodes in the rat optic nerve model reveal a substantial rise of extracellular K+ concentration ([K+]o) from a baseline of 3 mmol/L to a peak of ≈14 mmol/L within minutes of onset of anoxia (Ransom et al., 1992). A related study suggested that the source of K+ originated from both axons and glia (Ransom and Philbin, 1992), although subsequent direct measurements of axonal and glial K+ content during an anoxic challenge showed little loss of K+ from anoxic optic nerve glia (see below, also LoPachin and Stys, 1995). Although the rise in [K+]o is much smaller than that seen in cortex in which [K+]o can increase to ≈80 mmol/L (Hansen, 1985), the difference is likely because of diffusional properties, rather than a greater capacity of white matter to withstand anoxia. Experiments using electron probe microanalysis (EPMA), a quantitative electron microscopic technique capable of measuring elemental content in subcellular compartments (LoPachin and Saubermann, 1990), show a rapid and severe loss of axoplasmic K+, declining to approximately 10% of normal levels after 60 minutes of anoxia (Fig. 4B). A reciprocal rise of axoplasmic [Na+], from a resting level of ≈20 mmol/L (Stys et al., 1997a) to ≈100 mmol/L, also occurs in optic axons (LoPachin and Stys, 1995). As will be discussed below, Na+ entry occurs largely through tetrodotoxin (TTX)-sensitive voltage-gated Na+ channels. Interestingly, both electrophysiologic (Stys et al., 1992b) and EPMA (LoPachin and Stys, 1995) studies have shown that axonal Na+ accumulation continues throughout a 60-minute anoxic period, despite loss of excitability and strong membrane depolarization. Conventional voltage-gated Na+ channels would be expected to quickly inactivate with strong persistent depolarization (Chiu et al., 1979; Kostyuk et al., 1981), becoming impermeable to Na+ ions. The data therefore suggest that a noninactivating Na+ conductance may be responsible for the pathologic Na+ influx into anoxic axons. This hypothesis was further strengthened by experiments showing a finite, TTX-blockable Na+ permeability in resting optic axons, which persists even with strong depolarization in high K+ (Stys et al., 1993). In contrast to axonal responses to anoxia, changes in glial Na+ and K+ were far more modest, reaching statistical significance only after 60 minutes of anoxic exposure. Indeed, glial [K+] appeared to rise slightly during the early phase of anoxia (LoPachin and Stys, 1995), possibly indicating transient uptake of this ion (Newman, 1995) spilled by compromised axons.
Electrical and chemical responses of optic nerve axons to anoxia, with or without Na+ conductance inhibition. 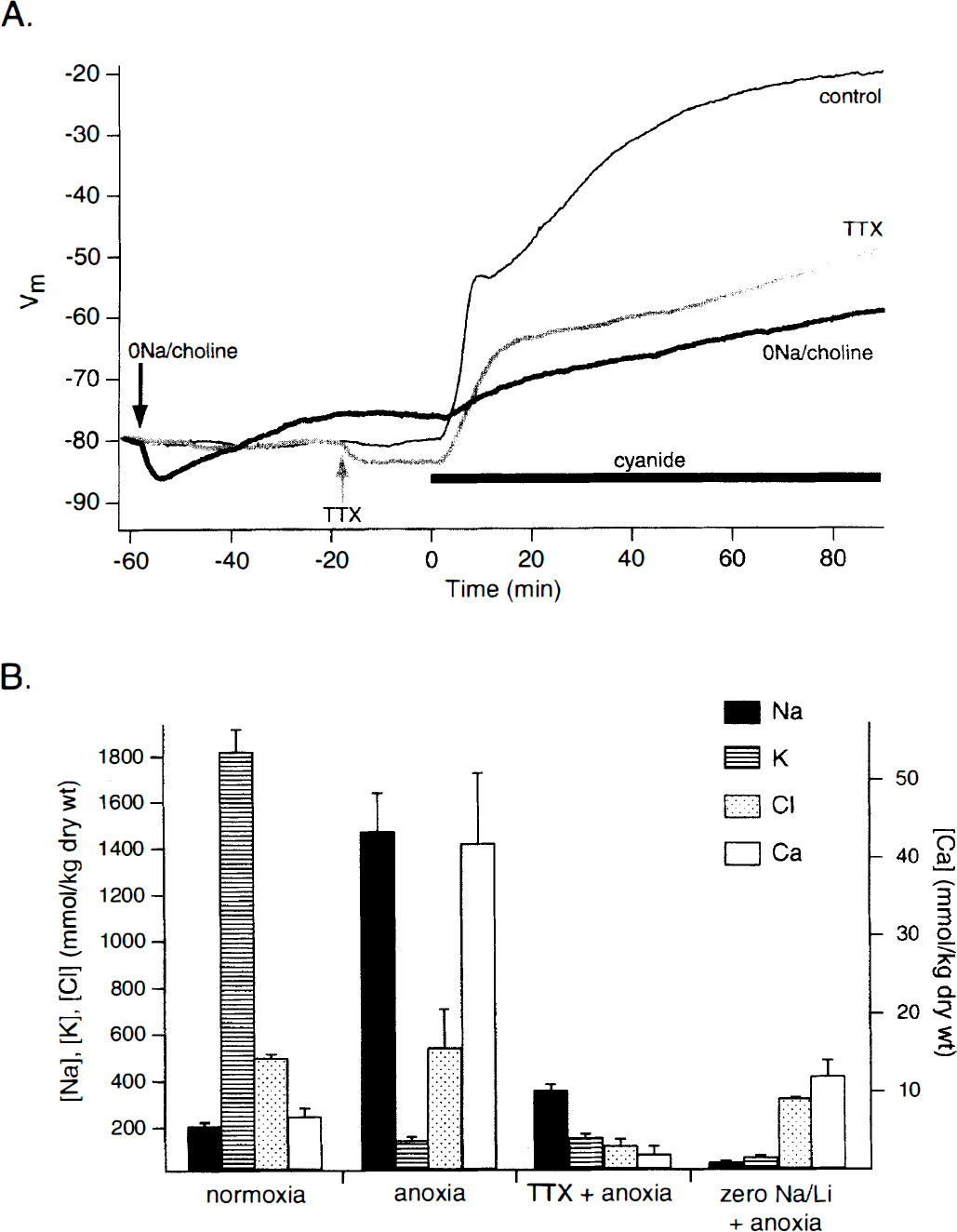
Along with a rise in [K+]o, there is a prompt acidification of the extracellular compartment in anoxic optic nerves, with a maximum drop of approximately 0.3 pH units in bathing medium containing 10 mmol/L glucose (Ransom et al., 1992). This indicates that optic nerves increase their rate of glycolysis with generation of lactic acid during anoxia. The acidification is exaggerated in solutions containing higher glucose concentrations, but is overall less than in cortex (Harris and Symon, 1984; Hansen, 1985). Despite a greater acidification in higher glucose, optic nerves are better able to withstand the deleterious effects of anoxia, displaying a smaller rise in [K+]o and delayed loss of excitability (Ransom et al., 1992), probably because increased glycolysis is able to contribute more ATP to energy-dependent homeostatic mechanisms under hyperglycemic conditions. This is in contrast to worsening of gray matter ischemic injury in high ambient glucose concentrations under certain conditions (Siesjö et al., 1996). Interestingly, acidification per se may be neuroprotective. When the pH of the external solution is artificially reduced, dorsal column axons are better able to withstand anoxic exposure (Imaizumi et al., 1997). The Na+–H+ antiporter may also be involved in the pathogenesis of injury because pharmacologic blockade of this transporter with harmaline or amiloride is protective in models of dorsal column anoxia (Imaizumi et al., 1997) and in vitro mechanical compression (Agrawal and Fehlings, 1996). These agents both have actions at Na+ channels, however (Velly et al., 1988; Deecher et al., 1992); thus, a protective effect secondary to reduced Na+ influx through Na+ channels cannot be ruled out.
Identification of the precise routes of abnormal transmembrane ion flux is of paramount importance for the rational design of therapeutic intervention, because deregulation of the major cations is largely responsible for subsequent cellular Ca2+ overload leading to irreversible damage (see Ca2+ overload, below). Blocking Na+ influx through voltage-gated Na+ channels with 1 μmol/L TTX greatly reduces optic nerve membrane depolarization caused by anoxia or glycolytic block (Leppanen and Stys, 1997b), indicating that the majority of Na+ influx occurs through these channels. Replacement of bath Na+ with an impermeant cation, such as choline or N-methyl-D-glucamine (NMDG), results in an even greater membrane potential sparing effect, suggesting an additional TTX-insensitive component of Na+ influx (Leppanen and Stys, 1997b) (Fig. 4A). Therefore, given the heavy dependence of axonal resting membrane potential on an adequate K+ gradient, one might infer that axoplasmic [K+] is relatively spared and that Na+ channel blockade indirectly prevents axonal K+ loss, i.e., in accordance with the law of electroneutrality, blocking Na+ influx will restrain K+ efflux. Direct measurements of axoplasmic composition using EPMA under these conditions revealed a surprising and unexpected result. Figure 4B summarizes dry weight elemental concentrations from large optic nerve axons exposed to anoxia and either Na+ channel blockade with 1 μmol/L TTX, or bath Na+ replacement with Li+ (which permeates Na+ channels [Hille, 1972; Richelson, 1977], in contrast to choline or NMDG [Stys and LoPachin, 1997]). Concentrations expressed in mmol/kg dry weight represent total amounts of an element in a cellular compartment, independent of the amount of water available for dissolution of this element into an ionized, biologically active state. This distinction becomes important as will be illustrated below. Under anoxic conditions in normal perfusate, axons lose K+ and gain Na+, while [Cl−] does not change. Totally unexpected was the observation that TTX, which significantly reduces resting membrane depolarization (Fig. 4A), does not alter the degree of severe axoplasmic K+ depletion. How then do we reconcile the paradox of relative preservation of membrane potential by TTX versus depletion of intracellular K+ concentration ([K+]i) that is equally as severe as in the absence of Na+ channel blockade? Results obtained by EPMA reveal that axoplasmic water content, although unaltered by anoxia alone, decreases dramatically in anoxic axons treated with TTX (Stys and LoPachin, 1997). The resulting ionized (biologically active) concentration of K+ might then be relatively high even with a sharply reduced total amount of K+, i.e., there would be less cellular water to dilute the residual K+. From the data of Stys and LoPachin (1997) the calculated ionized axoplasmic [K+] is ≈60 mmol/L at the end of a 60-minute anoxic exposure in the presence of TTX. This is enough to maintain K+ equilibrium potential (EK) at −80 mV, which would largely preserve resting membrane potential as was observed. Implicit is the fact that the large loss of axonal water and K+ can only occur with a parallel egress of an anion. As Fig. 4B illustrates, TTX did indeed induce a large loss of Cl− as would be expected with electroneutral flow of K+ and Cl− followed by osmotically obliged water. Taken together, these data demonstrate the movement of Na+ in electroneutral exchange for K+ across the axolemma of metabolically compromised axons in the absence of Na+ channel blockade, with the former ion largely (but not exclusively) flowing through TTX-blockable Na+ channels. Notably, Na+ channel inhibition induces a shift in the pattern of ion movement, from Na+–K+ exchange to K+–Cl−–water coefflux. Under the latter condition, the loss of water and electrolytes causes serious volume alterations with marked shrinkage of the axon cylinder (LoPachin and Stys, unpublished observations) and may cause mechanical disruption of axonal architecture, such as detachment of the paranodal myelin loops with compromise of the electrical integrity of the fiber (see Fig. 4 in Waxman et al., 1994); this may explain the persistently distorted waveshapes in postanoxic TTX-treated optic nerves despite excellent recovery of compound action potential area (see Fig. 1 in Stys et al., 1992b). The route of Cl− efflux from TTX-treated axons is presently unknown, but may be of great importance for therapeutic design, the most successful to date being Na+ channel blockade (see Mechanism-driven therapeutic design, below).
