
Abstract
A novel D2-like receptor-binding radioligand, [18F](N-methyl)benperidol ([18F]NMB), was evaluated via positron emission tomographic (PET) imaging studies of baboons. [18F]NMB rapidly localized in vivo within dopaminergic receptor-rich cerebral tissues, and striatum-to-cerebellum ratios as high as 35 were achieved after 3 hours. Pretreatment of an animal with unlabeled receptor-specific antagonists before injection of [18F]NMB confirmed that the radioligand bound specifically to central D2-like receptors in vivo, and not to S2- or D1-like receptors. Unlabeled eticlopride displaced striatal [18F]NMB in vivo, showing that D2-like binding is reversible. Receptor-binding by the radioligand was resistant to competitive displacement by synaptic dopamine, as illustrated by the lack of effect of intravenous d-amphetamine on the in vivo localization of [18F]NMB. Studies involving sequential intravenous administration of [18F]NMB, d-amphetamine, and eticlopride show that the radioligand does not undergo agonist-mediated internalization with subsequent trapping. The feasibility of applying a three-compartment non-steady state model for quantification of [18F]NMB receptor binding was demonstrated. These in vivo characteristics give [18F]NMB distinct advantages over the PET radiopharmaceuticals currently used for clinical investigation of D2-like receptor binding.
The involvement of cerebral dopaminergic D2-like receptors in Parkinson's disease (Reisine et al., 1977a; Lee et al., 1978a; Strange, 1993), schizophrenia (Burt et al., 1977; Lee and Seeman, 1978b, 1980; Owen et al., 1978), dystonia (Perlmutter et al, 1997), and Huntington's disease (Reisine et al., 1977b), as well as in the mechanism of action of antipsychotic drugs (Creese et al., 1976; Seeman et al., 1976; Seeman, 1992), has generated widespread interest in the use of positron emission tomography (PET) for noninvasive assessment of human D2-like receptor activity in vivo. For these applications of PET with human subjects, [11C]raclopride or radiolabeled analogues of spiperone have been used as D2-like receptor-binding radiopharmaceuticals (Baron et al., 1991).
[11C]Raclopride has advantages as a PET radioligand compared to radiolabeled analogs of spiperone, which have poor in vivo selectivity for D2-like receptor binding and require lengthy imaging protocols (Frost et al., 1987; Coenen et al., 1988; Perlmutter et al., 1991b). Raclopride has relatively low receptor-binding affinity, and it is susceptible to displacement by endogenous dopamine (Seeman et al., 1989; Dewey et al., 1993). Although this attribute has been useful for some PET applications (Dewey et al., 1992; Volkow et al., 1994), competitive displacement of the radioligand from D2-like receptor sites generally complicates the modeling and interpretation of PET data. A D2-like receptor-binding radioligand with both high receptor-binding selectivity and resistance to displacement by synaptic dopamine would thus be preferable to the PET radiopharmaceuticals currently used in human studies.
Benperidol is a dopamine antagonist of the butyrophenone class that has been used clinically as an antipsychotic drug (Reynolds, 1993). Unlike spiperone or raclopride, this ligand has both high affinity and high selectivity for binding to central D2-like receptors (Peroutka et al., 1980). These characteristics have led to the investigation of various positron-emitting analogs of benperidol as potential PET radiopharmaceuticals (Arnett et al., 1985; Moerlein et al., 1986; 1992b; 1995; Suehiro et al., 1990). In this work we have examined the utility of fluorine-18 labeled N-methylbenperidol ([18F]NMB) (Fig. 1) as a new radioligand for PET study of D2-like receptor activity. Our results show that [18F]NMB has unique characteristics in vivo that are not found with any of the PET radiopharmaceuticals that are presently used for this purpose in human subjects. In these investigations we examined NMB labeled with fluorine-18 because the half-life (t½ = 110 minutes) of this nuclide is convenient for both image acquisition and laboratory procedures. However, note that the receptor-binding characteristics determined in these investigations may also pertain to the carbon-11 labeled version of the ligand.