Although preserving the K+ gradient and resting membrane potential on the one hand, the loss of axoplasmic water will also tend to concentrate the small amounts of Na+ remaining in TTX-treated anoxic axons. Calculations show that the ionized axoplasmic [Na+] exceeds 100 mmol/L under these conditions, similar to concentrations measured in untreated anoxic fibers (LoPachin and Stys, 1995), despite a very modest increase in total Na+ (Fig. 4B). As will be discussed in the section on Ca2+ overload, below, a rise in intracellular Na+ concentration ([Na+]i) is a powerful stimulus for Ca2+ overload through reverse Na+–Ca2+ exchange, yet the Ca2+ entry is clearly prevented. This second paradox can be explained by the fact that Ca2+ entry mediated by Na+–Ca2+ exchange is promoted by both a rise in [Na+]i and membrane depolarization, the latter because of the electrogenic nature of the exchanger (Mullins et al., 1985; Fontana et al., 1995). It appears therefore, that neither Na+ overload nor depolarization alone are capable of driving the exchanger strongly enough to cause net Ca2+ accumulation; rather, both excess Na+ influx and collapse of membrane potential appear to be required to activate reverse exchange (Ca2+ import mode) sufficiently to result in axonal Ca2+ accumulation during anoxia. It is conceivable that during both anoxia and glycolytic block (mimicking ischemic conditions), the lesser stimuli individually might be sufficient to induce exchanger-mediated Ca2+ accumulation at a time when glycolytic ATP would be unavailable to fuel, for example, the axolemmal Ca2+-ATPase (Mata and Fink, 1989). For the above reasons, therefore, and in a more realistic clinical setting in which absolute Na+ channel blockade with potent toxins such as TTX is not feasible, adjunctive block of Cl− efflux may be a very important addition to Na+ channel antagonists as this would decrease the internal concentration of Na+ secondary to water loss, thus reducing the second potent stimulus for Ca2+ overload mediated by Na+–Ca2+ exchange, and additionally may help alleviate the mechanical injury caused by axonal volume changes.
Further support for Na+i-induced Ca2+ entry was obtained from anoxic optic nerves treated with Na+-depleted perfusate (Fig. 4B). When Na+ is replaced with Li+ ions, which are permeable at the Na+ channel (Hille, 1972; Richelson, 1977), anoxic Na+ accumulation is prevented as expected, whereas the K+ depletion is actually exacerbated compared with anoxia alone. Given that Na+i stimulates Na+–K+-ATPase activity (Shyjan et al., 1990; Sweadner, 1995), this latter observation is likely a result of the severe reduction of [Na+]i and the known inhibitory effects of Li+ on this pump (Halm and Dawson, 1983; Leppanen and Stys, 1997a). Under these connditions, the small component of Na+–K+-ATPase-mediated K+ accumulation fueled by glycolysis (Fig. 3B) (Leppanen and Stys, 1997b) may be inhibited and anoxic K+ depletion worsened. As with TTX treatment, Ca2+ accumulation is largely prevented, although not to the same extent as with Na+ channel block, suggesting a component of Ca2+ entry directly through TTX-sensitive Na+ channels, which are known to be slightly permeable to this divalent cation (Edwards, 1982). An unexplained observation is a loss of axoplasmic water as seen with TTX and a modest although significant reduction in intracellular Cl− concentration ([Cl−]i) (Stys and LoPachin, 1997), along with a parallel shrinkage of axons as occurs with exposure to TTX (LoPachin and Stys, unpublished observations). Axoplasmic [Cl−] in normoxic optic axons is maintained much higher than what would be expected from passive distribution (Stys et al., 1997a) implying one or more anion transport mechanisms. A plausible candidate for Cl− import would be the Na+–K+–2Cl− co-transporter (Alvarez-Leefmans and Russell, 1990), which could account for Cl− depletion and volume deregulation when a reversed Na+ gradient is artificially imposed.
Invariably, K+ is lost from the axoplasm in anoxic optic axons regardless of any experimental manipulations (Fig. 4B). The channels or carriers responsible for K+ efflux have not been identified. Given the multitude of K+ channel types identified on myelinated axons (Vogel and Schwarz, 1995; Waxman, 1995), it is highly likely that K+ escapes by several routes simultaneously. We have carried out recent experiments using a number of pharmacologic inhibitors of various K+ channel subtypes with little effect on optic nerve responses to anoxia (with one notable exception unlikely to be related to alteration of K+ fluxes; see Ca2+ overload, below) (Stys and Hubatsch, 1996; Stys et al., 1997b), suggesting that such manipulations affected the ionic changes very little. The multiplicity of K+ channels and transporters will make it difficult to identify with certainty which of these pathways mediates anoxic K+ loss from axons, and by extension, would not make a feasible approach for acute therapeutic intervention.
The present section dealt primarily with abnormal fluxes of monovalent ions that are largely responsible for the acute early failure of axonal excitability. The following section will discuss the mechanisms of movement and deleterious effects of Ca2+, arguably the most important ion dictating irreversible injury and recovery.
Ca2+ OVERLOAD: INDUCTION OF INJURY VERSUS AUTOPROTECTION
Loss of electrical excitability as a result of collapse of Na+ and K+ gradients will not necessarily induce irreversible injury. For example, a 10- to 15-minute anoxic exposure completely abolishes electrogenesis in optic nerve and causes marked loss of axoplasmic K+ (LoPachin and Stys, 1995), yet function recovers completely on reoxygenation. Longer anoxic exposures appear to induce additional changes in the fibers that promote irreversible functional damage (Fern et al., 1997). Cellular Ca2+ overload is generally thought to represent a key and fundamental step in the series of events leading to injury from a variety of conditions, such as anoxia and ischemia, or trauma. Indeed, excess Ca2+ accumulation is suggested to be the “final common pathway” leading to necrotic cell death (Schanne et al., 1979; Siesjö, 1986; Orrenius and Nicotera, 1996). Central axons, as well, are injured by excess accumulation of intracellular Ca2+. Studies in optic nerve show that injury can be completely prevented by removing Ca2+ from the perfusate (with the addition of the Ca2+ chelator EGTA) during a 60-minute anoxic exposure (Fig. 5A). The degree of injury is directly proportional both to the [Ca2+] in the bath and to the length of time that the anoxic nerve is maintained in high Ca2+ conditions (Stys et al., 1990b). Supportive ultrastructural studies indicate dissolution of the axonal cytoskeleton, swollen mitochondria, and retraction of paranodal myelin as correlates to physiologic injury (Waxman et al., 1992). Removal of Ca2+ completely protects the structural integrity of the axonal cytoskeleton, but mitochondria continue to exhibit abnormal swellings (Waxman et al., 1993). Physiologic studies suggest that Ca2+ influx into anoxic axons is gradual and continues throughout a 60-minute anoxic challenge. Subsequent experiments measuring axoplasmic Ca2+ with EPMA confirmed a substantial and continuous influx of Ca2+ during 60 minutes of anoxia in optic nerve fibers (Fig. 4B) (LoPachin and Stys, 1995). Although the above data are derived from optic nerve axons, the Ca2+-dependency of anoxic axonal injury is likely a generalized phenomenon. A recent study in dorsal columns of the rat spinal cord also confirmed the importance of extracellular Ca2+ for anoxic injury in this white matter tract (Imaizumi et al., 1997). We also have very recent data implicating intracellular Ca2+ stores as a potential source of damaging intracellular Ca2+ concentration ([Ca2+]i) increase (Steffensen and Stys, 1996). Together, these results indicate that excess accumulation of Ca2+ in the axoplasm, largely from influx across the axolemma, with possibly an additional component derived from uncontrolled release of internal Ca2+ stores, is a pivotal event in the induction of anoxic damage in CNS axons.
(A) Bar graph showing recovery of optic nerve CAP area recorded by suction electrodes after 1 hour of in vitro anoxia and 1 hour of reoxygenation. CAP area recovers to ≈25% of preanoxic values (“control”). Removing Ca2+ from the bath and adding the chelator EGTA (5 mmol/L) allows virtually complete recovery after 1 hour of anoxia, suggesting that extracellular Ca2+ flows across cell membranes and accumulates in the axoplasm (see Fig. 4). The route of Ca2+ entry does not involve L-type Ca2+ channels because dihydropyridines, such as nifedipine (“nif 5 μmol/L”) or nimodipine had no protective effects. Extracellular Na+ is required for injury because replacement with either the permeant Li+ ion or impermeant cations, such as choline or N-methyl-D-glucamine, were highly protective. The protective effect of TTX indicates that TTX-blockable channels play a major role in this type of injury; conversely, increasing permeability of these channels with the activator veratridine (“ver 1 μmol/L”) increases the amount of damage. Pharmacologic inhibition of the Na+–Ca2+ exchanger with bepridil (“bep 50 μmol/L”) is also protective, implicating this transporter in the injury cascade. Curiously, blocking inward rectifier channels with Cs+ is also beneficial. These channels may play a role by virtue of their known Na+ permeability. See text and Fig. 8 for a synthesis of these observations. All manipulations were begun before the start of anoxia, and continued during the anoxic period. 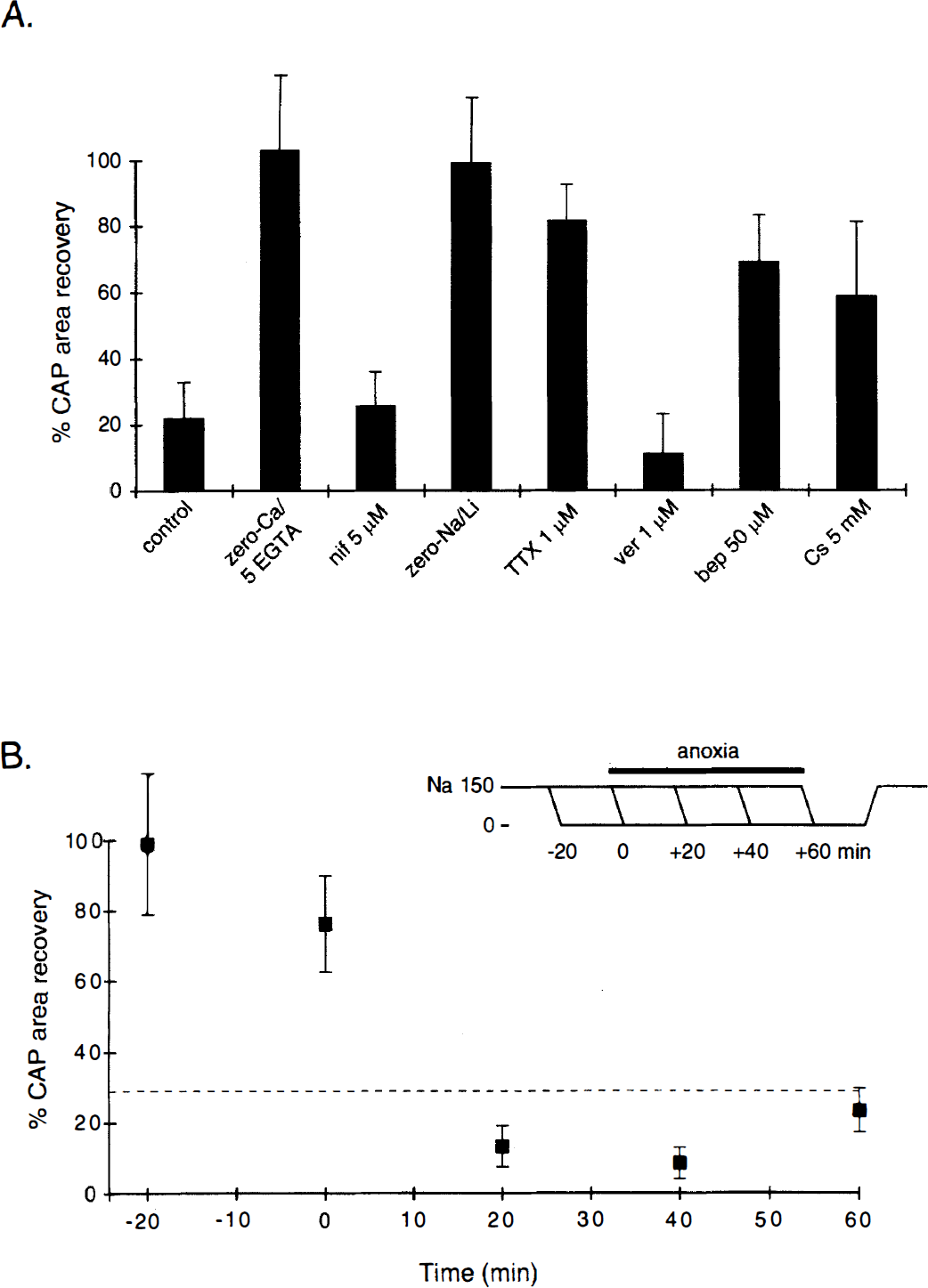
Given the unique structure of myelinated fibers and the lack of synaptic machinery, extrapolating to white matter what is known about Ca2+ influx in anoxic and ischemic gray matter may not be applicable. In general terms, Ca2+ can enter a cell across the plasma membrane through several routes: (1) voltage- or transmitter-gated Ca2+ channels, (2) leakage through imperfectly selective channels nominally permeable to other ions, (3) coupled transporters such as the Na+–Ca2+ exchanger, or (4) nonspecific leakage through the cell membrane. In contrast to the neuronal soma and dendrite, in which a large portion of pathologic Ca2+ entry has been shown to occur through NMDA and certain subtypes of α-amino-3-hydroxy-5-methyl-4-isoxasole propionic acid (AMPA) receptors (Choi, 1988; Silver and Erecinska, 1990; Pellegrini-Giampietro et al., 1992; Tymianski et al., 1993; Brorson et al., 1994), optic nerve axons are not injured even after prolonged exposure to high glutamate concentrations (with glycine added and in the absence of Mg2+, to maximize permeability through NMDA receptors) (Stys, Ransom, and Waxman, unpublished observations). The reported protective effect of ketamine (Ransom et al., 1990), an NMDA antagonist (Thomson et al., 1985), in anoxic optic nerve was likely nonspecific and related to this agent's known blocking effects of voltage-gated Na+ channels at higher concentrations (Frenkel and Urban, 1992) (see below). An effect of ketamine on glia (Chan and Chu, 1989), which exhibit [Ca2+]i fluctuations in response to glutamate in optic nerve (Kriegler and Chiu, 1993; Chiu and Kriegler, 1994), cannot, however, be excluded (see Glial cell injury, below). Interestingly, protective effects of AMPA or NMDA receptor antagonists have been reported in models of in vivo spinal cord ischemia (Madden et al., 1993; Bowes et al., 1996). Because of the increased complexity of an in vivo system, it is unclear whether these antagonists exerted their actions on the spinal gray or white matter. Interestingly, it has been suggested that the juxtaposition of gray and white matter alters the susceptibility of the latter tissue to anoxia and ischemia, possibly because of diffusion of factors such as γ-aminobutyric acid (GABA) (Lee et al., 1993). Therefore glutamate antagonists may improve ischemic spinal cord function indirectly by protecting the spinal gray matter.
On balance, it appears unlikely that CNS axons are directly damaged by excitatory neurotransmitters during anoxia and ischemia, in stark contrast to neuronal cell bodies and synaptic elements. Alternatively, Ca2+ influx may occur through voltage-gated Ca2+ channels. However, the existence of these channels has never been shown convincingly in myelinated fibers. Voltage-gated Ca2+ channels have not been detected by patch clamp studies in peripheral axons (Vogel and Schwarz, 1995) nor by electrophysiologic experiments in CNS fibers (Foster et al., 1982). Although activity-dependent Ca2+ transients have been demonstrated in optic nerve (Lev-Ram and Grinvald, 1987), it is unclear to what extent glia contributed to the signals; even axonal signals may have been related to depolarization-induced Ca2+ entry mediated by Na+–Ca2+ exchange, as has been shown in squid giant axons (Mullins et al., 1985). Consistent with the lack of physiologic evidence for Ca2+ channels in myelinated fibers, a study on anoxic rat optic nerve revealed no protection using nonspecific Ca2+ channel blockers, such as Co2+, Mn2+, or La3+, nor did more specific inhibition of L-type Ca2+ channels with dihydropyridines (nifedipine, nimodipine) improve outcome (Stys et al., 1990a) (Fig. 5A). Although the results with the organic blockers convincingly show that L-type Ca2+ channels do not contribute significantly to optic nerve anoxic injury in this study, normoxic controls with the polyvalent inorganic ions were not performed, and so any potential toxic effects of these transition elements (Fern et al., 1995a) may have masked a modest protective action. In contrast, a subsequent study reexamined the effects of voltage-gated Ca2+ channel blockers in greater detail and reported beneficial effects (Fern et al., 1995a). These investigators found that three types of L-type Ca2+ channel antagonists (diltiazem, verapamil, and nifedipine) each conferred protection to anoxic optic nerve. In addition, protective effects of ω-conotoxins implicated N-type Ca2+ channels. Unfortunately, none of these L-channel blockers is specific, having effects on other mechanisms that may be directly related to the genesis of axonal injury. For instance, all three agents block Ca2+ release from a subtype of intracellular Ca2+ store (Genazzani et al., 1996); if release from this pool contributes to axonal damage (Steffensen and Stys, 1996), then this may at least partially explain the observed effects. These agents also block Na+–Ca2+ exchange (Hata et al., 1988; Canzoniero et al., 1993), which is a key mechanism for Ca2+ overload in anoxic axons (see below). Also, verapamil has been shown to be a potent use-dependent blocker of voltage-gated Na+ channels (Ragsdale et al., 1991), which is a key step in the anoxic cascade (see further discussion below). Finally, we have recently shown with EPMA that neither nifedipine nor nimodipine reduces the accumulation of axonal Ca2+ during anoxia (Stys and LoPachin, 1997). Surprisingly, nifedipine reduces Ca2+ entry in some axons, but only in the context of a diminished Na+ accumulation. This likely represents an indirect effect on the Na+–Ca2+ exchanger, and also raises questions about the specificity of this compound (Dargent et al., 1996). Taken together, although the data available to date on the role of voltage-gated Ca2+ channels in permeating significant amounts of Ca2+ in anoxic central myelinated fibers are suggestive (e.g., ω-conotoxin-induced protection through N-type Ca2+ channel block), the contribution of other Ca2+ channel subtypes is controversial and inconclusive, and a final determination requires further study.
What other mechanism might be responsible for Ca2+ overload in metabolically compromised fibers? Studies on rat optic nerves have shown that influx of extracellular Na+ is also of great importance for anoxic injury (Fig. 5A). Replacing bath Na+ with choline or Li+ ions allows virtually complete recovery of compound action potential magnitude after a 60-minute anoxic exposure, although waveshapes remain somewhat distorted (Stys et al., 1992b). Although choline ions cannot pass through Na+ channels, Li+ is permeable, but cannot substitute for Na+ at the Na+–Ca2+ exchanger (Fontana et al., 1995). Together with the Ca2+-depleted experiments, these findings indicate that Na+ and Ca2+ movements may be causally linked, and the common mechanism might be Na+–Ca2+ exchange. Subsequent EPMA studies confirmed that Na+-depleted conditions greatly (but not completely) reduce axoplasmic Ca2+ accumulation in anoxic axons, providing strong evidence for Na+-coupled Ca2+ entry by the Na+–Ca2+ exchanger, a ubiquitous ion transporter that has been demonstrated immunohistochemically in peripheral and central myelinated fibers (Steffensen et al., 1997). As shown further in Fig. 5B, applying zero-Na+ solution after the above onset of anoxia, when axoplasmic [Na+] has risen to normal (LoPachin and Stys, 1995), increases the degree of injury because a strong reversed [Na+] gradient is now imposed, driving Na+–Ca2+ exchange even more strongly to import deleterious Ca2+ (Stys et al., 1992b). Together with studies using pharmacologic blockers of Na+–Ca2+ exchange (Stys et al., 1992b; Imaizumi et al., 1997; Stys and LoPachin, 1997), these observations provide strong support for this antiporter as the main, but not the sole, mechanism of Ca2+ overload in anoxic CNS axons.