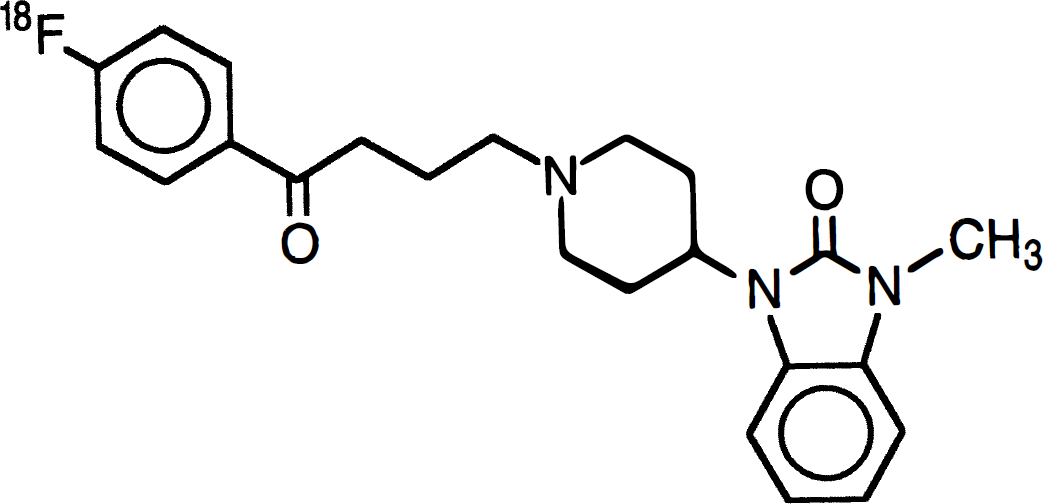
Molecular structure of [18F](N-methyl)benperidol ([18F]NMB).
METHODS
Radiotracer preparation
[18F]Fluoride was produced with use of the 18O(p,n)18F nuclear reaction induced on an isotopically-enriched [18O]water target with the Washington University JSW BC-16/8 cyclotron. [18F]NMB was synthesized from [18F]fluoride using a three-step reaction sequence (Moerlein et al., 1992a). The radiochemical purity of the final product exceeded 99%, and the end-of-synthesis specific activity was greater than 2000 Ci/mmol (74 TBq/mmol) as measured by ultraviolet high-pressure liquid chromatography detection validated by receptor-binding assay (Moerlein et al., 1992a). The radioligand was dissolved in mmol/L lactic acid in 0.9% saline for intravenous injection into the test animals.
[15O]Oxygen was produced via the 14N(d,n)15O reaction on naturally-abundant [14N]nitrogen gas using either the Washington University JSW BC-16/8 or CS-15 cyclotron. [15O]Oxygen was converted into [15O]water for measurement of regional cerebral blood flow (rCBF) or [15O]carbon monoxide for measurement of regional cerebral blood volume (rCBV) with use of an automated, online production system (Welch and Kilbourn 1985).
Unlabeled drugs
The unlabeled receptor antagonists eticlopride, SCH 23390, and ketanserin were purchased from Research Biochemicals International (Natick, MA, U.S.A.), and d-amphetamine was obtained from Sigma Chemicals (St. Louis, MO, U.S.A.). These drugs were dissolved in 5 to 10 mL of sterile water for intravenous injection into the test animals.
Instrumentation
Imaging studies were performed on baboons using either a Siemens 953B PET scanner in the 2-D mode, or the PETT VI system in the high-resolution mode. For the 953B scanner, the reconstructed resolution was approximately 6 mm in the transverse plane and about 4.5 mm in the axial direction; axial sampling was about 3.4 mm (Spinks et al, 1988; Mazoyer et al, 1991; Spinks et al, 1993). For PETT VI, transverse reconstructed resolution was about 12 mm, and axial resolution was approximately 14 mm (Ter-Pogossian et al., 1982; Yamamoto et al., 1982). A head-holder and acrylic skull caps permanently affixed to the skull (Perlmutter et al., 1991a) facilitated precise repositioning of the animal within the PET and magnetic resonance imaging (MRI) scanners for the different imaging sessions.