Interestingly, there appear to be parallel routes for both Na+ and Ca2+ entry into compromised axons. Although the majority of Na+ enters through TTX-inhibitable voltage-gated Na+ channels (Stys et al., 1992b; Stys and LoPachin, 1997), a small component permeates through a TTX-insensitive route (Stys and LoPachin, 1997), possibly a Cs+-sensitive inward rectifier (Stys et al., 1997b), which is known to permeate both K+ and Na+ ions (Eng et al., 1990; Poulter et al., 1993; Solomon and Nerbonne, 1993). Conversely, whereas most of the inward Ca2+ movement is mediated by Na+–Ca2+ exchange operating in the Ca2+ import mode, a small proportion enters directly through Na+ channels (Stys and LoPachin, 1997), which are known to possess some permeability to Ca2+ ions (Edwards, 1982); in anoxic optic nerve, TTX block of Na+ channels results in less axonal Ca2+ accumulation than exposure to zero-Na+ perfusate (when Na+ channels would remain unblocked), even though axonal [Na+], particularly ionized [Na+] (see Disruption of axonal ion homeostasis, above), rises much more with Na+ channel block (which should promote more Ca2+ entry by reverse Na+–Ca2+ exchange) than with zero-Na+ exposure (Stys and LoPachin, 1997). A recent study in anoxic dorsal columns seems to support a component of Ca2+ influx through Na+ channels as well. Injury in this tissue is also largely produced by a Na+-dependent Ca2+ influx, but the degree of physiologic recovery is substantially greater with Na+ channel block using TTX than with removal of extracellular Na+ (Imaizumi et al., 1997); this finding is consistent with significant components of Ca2+ entry occurring through both TTX-blockable Na+ channels, as well as reverse Na+–Ca2+ exchange.
The subcellular axoplasmic distribution of Na+ and Ca2+ in anoxic fibers has not been measured, although axial gradients of both ions extending from nodes of Ranvier might be expected given the high density of Na+ channels in this region. Interestingly, neonatal optic nerves that are not yet myelinated and have continuously distributed Na+ channels (versus high-density collections at nodes of myelinated adult fibers) are resistant to anoxic injury (Davis and Ransom, 1987; Fern et al., 1997). Similarly, amyelinated optic nerve axons from myelin-deficient mutants are also able to withstand anoxic insults (Waxman et al., 1990). Although a number of factors may contribute to the amyelinated axon's resistance to injury, a common feature is lack of myelin and corresponding low-density, continuous distribution of Na+ channels, which have been shown to be pivotal in the genesis of axonal damage. It is instructive to consider how Na+ channel densities may play an important role in determining the site and extent of injury. Comparing an amyelinated neonatal rat optic axon (mean diameter, 0.22 μm; Na+ channel density, 2 per μm2 [Waxman et al., 1989]) with a larger myelinated adult optic nerve fiber (diameter, 2 μm; nodal length and Na+ channel density, 1 μm and 1,500 per μm2; internodal length and density, 200 μm and 20 per μm2 (Hildebrand and Waxman, 1984; Waxman, 1995)) we can calculate the number of Na+ channels per unit axoplasmic volume (as these channels will cause Na+ loading into the axon cylinder's volume causing a [Na+] change). Surprisingly, both neonatal and adult fibers (nodal and internodal regions considered together in the latter) have identical mean densities of ≈35 Na+ channels per μm3 axoplasm. However, myelinated fibers display high nodal densities (3,000 channels per μm3 nodal axoplasm), about 100-fold greater than amyelinated axons or internodal regions of adult fibers. Considering that there likely exist significant barriers to ionic diffusion (Bergman, 1970), localized ionic disturbances at the node of Ranvier may render this region particularly vulnerable to injury, and may at least partially explain why fibers with homogeneous distributions of Na+ channels at lower densities are more resistant.
Although the specialized architecture of myelinated axons may render them especially sensitive to injury, there is some evidence that these fibers possess autoprotective mechanisms, possibly designed to limit irreversible damage under conditions of metabolic stress. Fern and colleagues discovered that GABA-B receptor activation in adult rat optic nerves was partially protective against anoxic injury (Fern et al., 1995b). This is in contrast to GABA-A stimulation, possibly related to diffusion of GABA from hypoxic gray matter in the in vitro hemisected spinal cord preparation, which increases functional injury (Lee et al., 1993). The favorable GABA-B effect is caused by endogenously released transmitter, since it can be reproduced with nipecotic acid, an inhibitor of GABA uptake. This signal transduction pathway appears to involve a G-protein and protein kinase C-mediated mechanism, although neither the final phosphorylation targets nor the source (axonal or glial) of the transmitter has been conclusively identified. These investigators also demonstrated that a second neuromodulator, adenosine, is also released during white matter anoxia and confers partial neuroprotection through a protein kinase C-dependent pathway (Fern et al., 1994). The putative signal transduction pathways involved in white matter autoprotection are illustrated in Fig. 6 (Fern et al., 1996b). Data from our laboratory also point to potential autoprotective mechanisms. Optic nerves exhibit hyperpolarizing deflections of compound resting membrane potential during glycolytic inhibition (but not anoxia alone) that may be related to activation of a Ca2+-dependent K+ conductance (Leppanen and Stys, 1997b). Moreover, the voltage trajectory of resting membrane potential during anoxia or glycolytic block consistently undergoes an abrupt, Ca2+-dependent reduction of the rate of depolarization after several minutes, which may be the result of a downregulation of axonal Na+ conductance (Leppanen and Stys, 1997b). Indeed, Ca2+-dependent activation of protein kinase C has been shown to reduce Na+ currents in reconstituted rat brain Na+ channels (Murphy and Catterall, 1992; Li et al., 1993). Reducing the extent of depolarization would limit the amount of deleterious Ca2+ admitted through the electrogenic Na+–Ca2+ exchanger. One could further speculate that the protein kinase C-mediated GABA-B- and adenosine-induced protection described above might be the result of Na+ channel downregulation, thus unifying the above observations. Proof of such an intriguing mechanism must await further study. Collectively, these results raise the interesting concept of several or many endogenous neuromodulators, normally inactive with respect to axonal conduction, destined to be released during pathologic conditions in an attempt to limit injury. Pharmacologic modulation of such endogenous mechanisms may represent an intriguing therapeutic strategy.
Diagram illustrating potential signal transduction pathways mediating autoprotection in anoxic white matter. During anoxia, Na+ influx occurs through noninactivating Na+ channels and, together with depolarization caused by internal K+ depletion, promotes reverse Na+–Ca2+ exchange-mediated Ca2+ overload. Anoxia also induces release of γ-aminobutyric acid (GABA) and adenosine from as yet unidentified sources in optic nerve. Extracellular accumulation of these neuromodulators causes receptor-coupled activation of a G-protein and protein kinase C cascade that confers partial resistance to anoxia. The targets of protein kinase C phosphorylation are unknown but could involve the Na+–Ca2+ exchanger, Na+ channels, inward rectifier, or any number of other components in the sequence of events leading to irreversible anoxic and ischemic injury (see Fig. 8). This raises the interesting possibility of harnessing endogenous mechanisms to improve outcome after anoxia and ischemia. Modified from Fern et al., 1996b with permission from Karger, Basel, Switzerland.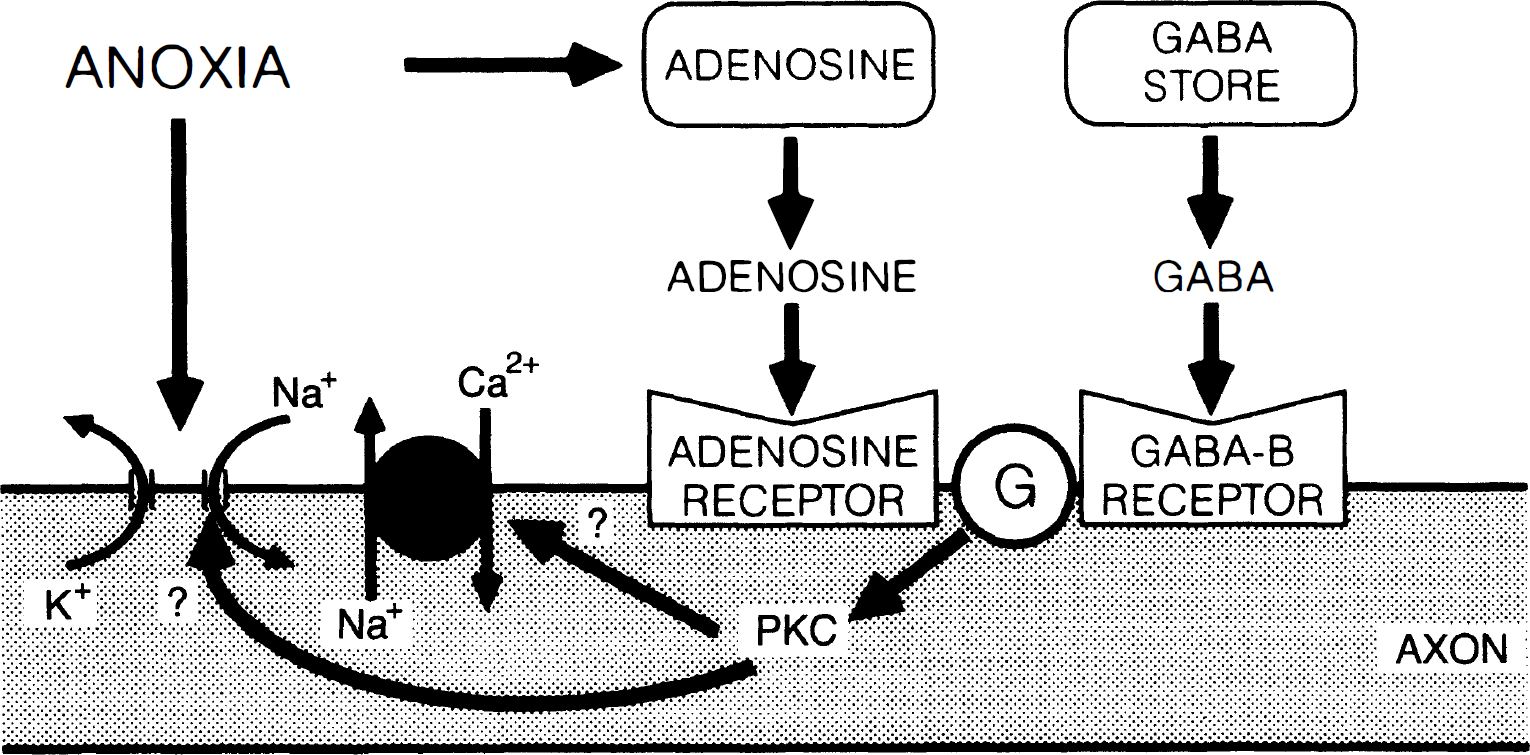
REOXYGENATION: RECOVERY VERSUS MATURATION OF INJURY
Inhibition of energy-producing metabolic pathways, particularly of oxidative phosphorylation, causes a rapid loss of excitability in central axons, with a parallel depolarization of resting membrane potential and collapse of ionic gradients (LoPachin and Stys, 1995; Stys, 1996; Fern et al., 1997; Imaizumi et al., 1997; Leppanen and Stys, 1997b). However, neither the rapidity nor the degree of acute loss of electrogenesis is necessarily predictive of functional recovery after reoxygenation after an anoxic exposure. For instance, brief exposures of optic nerves to anoxia with or without hypoglycemia produce total loss of excitability, yet allow complete recovery when a normal environment is reintroduced. Moreover in the same model, loss of excitability is more rapid in low-Ca2+ conditions (Stys, Waxman, and Ransom, unpublished observations), and deregulation of axonal [K+] and [Na+] is as severe as during normocalcemic conditions (Stys and LoPachin, 1997), yet recovery is nearly complete even after 60 minutes of anoxic exposure (Stys et al., 1990b). Therefore, acute loss of excitability is more symptomatic of energy failure, and does not necessarily predict outcome. Other processes that occur during anoxia, namely the gradual and significant accumulation of cellular Ca2+, initiate a number of destructive mechanisms that may be paradoxically stimulated at the time of reoxygenation and reperfusion.
After a period of in vitro anoxia, electrophysiologic function of adult rat optic nerves gradually recovers during the reoxygenation phase; the magnitude of the compound action potential (CAP) attains a plateau about 30 to 40 minutes after anoxia, with shorter anoxic exposures promoting a more rapid and extensive recovery of CAP magnitude (Stys, 1996; Fern et al., 1997). The area under the CAP after a standard 60-minute in vitro anoxic challenge typically recovers to 20% to 30% of preanoxic control area, and fails to improve further even with prolonged in vitro reoxygenation (Fig. 7A) (Stys, 1996). Given the rather steep relationship of Na+ channel inactivation as a function of membrane potential (Chiu et al., 1979; Kostyuk et al., 1981), it is more likely that the fractional CAP recovery reflects populations of fibers that function almost normally compared with others that are completely dysfunctional and unable to propagate action potentials; it is unlikely that impaired fibers, with intermediate resting potentials, would contribute “subnormal” spikes to form a reduced CAP. Indeed, EPMA studies have revealed intriguing differences in anoxic susceptibility among populations of axons even within a fairly homogeneous tract such as the optic nerve. Large optic axons (>2 μm), which are most resistant to acute loss of excitability at the onset of anoxia (Fig. 3A), appear to be most sensitive to exacerbation of injury after reoxygenation (Stys, 1996; Stys and LoPachin, 1996). Although a significant number of large axons make an initial attempt at restoring K+ gradients toward normal within 20 to 40 minutes of reoxygenation, they invariably undergo a secondary deterioration as reoxygenation continues, so that after 1 to 3 hours of reoxygenation, the degree of deregulation of axoplasmic Na+, K+, and Ca2+ far exceeds even that seen during anoxia (Fig. 7B). Ischemic spinal cord axons have also been shown to accumulate large quantities of Ca2+ in the axoplasm and paranodal myelin during reperfusion (Jalc et al., 1995). The reasons for this secondary deterioration are not understood, but may represent important mechanisms of reoxygenation-induced injury. Generation of free radicals at the time of reoxygenation may cause additional injury to critical cellular components, such as cell membranes, key ion transporters (Rohn et al., 1993), and mitochondria (Crompton and Andreeva, 1993). Paradoxically, restoration of cellular ATP levels in an abnormal ionic environment may be deleterious. For instance, Ca2+ import through reverse Na+–Ca2+ exchange is significantly enhanced by intracellular ATP (DiPolo and Beauge, 1987; DiPolo and Beauge, 1988). In the presence of high axonal [Na+]i (LoPachin and Stys, 1995) and marked depolarization (Leppanen and Stys, 1997b) (both of which will strongly favor Na+–Ca2+ exchange-mediated Ca2+ entry), resumption of ATP synthesis at the time of reoxygenation and reperfusion in a potentially viable fiber will upregulate exchanger operation, thus potentially causing an additional Ca2+ load from which the axon is unable to recover; this may result in a secondary deterioration of ionic gradients and further Ca2+ entry (Stys and LoPachin, 1996). This in turn might incite further mitochondrial compromise leading to secondary, and final, cellular energy failure (see below).
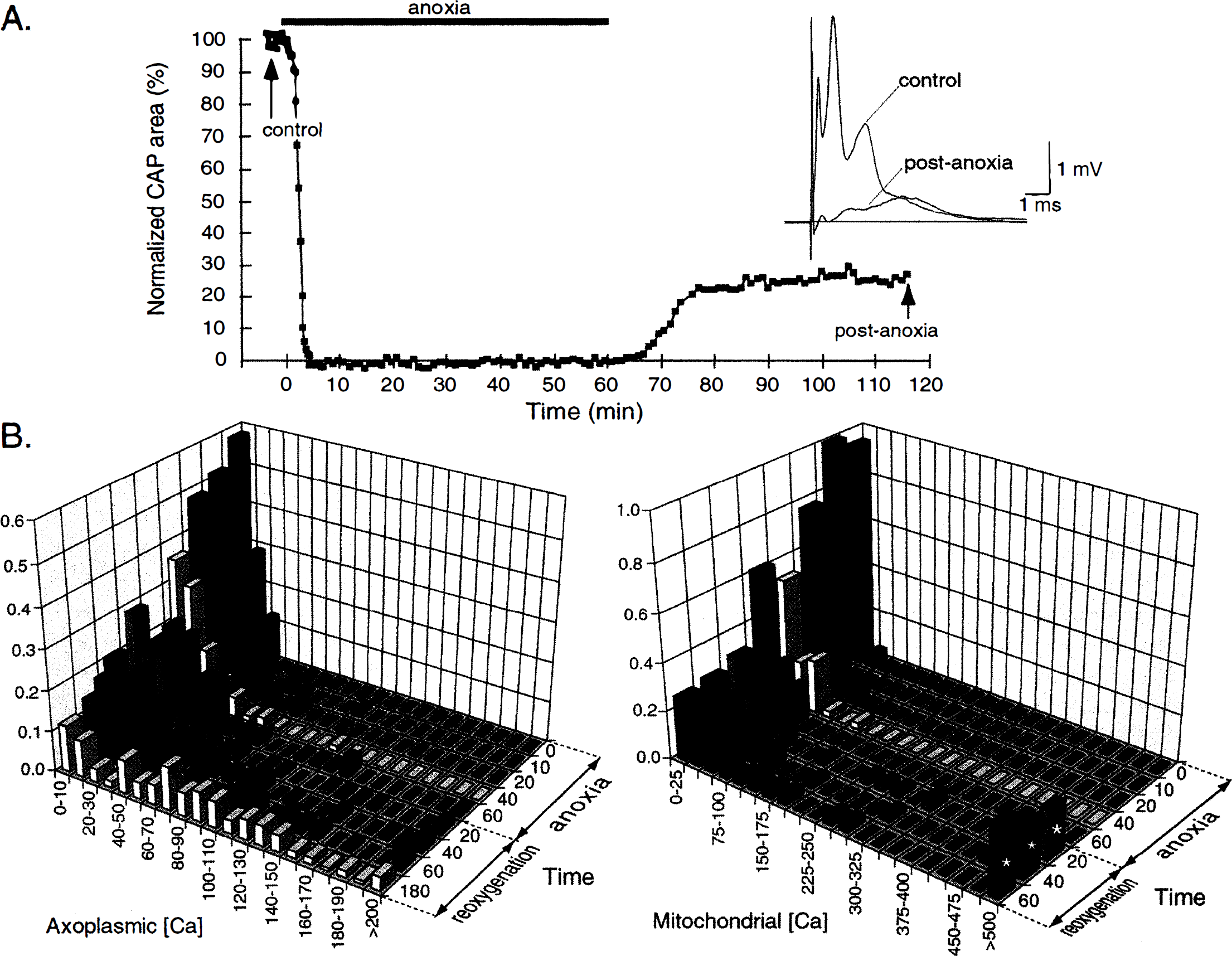
In contrast to large optic axons, about half of the small (<1 μm) fibers achieve normal or near normal ionic composition after reoxygenation (Stys and LoPachin, 1996). Interestingly, these small fibers are the first to be acutely silenced by anoxia (Fig. 3A). Electrophysiologic studies are less capable of discerning population differences within an axon bundle such as the optic nerve, but compound recordings often suggest a better outcome for smaller, slower fibers (Fig. 7A), in keeping with the EPMA data. The reasons for these population differences are unknown, but may be fundamentally important for understanding how to render axons more resistant to anoxia and reoxygenation cycles and to allow larger fibers to take better advantage of reoxygenated conditions.
Given the overwhelming reliance of myelinated axons on ATP generated through oxidative phosphorylation, the rapid collapse of resting membrane potential and loss of excitability within minutes of anoxia in turn attests to a rapid depletion of axonal ATP and discharge of the proton gradient across the inner mitochondrial membrane. Mitochondrial Ca2+ accumulation during anoxia in optic axons is surprisingly modest, increasing only about three- to fourfold after 60 minutes (LoPachin and Stys, 1995). These organelles accumulate Ca2+ through an electrophoretically driven uniporter (Crompton, 1985; Nicholls, 1985); therefore, collapse of the potential across the inner mitochondrial membrane during anoxia probably limits the amount of Ca2+ influx into the matrix. In contrast, reoxygenation promotes an extreme rise of mitochondrial Ca2+, increasing to over 100 times the amount of total Ca2+ measured in preanoxic organelles (LoPachin and Stys, 1995; Stys and LoPachin, 1996), in agreement with observations in other tissues (Allen et al., 1993). This Ca2+ increase is accompanied by a rise in phosphorus concentration, likely representing entry of phosphate ions, which then complex with Ca2+, forming insoluble precipitates that may damage the mitochondria (Carafoli, 1986). Ca2+ is preferentially accumulated into mitochondria at reoxygenation, rather than during anoxia, because reintroduction of oxygen will restart electron transport, thus restoring the mitochondrial proton gradient and membrane potential, which is the major driving force for Ca2+ uptake by the uniporter. The high cytosolic Ca2+ environment to which mitochondria are exposed during reoxygenation may be deleterious for more subtle reasons. It has been shown that mitochondrial Ca2+ accumulation occurs at the expense of ATP synthesis, because compensatory protons must be extruded by the electron transport chain to neutralize the positive charge carried in by Ca2+, thereby dissipating the electrochemical proton gradient required for ATP production by ATP-synthase. Indeed, with a sufficiently elevated extramitochondrial [Ca2+], brain mitochondria divert all of their respiratory effort from ATP synthesis to Ca2+ accumulation (Nicholls, 1978; Nicholls, 1985). Moreover, the high extramitochondrial [Na+] will strongly drive the mitochondrial Na+–Ca2+ exchanger (Cox and Matlib, 1993; Jung et al., 1995; White and Reynolds, 1997) to remove Ca2+ from the matrix, thus promoting a wasteful Ca2+ cycling across the inner mitochondrial membrane at the expense of the proton gradient that is now vital for the prompt resumption of aerobic ATP synthesis. Even worse, the influx of Ca2+ through the high-capacity uniporter may be large enough to lower the proton electrochemical gradient to such an extent that the back-pressure on the ATP-synthase is relieved to the point that glycolytic ATP is consumed by this enzyme to transport protons, rather than to generate ATP (Rossi and Lehninger, 1964; Nicholls, 1985). This could conceivably result in an energy deficit that is worse than during the anoxic period itself. In addition, high matrix Ca2+ levels have been implicated in the mitochondrial pore transition, a phenomenon whereby the inner membrane becomes nonselectively permeable to many solutes, further accelerating the rundown of the proton gradient (Gunter and Pfeiffer, 1990; Crompton and Andreeva, 1993). Taken together, these arguments propound the following hypothesis: pathologically elevated cytosolic [Na+] and [Ca2+] produced during the anoxic period conspire to prevent reoxygenated mitochondria from generating sufficient ATP to allow potentially viable axons to recover from the original insult, committing these fibers to an inexorable cycle of “chemically induced” anoxia despite adequate oxygen availability, thus perpetuating the damaging events set in motion by the initial oxygen deprivation.