Animal studies
Studies were performed on Papio anubis baboons weighing 8.2 to 18.2 kg. All experimental procedures (Moerlein et al., 1995; Perlmutter et al., 1989; 1991a; 1991b) were approved by the Animal Studies Committee of Washington University. Surgical procedures were performed with appropriate anesthesia and postoperative analgesia. Magnetic resonance imaging studies were performed with the animal fully anesthetized. Careful monitoring of the animal's condition including temperature, end-tidal CO2, respiratory rate, electrocardiogram, and pulse provided online information during the MRI scans. Positron emission tomographic studies, with the animal's head placed in the headholder, were performed with the animal sedated with nitrous oxide. Review of the literature and more than 15 years experience with this regimen showed the adequacy of sedation and the favorable postprocedure recovery, and suggested that nitrous oxide produces fewer changes in brain blood flow and metabolism compared with other available agents. During these procedures, blood pressure, pulse, end-tidal pCO2, and rectal temperature were carefully monitored to identify undue animal distress. There were no signs of distress throughout the PET or MRI procedures.
Magnetic resonance imaging
Each animal was scanned by MRI after placement of the acrylic skull cap. The animal was fasted overnight but allowed free access to water up to 2 hours before the MRI procedure. Anesthesia was induced by ketamine 10 to 20 mg/kg intramuscularly and xylazine 2 mg/kg intramuscularly with glycopyrrolate 2 to 3 µg/kg administered to decrease secretions. If necessary, a 20-gauge plastic catheter was inserted into a limb vein to permit drug administration. A soft-cuffed endotracheal tube was placed to protect the airway. Lacrilube (Allergan Inc., Irvine, CA, U.S.A.) was placed into the eyes to protect the corneas. The animal was placed into the same headholder that was used with the PET scanner, and was monitored constantly by video and electrocardiographic recording. Six pins protruding from the truncated cylinder fixed in the acrylic cap permitted precise positioning within the headholder. This headholder had embedded within it thin tubing forming three “N's” in three orthogonal planes. The tubing was filled with copper sulfate or magnesium chloride solution for the MRI procedure.
Positron emission tomographic imaging
Baboons were fasted overnight, but allowed free access to water up to 2 hours before each study. Just before the study, the animal was initially anesthetized with 10 to 15 mg/kg ketamine intramuscularly and given glycopyrrolate 2 to 3 µg/kg intramuscularly to decrease secretions. Two 20-gauge plastic catheters were inserted into limb veins to permit radiotracer and drug administration, as well as the return of arterial blood after circulation through a plastic scintillator. The animal was paralyzed with gallamine 2 to 4 mg/kg intravenously, intubated with a soft-cuffed endotracheal tube, and ventilated with 70% nitrous oxide and 30% oxygen to maintain heavy sedation. Lacrilube was placed into the eyes to protect the corneas. A 20-gauge plastic catheter was also inserted into a femoral artery after local lidocaine anesthesia to permit arterial-blood sampling during the PET scans. Blood pressure, pulse, end-tidal pCO2, and rectal temperature were monitored throughout the study. Periodic arterial-blood gases confirmed that carbon dioxide and oxygen tensions were constant throughout the procedures. The animal was positioned in the scanner using a modified stereotaxic head holder that permitted precise repositioning (Perlmutter et al., 1991b). The position of the head within the scanner was recorded with a lateral skull radiograph. A transmission scan was made for individual attenuation correction. The fiducial tubing was filled with 18F after the transmission scan and before the emission scans.
Arterial-blood sampling required as much as 15 mL for each radioligand-receptor binding measurement. Blood loss was reduced considerably by recirculating blood through a closed loop (from artery to vein) that passed through a detector to measure blood radioactivity. This was done for the rCBF and rCBV scans, but not for the radioligand scans, as we made measurements of radiolabeled metabolites on the arterial blood samples.
Control studies
Control measurements were performed for each animal used in the interventional studies described below. After a tissue attenuation scan was acquired, rCBF was measured by PET after the intravenous bolus injection of 15 to 40 mCi (0.55 to 1.48 GBq) of [15O]water (Videen et al., 1987), and rCBV was determined after inhalation of 45 to 65 mCi (1.66 to 2.40 GBq) of [15O]carbon monoxide (Videen et al., 1987).