The unfortunate combination of high axoplasmic [Ca2+] and [Na+], with oxygen available for mitochondrial respiration, will additionally promote formation of large amounts of highly reactive oxygen species, such as hydroxyl radicals (Dykens, 1994). Elevated [Ca2+] will also stimulate xanthine oxidase with the production of superoxide and hydrogen peroxide (Patt et al., 1988; Kinuta et al., 1989). Such free radical species can cause significant damage through peroxidation of membrane lipids with release of injurious arachidonic acid and inhibition of a variety of proteins, including ion channels and transporters, enzymes involved in energy metabolism, and even those designed to detoxify free radical molecules, as well as causing damage to nuclear DNA (Haddad and Jiang, 1993; Hall, 1996). For these reasons, antioxidants have been studied in the hope of limiting this portion of cellular damage induced by anoxia and reoxygenation. Although the role of free radical generation has not been specifically examined in detail in ischemic central axons, a contribution of this type of injury is strongly suggested by results with spinal cord trauma and posttraumatic ischemia, which show that agents that interfere with free radical generation and reactivity are beneficial (Hall and Braughler, 1986). Collectively, it is evident that the events triggered by reoxygenation and reperfusion may be as important as those occurring during the anoxic and ischemic period. From a therapeutic perspective, in which compromised tissue must be successfully reperfused to restore function, it will be very important to study further the precise details of how to minimize the deleterious effects that are initiated by anoxia and ischemia, and then paradoxically exacerbated through additional mechanisms at the termination of the insult.
GLIAL CELL INJURY
Most of the previous discussions have focused on events affecting the axon per se. However, myelinated axons of white matter are dependent on a stable and intimate relationship with their surrounding oligodendrocytes and astrocytes. Therefore compromise of the supporting glia by anoxia or ischemia would be expected to adversely affect the structure and function of the fiber. Although mammalian astrocytes and neurons have comparable metabolic rates (Hertz and Peng, 1992), the former are far more resistant to oxygen deprivation than neurons or axons. For instance, cultured murine astrocytes are able to withstand hypoxia for several days, but become injured after only one day of aglycemia under normoxic conditions (Goldberg and Choi, 1993). This is in stark contrast to central neurons and axons, which suffer severe injury after much shorter periods of anoxia or aglycemia (Facci et al., 1990; Stys et al., 1990b; Tasker et al., 1992; Fern et al., 1997; Silver et al., 1997). Even combined anoxia and aglycemia damages astrocytes only after more than an hour of exposure, whereas this same treatment causes severe damage to neurons and axons after several tens of minutes (Fern et al., 1997; Silver et al., 1997). Studies on intracellular ionic balance in white matter glia are consistent with the relative resistance of these cells to anoxic exposure. Whereas anoxic central axons accumulate large quantities of Na+ and Ca2+ in parallel with a severe loss of K+, glia display far more modest changes, which become significant only after 60 minutes of exposure; indeed, only Na+ and K+ contents are affected, with no net Ca2+ accumulation, indicating that these cells are able to control internal levels of this important cation even during impaired oxidative metabolism (LoPachin and Stys, 1995). Similar observations were made in cultured glia exposed to the mitochondrial inhibitor rotenone (Silver et al., 1997). EPMA examination of myelin during anoxia also shows a relatively modest rise in Na+ and loss of K+, but in contrast to glial cell bodies and proximal processes, myelin accumulates significant Ca2+, which increases nearly fourfold at the end of a 60-minute anoxic exposure (LoPachin and Stys, 1995). This may point to the more vulnerable nature of oligodendrocytes and related myelin, likely because of this cell's susceptibility to free radical damage, sensitivity to glutamate toxicity (possibly by activation of AMPA or kainate receptors) and a greater dependence on oxidative phosphorylation (Wender et al., 1988; Oka et al., 1993; Husain and Juurlink, 1995; Juurlink, 1997; McDonald et al., 1998).
Although glia are able to withstand inhibition of oxidative phosphorylation, especially in high ambient glucose (Kelleher et al., 1993), concomitant impairment of glycolysis greatly increases glial dysfunction (Ransom and Fern, 1996). This is particularly pronounced with treatments that impair metabolism of glucosyl units derived from glycogen. For instance, the deregulation of glial ion homeostasis is accelerated from hours under conditions of aglycemia alone, to minutes when the inhibitor 2-deoxyglucose is added to the glucose-free medium (Silver et al., 1997). This nonmetabolizable glucose analog impairs the utilization of glycogen (Dringen and Hamprecht, 1993), underscoring the importance of this energy reserve known to be present in large quantities in glial cells, but not in neurons (Koizumi, 1974; Swanson et al., 1989). The mechanisms of glial cell injury from energy deprivation are not well understood, but appear to involve entry of extracellular Ca2+, together with peroxidation of membranes, cellular edema, and extracellular acidification leading to intracellular acidosis (Yu et al., 1989; Ransom et al., 1992; Pappas and Ransom, 1995; Ransom and Fern, 1996). The precise mechanisms of Ca2+ entry into astrocytes are not known, but may involve permeation of this ion through L-type voltage-gated Ca2+ channels and possibly release from internal stores (MacVicar, 1984; Yu et al., 1989; Pappas and Ransom, 1995; Duffy and MacVicar, 1996). Reverse operation of the Na+–Ca2+ exchanger (Takuma et al., 1994; Holgado and Beauge, 1995; Holgado and Beauge, 1996), may also contribute to Ca2+ entry, especially under conditions of elevated [Na+]i and depolarization documented in glia subjected to severe energy depletion (Silver et al., 1997). As with astrocytes, oligodendroglia are known to be sensitive to injury from Ca2+ influx (Scolding et al., 1992), and the presence of extracellular Ca2+ promotes structural injury in oligodendrocytes of anoxic optic nerve (Waxman et al., 1992; Waxman et al., 1993). The mode of Ca2+ entry into these cells, however, is not well understood.
In contrast to neurons, NMDA receptors probably do not contribute to glial [Ca2+]i increase, but AMPA-kainate, as well as metabotropic glutamate, receptors likely do (Kriegler and Chiu, 1993; Blankenfeld et al., 1995). In this regard, Kriegler and Chiu observed an influence of optic nerve axons on glial [Ca2+] (Kriegler and Chiu, 1993; Chiu and Kriegler, 1994). These investigators proposed a physiologic role for axoglial signaling during normal action potential propagation, whereby glutamate released from activated fibers would in turn evoke glial Ca2+ transients through AMPA-kainate and metabotropic glutamate receptors. This nonsynaptic release of glutamate from the axon is likely mediated by reversal of the electrogenic Na+–K+–glutamate transporter (Nicholls and Attwell, 1990). One might speculate that during anoxia and ischemia, axonal depolarization, together with collapse of the transaxolemmal Na+ and K+ gradients, will strongly promote glutamate efflux through this transporter, adversely affecting surrounding glia (Fig. 8). It is interesting to note that myelin regions in particular tend to accumulate Ca2+ during anoxic exposure, rather than more proximal glial processes or somata (LoPachin and Stys, 1995). If the above model of glutamate release from impaired axons is correct, it is precisely the enveloping myelin sheath that would be exposed to potentially large concentrations of transmitter in the restricted internodal submyelinic and interlamellar spaces; this may explain the observed accumulation of net Ca2+ in the sheath. This mechanism could partially underlie the subcortical ischemic white matter lesions (so-called leukoaraiosis) seen in elderly patients, in which oligodendroglia and the myelin sheath are preferentially destroyed (Akiguchi et al., 1997; Pantoni and Garcia, 1997), and perhaps also the demyelination seen in less frequent conditions such as carbon monoxide or cyanide poisoning (Hirano et al., 1967; Korthals et al., 1973). Although quite plausible, the concept of axon-derived glial injury is theoretical only, based on observed physiologic axoglial relationships; further study will be required to confirm such events during actual pathologic conditions. Irrespective of the exact mechanisms of glial damage, it is evident that a detailed understanding of how glia are injured will be important for maximizing recovery of white matter tracts after injury. For a more detailed discussion of anoxic and ischemic glial injury, the reader is referred to an excellent recent review (Ransom and Fern, 1996).