At least 3 hours after induction of anesthesia, 4.8 to 15.2 mCi (0.18 to 0.56 GBq) of [18F]NMB were injected intravenously over 30 seconds into an antecubital vein. Immediately before and after injection, the syringe containing the sample was measured in a well-type NaI(T1) scintillation counter that was cross-calibrated with the PET scanner (Mintun et al., 1984). Sequential scans were obtained for 3 hours (acquisition frames: 10 × 60 seconds; 6 × 200 seconds; 30 × 300 seconds). The midpoint (weighted for radioactive decay of fluorine-18) of each scan was recorded as the time elapsed since the start of radioligand injection. Note that the 3-hour imaging period was selected to facilitate direct comparison with our previous studies performed with [18F]spiperone, as well as to provide sufficient time for parameter estimation with our tracer kinetic model.
Data analysis
Regions of interest for striatal and cerebellar tissues were identified for each baboon from baseline blood flow images using corresponding sections from a stereotaxic atlas of the baboon brain (Davis and Huffman, 1968). Once identified, the regions of interest remained identically placed for all subsequent analyses for that animal. Regional values of tissue activity after [18F]NMB injection were decay-corrected to time of injection. For comparisons of tissue activity, curves, the regional values were corrected for body weight and amount of radioactivity injected (percent injected dose per milliliter of tissue times total body weight [(%ID/mL) × kg]).
Receptor specificity studies
These imaging experiments were performed on 8.2- to 10.0-kg animals with PETT VI. Positron emission tomographic studies were performed in which unlabeled receptor antagonists were injected intravenously into the animal before injection of [18F]NMB. The dosages and administration times were appropriate for saturation of the respective receptor sites in primates. These were eticlopride 4 mg/kg 1 hour before (Perlmutter et al., 1991b), ketanserin 0.6 mg/kg 30 minutes before (Wong et al., 1987), and SCH 23390 1 mg/kg 1 hour before (Sedvall et al., 1991) [18F]NMB. Control studies were also performed on each animal using [18F]NMB without pretreatment.
Displacement studies
The PETT VI scanner was used to image two animals weighing 8.2 and 9.1 kg, respectively. For each animal, control PET studies were initially performed with only [18F]NMB administered intravenously. A displacement study was then performed in which the tracer kinetics of the radioligand were determined over a 3-hour interval, during which eticlopride 4 mg/kg was administered intravenously 45 minutes after the injection of [18F]NMB.
Dopamine challenge studies
These imaging experiments were performed on an 18.2-kg animal with use of the 953B scanner. Control studies were initially performed on the animal with [18F]NMB alone. These were followed by experiments in which the tracer kinetics of [18F]NMB were measured when different dosages of d-amphetamine were injected intravenously at various times after injection of the radioligand. Specifically, one 3-hour imaging session was performed in which d-amphetamine was injected at a dose of 0.01 mg/kg 60 minutes after [18F]NMB, as well as a second dose of 0.1 mg/kg 85 minutes after the radioligand. A follow-up, 3-hour study was also made, in which high-dose d-amphetamine (1.0 mg/kg, intravenously) was given to the animal 30 minutes after injection of [18F]NMB. Microdialysis studies have shown that this intravenous dose of d-amphetamine increases synaptic dopamine concentrations in the caudate-putamen of primates by more than 20-fold (Moghaddam et al., 1993).
Ligand internalization experiment
This study was performed on an 18.2-kg animal with use of the 953B scanner. A preliminary PET imaging session was dedicated to the determination of the control kinetics for [18F]NMB in this test subject. In a subsequent session, [18F]NMB was administered intravenously and the tracer kinetics measured by PET over a 3-hour interval. For this experiment, d-amphetamine (1.0 mg/kg, intravenously) was injected 30 minutes after giving the [18F]NMB, and eticlopride (4.0 mg/kg, intravenously) was administered 70 minutes after injection of the radioligand. This intravenous dose of d-amphetamine increases synaptic dopamine levels in caudate-putamen 20-fold (Moghaddam et al., 1993), and this dose of unlabeled eticlopride was shown to displace receptor-bound [18F]NMB in vivo (see Results section). The intent of this study was to administer amphetamine and thereby increase synaptic dopamine that could potentially increase dopamine-mediated internalization. If this situation were to occur, then it would be anticipated that a change in the kinetics of eticlopride displacement would take place.
Analysis of blood samples
The free fraction of [18F]NMB was measured in triplicate from blood obtained from the test animals immediately before each imaging experiment. Details of the in vitro microfiltration technique have been described previously (Perlmutter et al., 1991b). After the injection of [18F]NMB, at least thirty 0.5 mL arterial blood samples were collected, initially at 5-second intervals then decreasing in frequency to every 30 minutes for the last 2½ hours. Most of the samples were taken in the first 2 minutes to ensure adequate description of the arterial input of radioactivity. Total radioactivity content in each blood sample was measured in a well-type NaI(T1) detector cross-calibrated with the PET scanner.
Eleven arterial blood samples (0.5 to 1.0 mL) were drawn in duplicate at intervals ranging from 1 minute to 3 hours after injection of [18F]NMB for measurement of radiolabeled metabolites. Two mL of methanol was added to the blood samples, the mixture vortexed for 1 minute, and the solid precipitate separated by centrifugation at 500 rpm for 10 minutes. The precipitate was resuspended in 1 mL of methanol, vortexed for 2 minutes, and centrifuged at 500 rpm for 10 minutes. The two methanol fractions and the solid pellet were assayed for radioactivity content.
The peripheral radiometabolites in the methanol extract were analyzed using radio-TLC. A chromatographic sample (50 µL of the extract) was developed with a stationary phase consisting of a 5 cm × 20 cm plate of silica gel G (Fisher Scientific, Pittsburgh, PA, U.S.A.) and a mobile phase of CH3OH/CH2Cl2 = 1/20 (v/v). The Rf value corresponding to [18F]NMB (Rf = 0.5) was determined using an authentic sample of NMB.
After chromatographic development, the radiolabeled metabolite spectra were measured by scanning the TLC plates with a Tracemaster 20 Automatic TLC Linear Analyzer (Berthold Systems Inc., Pittsburgh, PA, U.S.A.) interfaced with an Equity III+ computer (Seiko Epsom Corp., Nagano, Japan). For each sample, the system integrated the peak areas of the radioactive metabolites to give the percent conversion of [18F]NMB to metabolic byproducts.
Tracer kinetic modeling
A tracer kinetic model describing the in vivo behavior of [18F]NMB after intravenous injection and parameter estimation was used to analyze the PET and arterial blood data (Mintun et al, 1984; Perlmutter et al, 1986, 1989, 1991). This method includes PET-derived measures of regional tissue activity, rCBF, and rCBV, as well as independently measured total blood radioactivity, blood radioactivity corrected for radiolabeled metabolites, and the free fraction (f1; dimensionless) of [18F]NMB in blood. A two-compartment model (Mintun et al., 1984) was applied to the cerebellar PET data and the arterial blood data to calculate the free fraction of radioligand in brain tissue (f2; dimensionless). The tissue free fraction f2, which is a measure of nonspecific binding, is assumed to be the same in the striatum. A three-compartment model (Perlmutter et al., 1986) was then applied to the striatal data to estimate the following three variables for [18F]NMB in a 1 mL tissue volume: the regional permeability-surface area product (sec−1), the combined forward rate constant (CFRC; sec−1), and the dissociation rate constant (DRC; sec−1). The CFRC is the product of the association rate constant of [18F]NMB for specific binding sites times the apparent maximum number of available binding sites (Bmax). Dividing the CFRC by the DRC gives the binding potential (BP; dimensionless), which represents the capacity of the specific binding sites to interact with the radioligand (Perlmutter et al., 1986).
Because the estimates of the CFRC and the DRC are not completely independent, BP can be used as an index that is less sensitive to errors that equally affect both the association rate and the dissociation rate constants. If the estimate of the DRC is poor, however, the variance of the BP estimates may be increased, and a better index for measurement of receptor binding may be the CFRC. The assumptions and limitations of this modeling approach have been described in detail (Perlmutter et al., 1986, 1987, 1989, 1991).
RESULTS
Radioligand metabolism
The metabolic spectrum for [18F]NMB in the periphery is shown for a control study in Fig. 2. The degradation of the radioligand is relatively rapid; after 30 minutes only 10% of the total radioactivity in the blood is in the form of the intact radioligand. Based on chromatographic behavior, only polar (Rf < 0.15) radiometabolites were formed from [18F]NMB. Polar metabolites accounted for more than 90% of the total radioactivity in the blood after 60 minutes. This metabolic pattern did not change in any of the interventional studies that were performed.