Diagram illustrating sequence of interrelated events leading to anoxic injury of a central myelinated axon. Interruption of oxygen or glucose supply, or both, leads to depletion of ATP stores, resulting in failure of ATP-dependent pumps, such as axolemmal Na+–K+-ATPase (1) and endoplasmic reticulum Ca2+-ATPase (2). There is preliminary evidence that uncontrolled release of Ca2+ from internal stores may contribute to axonal injury. Reduction of Na+ pumping across the axolemma leads to accumulation of axoplasmic Na+ mainly through noninactivating Na+ channels (3). These channels probably also allow entry of Ca2+ directly. The rise in [Na+]i coupled with depolarization caused by K+ efflux through a variety of K+ channels (8), stimulates the Na+–Ca2+ exchanger to operate in the Ca2+ import mode, overloading the axon with damaging amounts of this cation (4). The accumulation of axonal Ca2+ in turn leads to mitochondrial injury (5), especially during reoxygenation, and activation of a number of Ca2+-dependent enzyme systems that result in irreversible structural and functional injury to the fiber (6). Protein kinase C may feed back to several upstream elements as a means of limiting the deleterious ionic deregulation (see section on Ca2+ overload). A significant component of pathologic Na+ entry may also occur through Cs+-sensitive inward rectifier channels (7). Cl− efflux plays a significant role, but only under conditions of Na+ conductance blockade; it is not known whether Cl− moves through a channel or a coupled transporter (9). Glial injury, particularly involving oligodendrocytes and the myelin sheath (MY), may be exacerbated by glutamate export through the Na+–K+-glutamate exchanger (10), which will be stimulated to release this transmitter under conditions of axoplasmic Na+ loading and depolarization. The locations of the various channels and transporters are drawn for convenience and do not necessarily reflect their real distributions in central axons.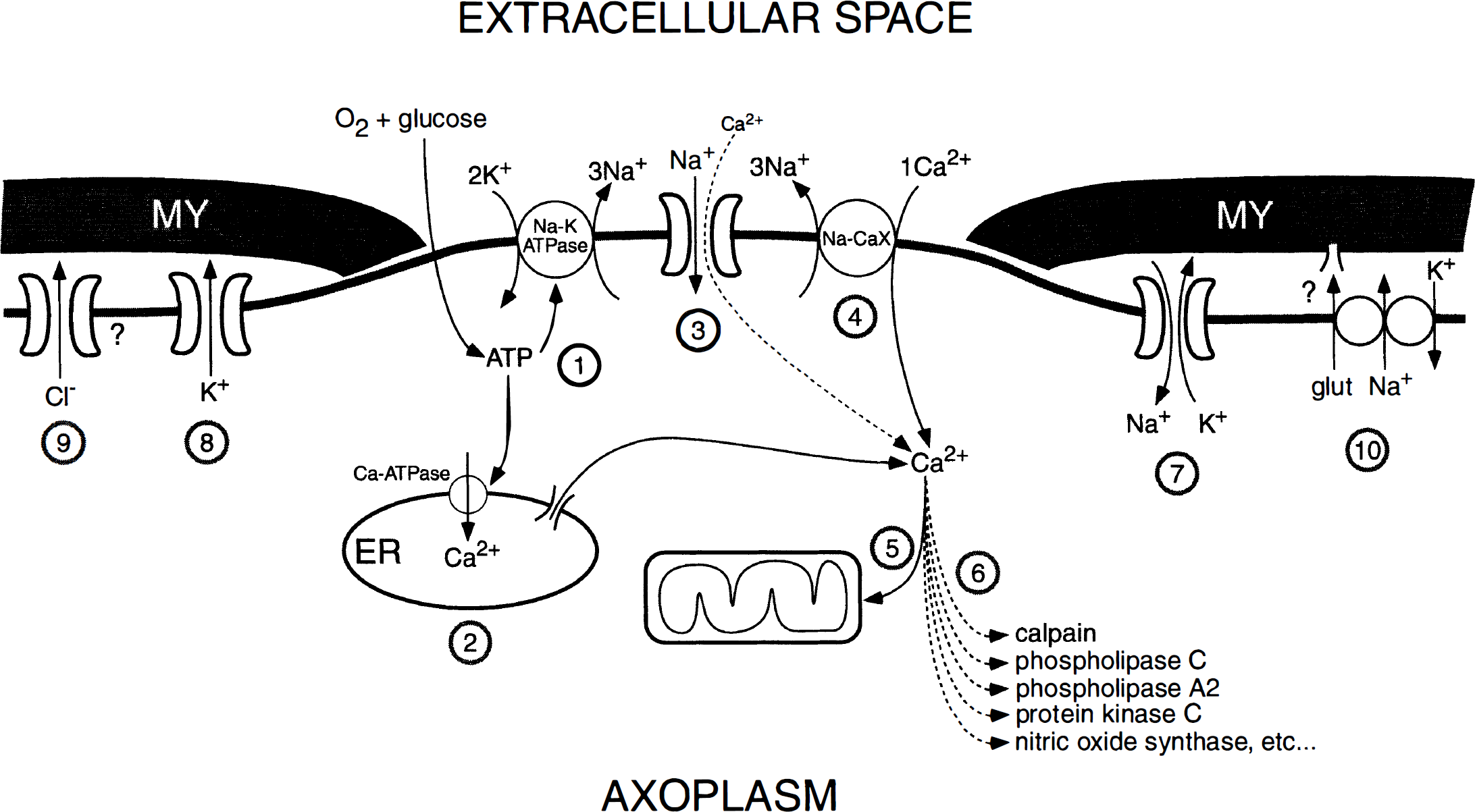
MECHANISM-DRIVEN THERAPEUTIC DESIGN
Based on the previous discussions, we can propose a synthesis of the many events triggered by energy impairment in white matter resulting from anoxia and ischemia. The number of steps involved in this type of white matter injury continues to increase as our understanding of the subject progresses. Figure 8 summarizes our most current knowledge to date. An impairment of glucose or oxygen delivery, or both, causes ATP levels to decrease, resulting in failure of several key ATP-dependent ion pumps, such as the axolemmal Na+–K+-ATPase and endoplasmic reticulum Ca2+-ATPase. Failure of the former causes accumulation of Na+ within, and loss of K+ from, the axoplasm, whereas impairment of the latter allows Ca2+ to escape from internal stores. A rise in [Na+]i mediated by noninactivating TTX-blockable channels, and probably by inward rectifier channels as well, coupled with axonal depolarization caused by K+ loss, promotes reverse operation of the Na+–Ca2+ exchanger, admitting damaging quantities of Ca2+ into the fiber. Accumulation of Ca2+ in turn leads to mitochondrial injury (especially during reoxygenation), and activation of a variety of Ca2+-dependent enzyme systems, such as calpain, protein kinase C, and others. There is also evidence of Cl− efflux from axons, but only under conditions of Na+ conductance blockade. A postulated but as yet unproven component may involve reversal of the Na+–K+–glutamate transporter (Kriegler and Chiu, 1993), leading to activation of glial glutamate receptors (Kettenmann and Schachner, 1985; Berger et al., 1992; Butt and Tutton, 1992) and possible injury to surrounding glia and the myelin sheath (McDonald et al., 1998).
Nonspecific downregulation of many of the pathways illustrated in Fig. 8 can be achieved by manipulations such as hypothermia. Cooling optic nerves during anoxic exposure in vitro results in highly significant protection; reducing bath temperature by only 2.5°C doubles the degree of physiologic recovery, and a further 2.5°C reduction to 32°C completely protects optic nerves from a 60-minute exposure (Stys et al., 1992a). Conversely, raising the temperature from 37° to 42°C greatly exacerbates injury. The estimated Q10 for anoxic white matter injury is high (≈10), i.e., the amount of physiologic injury increases 10-fold for a 10°C change in temperature, emphasizing the brain's exquisite sensitivity to even minor changes in temperature, which may have important clinical implications for febrile patients. Importantly, no protection was observed if hypothermia was applied after the anoxic period.
Based on the model in Fig. 8, more specific therapeutic intervention can be proposed. Various points along the injury cascade can be specifically targeted, such as Na+ channel or inward rectifier inhibition to reduce Na+ influx, Na+–Ca2+ exchange blockade, reduction of Ca2+ release from internal stores, and suppression of any one of the variety of enzyme systems that are presumably overactivated by Ca2+ ions. Closer study of Fig. 8 shows that Na+ channels, particularly the noninactivating subtype (step 3 in Fig. 8), occupy a central position in the injury cascade (possibly along with other Na+ influx routes, such as the Na+-permeable inward rectifier channels, step 7) for the following reasons. Blocking this channel will: (1) reduce Na+ influx and retard depolarization, both of which will restrain the Na+–Ca2+ exchanger from admitting excess Ca2+, (2) reduce the component of Ca2+ entry that occurs directly through this channel (Edwards, 1982; Stys and LoPachin, 1997), (3) limit axoplasmic Na+ accumulation, thus reducing consumption of limited energy reserves by the Na+–K+-ATPase, and (4) limit reverse Na+–K+–glutamate transport, which may contribute to glial damage. Moreover, there exists a variety of “use-dependent” Na+ channel blockers from the local anesthetic and antiarrhythmic classes that are preferentially active at the open conformation of the voltage-gated Na+ channel (Yeh and Tanguy, 1985; Wang et al., 1987; Khodorov, 1991), and should therefore be relatively selective for the noninactivating Na+ channel subtype implicated in axonal injury.
Figure 9 summarizes experiments with a number of Na+ channel blockers studied during normoxia and anoxia and reoxygenation in rat optic nerve in vitro. Panel A shows a bar graph of relative CAP area after 1 hour of exposure to a Na+ channel blocker during normoxia, to determine the extent of “anesthetic” effect of each compound (gray bars). Corresponding black bars show the amount of CAP area recovery after 1 hour of anoxia followed by reoxygenation and wash to remove any inhibitory effects of these agents. Tetrodotoxin (1 μmol/L), a specific, state-independent blocker of voltage-gated Na+ channels (Catterall, 1980) is an effective neuroprotectant, but completely abolishes excitability at this concentration. There is a wide variability in the effectiveness of various Na+ channel blockers, with QX-314 displaying a nearly ideal profile at 300 μmol/L, with negligible depression of excitability and nearly complete protection. Thus, the ideal compound would have no anesthetic effect under physiologic conditions (gray bar at 100%), yet would confer total protection (black bar also at 100%). These requirements can be combined into an “efficacy index” by multiplying the preanoxic and postanoxic relative CAP area recoveries (Stys, 1995); the ideal compound would have an index of 1.0. Any anesthetic effect or incomplete anoxic protection would lower the index. This is a convenient way to evaluate the effectiveness of various Na+ channel blockers that allows comparison between agents. Figure 9B summarizes the efficacy index for each of the compounds plotted in panel A. The very favorable properties of QX-314 (300 μmol/L) are immediately apparent, but in contrast, a drug such as lidocaine, which is almost as protective at 1 mmol/L as QX-314, has an index of zero because it completely abolishes the preanoxic response at this concentration. There appears to be a wide variability between Na+ channel blockers, emphasizing the point that examining one inhibitor will not necessarily predict effectiveness of another, even structurally related, compound (in Fig. 9 for example, compare procaine and procainamide that are nearly identical molecules, differing only in an ester versus amide group).