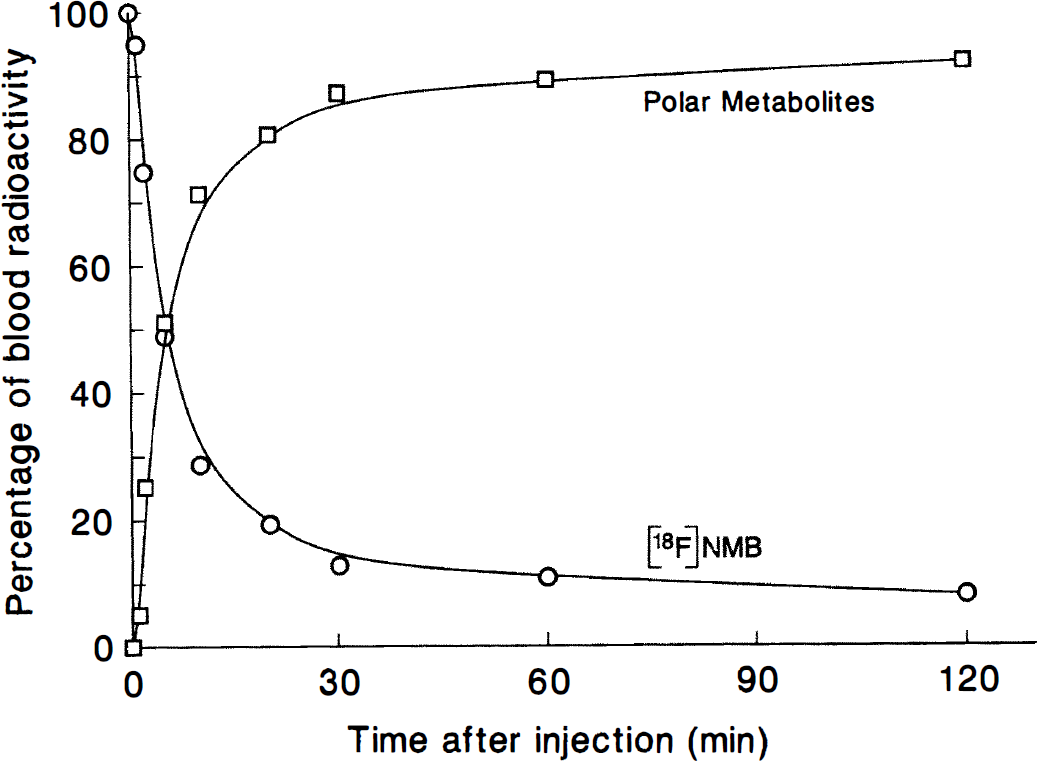
Metabolism of [18F]NMB in vivo as a function of time.
Control studies
Typical images of [18F]NMB acquired in a control study using the 953B scanner are presented in Fig. 3. These tomographic slices include the base of the frontal lobes and the striata; images were reconstructed from data obtained for 5 minutes starting 30 minutes, 1 hour, 2 hours, and 3 hours after intravenous injection of [18F]NMB. Selective localization and retention of the radioligand in D2-like receptor-rich striatum is remarkable in these images, with relatively low accumulation of nonspecifically bound radioactivity in D2-like receptor-poor brain tissue.