Bar graph summarizing effects of various Na+ channel blockers on preanoxic and postanoxic optic nerve CAP. 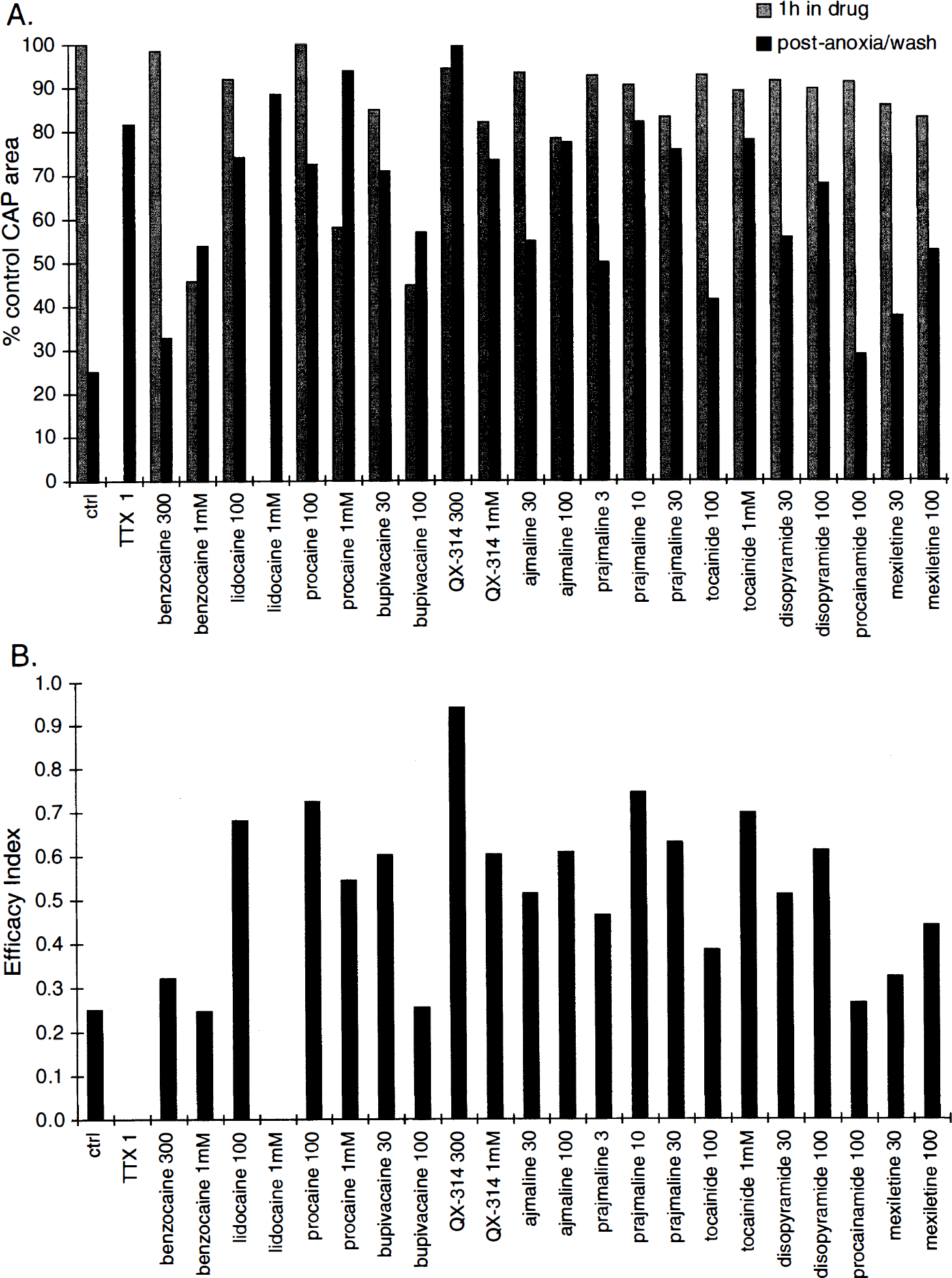
An interesting pattern emerges among the local anesthetics (Fig. 9). The permanently neutral benzocaine (Strichartz, 1987) displays a rather poor profile, with significant depression of the preanoxic CAP before neuroprotection becomes evident, reminiscent of the state-independent block of TTX. The ionizable amides (lidocaine, procaine, and bupivacaine), with pKa between 7.7 and 8.9 at physiologic temperatures (Butterworth and Strichartz, 1990), possess better characteristics, whereas permanently charged quaternary lidocaine analogs such as QX-314 and QX-222 (Stys et al., 1992c) are even more effective. Charged molecules such as QX-314 and the protonated forms of ionizable species are thought to be more selective for the open conformation of Na+ channels (Yeh and Tanguy, 1985; Wang et al., 1987; Khodorov, 1991). If the hypothesis that noninactivating voltage-gated Na+ channels permeate Na+ during anoxia is correct, then it follows that charged anesthetics would be more active at the open conformation of noninactivating channels, leaving conventional Na+ channels, those responsible for normal signaling and which exist in the open state only briefly, relatively unaffected. Moreover, it has been proposed that neutral forms of local anesthetics bind and unbind from Na+ channels more quickly than charged species, possibly because the uncharged form can gain access to and escape from the channel through hydrophobic pathways in the membrane, whereas charged molecules can only travel through hydrophilic regions at the inner mouth of the channel; the latter pathway is available only when the gates of the Na+ channel are open (Hille, 1977). As Na+ channels flicker between open and closed states, with the probability of late openings increasing with depolarization (Alzheimer et al., 1993) (as would be encountered in an anoxic cell), charged molecules would tend to be trapped in the Na+ channels for longer periods compared with uncharged species. Therefore, there would be a greater probability that a blocking molecule would be present in the channel at the time of subsequent openings. Overall, this would translate into a reduction of persistent Na+ permeability, which is precisely what is required. In contrast, conventional channels that are largely deactivated at rest, or inactivated during sustained depolarization, would be less susceptible to charged blockers because these molecules would not have ready access to the inner mouth of the channel. This hypothesis of drug interaction with various states of the Na+ channel may explain the clear superiority of charged compounds, and raises interesting issues for rational drug design.
Although charged molecules appear more effective in vitro, their hydrophilic nature defeats their usefulness in vivo, preventing penetration across the blood–brain barrier. Using axonal recovery cycle measurements based on paired stimulation protocols (Stys and Waxman, 1994; Stys and Lesiuk, 1996), a technique well suited to detect effects of use-dependent Na+ channel blockers, we have found that systemically administered QX-314 in rats does not enter the CNS to any measurable degree (Stys, unpublished observations). We are then faced with the paradoxical requirement of charge for maximal open Na+ channel inhibition versus neutrality to allow CNS penetration. Bypassing the blood-brain barrier by intrathecal administration of QX-314 was shown to significantly improve neurological recovery after spinal cord ischemia in the rat (F. Follis, personal communication). Alternatively, selecting an ionizable compound with a pKa near physiologic pH, which will exist in both neutral and charged forms, will aid penetration into the CNS after systemic administration. Studies with the antiarrhythmic mexiletine, a primary amine with a pKa of 8.4, showed that this drug is reasonably effective in vitro against optic nerve anoxia (Fig. 9). More importantly, after intraperitoneal injection, mexiletine was found to penetrate into the CNS at concentrations sufficient to significantly protect optic nerves from in situ ischemia (Stys and Lesiuk, 1996). Moreover, the intracellular acidosis that occurs during anoxia and ischemia will increase the proportion of the protonated form, thus trapping more drug in the cytosol and raising the concentration of charged drug that will preferentially interact with noninactivating Na+ channels.
In addition to local anesthetics and antiarrhythmics, certain anticonvulsants, such as phenytoin, carbamazepine, and diazepam, which are in common clinical use, are also protective against in vitro optic nerve anoxia, presumably as a result of their Na+ channel blocking properties (Fern et al., 1993). Closer scrutiny of the dose-response relationship with diazepam reveals that this agent confers maximal protection at concentrations well below those required for Na+ channel block (Fern et al., 1993; Fern et al., 1996a), and may be related to diazepam's conventional interaction with GABA receptors. This prompted an investigation into the possible role of GABA in white matter injury, the results of which are discussed in the section on Ca2+ overload. In addition to Na+ channels, modulation of other steps in the anoxic cascade might also be beneficial. Blocking the Na+–Ca2+ exchanger with bepridil or benzamil is significantly protective against optic nerve anoxia in vitro (Stys et al., 1992b). However, this transporter may operate to extrude Ca2+ from compromised neurons; thus, blocking this exchanger may in fact exacerbate neuronal injury (Mattson et al., 1989; Andreeva et al., 1991; Kiedrowski et al., 1994). A selective blocker of reverse Na+–Ca2+ exchange would be ideal (Iwamoto et al., 1996). Inhibition of the inward rectifier, anion transport, Ca2+ release from stores, or glutamate release to protect the myelin sheath, or downregulation of various Ca2+-dependent enzymatic pathways might also be viable therapeutic options. Because of the complexity of the interrelated mechanisms it is likely that combination therapy will be more effective, minimizing unwanted side-effects in vivo, yet maximizing functional axonal recovery.
CONCLUSION AND FUTURE DIRECTIONS
On the one hand, the specialized architecture of white matter portends unique pathophysiologic mechanisms. Yet the many similarities between white and gray matter with respect to ion channels and transporters, enzymatic pathways, intracellular Ca2+ dynamics, and mitochondrial physiology indicate that at least some mechanisms may be common to both regions, and information obtained from white matter studies may be helpful in understanding injury cascades and devising therapy in gray matter, and vice versa. Moreover, certain details pertaining to axonal anoxic and ischemic injury may be applicable to other white matter disorders, indicating overlap not only between different CNS tissues for a single condition, but also between different pathophysiologic conditions in a single region. For example, a significant component of traumatic spinal cord injury involves ischemia of the damaged tracts (Sandler and Tator, 1976; Li and Tator 1997), which may be amenable to neuroprotection based on mechanisms and therapeutic strategies as discussed in this article. Even in vitro spinal cord injury models, in which the influence of “conventional” ischemia has been removed, show certain similarities to anoxia and ischemia, in that voltage-gated Na+ channels occupy a central role in the pathophysiologic sequence of events leading to axonal dysfunction (Agrawal and Fehlings, 1996). Another example includes head trauma, with nondisruptive axonal injury causing perturbation of the axolemma, disruption of ionic homeostasis, and Ca2+ overload with activation of Ca2+-dependent enzymes, such as calpains and kinases (Maxwell et al., 1997).
Most of the mechanistic studies described in this review have been performed in optic nerve, the results of which may not necessarily be generalizable to other white matter regions. However, recent corroborative studies in anoxic spinal cord (Imaizumi et al., 1997), similarities with peripheral axons (Lehning et al., 1996a), and overlapping mechanisms in other paradigms, such as mechanical injury, all strongly suggest that although the primary insults may vary, downstream events (see Fig. 8) are funneled into a common progression, culminating in irreversible cellular injury. Figure 10 summarizes the unique as well as overlapping events responsible for gray and white matter anoxic and ischemic injury. The complexity of the many events set in motion demands a logical dissection of the pathophysiology of cerebral ischemia if successful therapy is to be devised. With a greater understanding of the details of these steps, whether common to different regions of the CNS, or unique, we will be in a better position to design combinations of interventions specifically tailored to each tissue type (e.g., glutamate antagonists for gray matter with Na+–Ca2+ exchange blockers for white matter), or to select common events for inhibition, such as Na+ channel blockade. As our knowledge of the molecular structure of the various components progresses, advances in “molecular pharmacology” will hopefully guide the rational development of more potent and selective agents, e.g., specific reverse Na+–Ca2+ exchange inhibitors, or blockers specific for noninactivating brain Na+ channels. Although this field is still in its infancy, we have already begun to probe the nature of drug-channel interactions at a very detailed molecular level (Qu et al., 1995). A thorough understanding of the fundamental cellular and molecular mechanisms of tissue injury that will guide the logical choice of targets for intervention, coupled with advances in molecular pharmacology, will greatly accelerate the design of successful neuroprotective strategies for CNS injury.
Comparison of events leading to anoxic and ischemic injury of CNS tissue. Both gray and white matter are vulnerable to interruption of energy supply, which leads to impairment of ion transport and collapse of transmembrane ion gradients. Glutamate-gated channels play a central role in gray matter pathophysiology, allowing Na+ and Ca2+ entry, whereas Na+ channels are the main Na+ influx pathway in myelinated axons. The resultant cellular Na+ overload and membrane depolarization may further activate certain subtypes of voltage-sensitive Ca2+ channels (VSCC) in gray matter, while promoting Ca2+ overload through reverse Na+–Ca2+ exchange in white matter. Downstream injury mechanisms likely then converge, involving excess activation of Ca2+-dependent biochemical pathways, generation of free radicals, and mitochondrial injury, culminating in cell death. Optimal protection of the CNS as a whole will therefore require combination therapy aimed at unique steps in gray and white matter regions, or intervention at common points in the injury cascades.