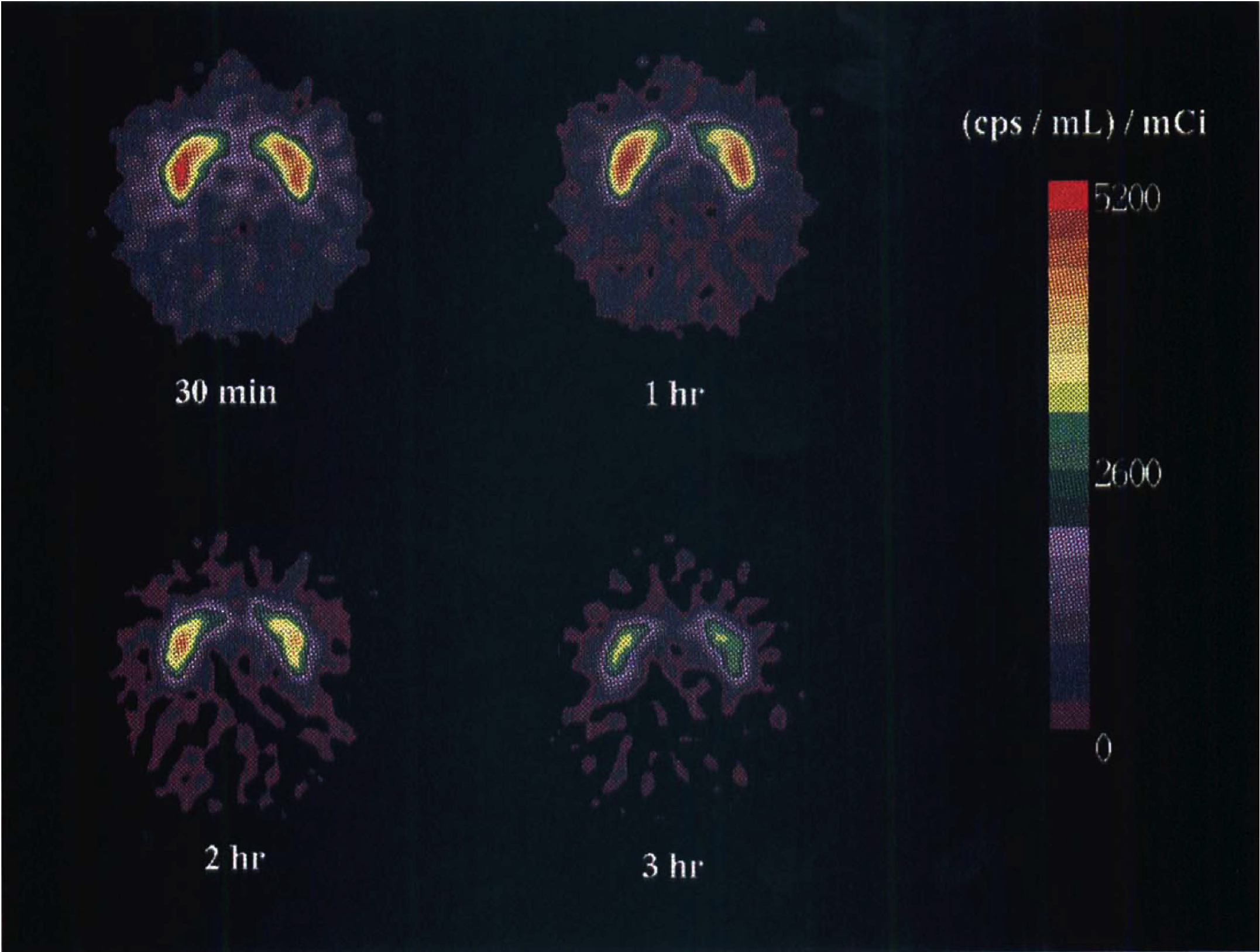
Transverse positron emission tomographic (PET) images of the same tomographic slice of a baboon brain after intravenous injection of [18F]NMB. The same color scale applies to all images. Each image was reconstructed from data acquired with the 953B scanner over 5 minutes. The anterior side of the brain is at the top of each image; the right side of each image corresponds to the right side of the animal's brain. Note the clear definition of basal ganglia after only 30 minutes; specific binding by the radioligand remains high for 3 hours.
The tissue-activity curves (normalized for animal body weight) of [18F]NMB in the striatum and cerebellum of a control study using the PETT VI scanner are given as a function of time in Fig. 4. Radioactivity preferentially localized within D2-like receptor-rich striatum compared with nonspecific binding in the cerebellum, which has very low concentrations of D2-like receptors. Binding to receptor sites in striatum is robust; striatal enrichment is significant after only 15 minutes and selective localization remains high over the 3-hour imaging interval. Although radioactivity clears slowly from the striatum, it does so at a much slower rate than from the cerebellum. With PETT VI, the striatum/cerebellum radioactivity ratio was 1.9 after 15 minutes, and gradually increased to a value of 12 at 3 hours.
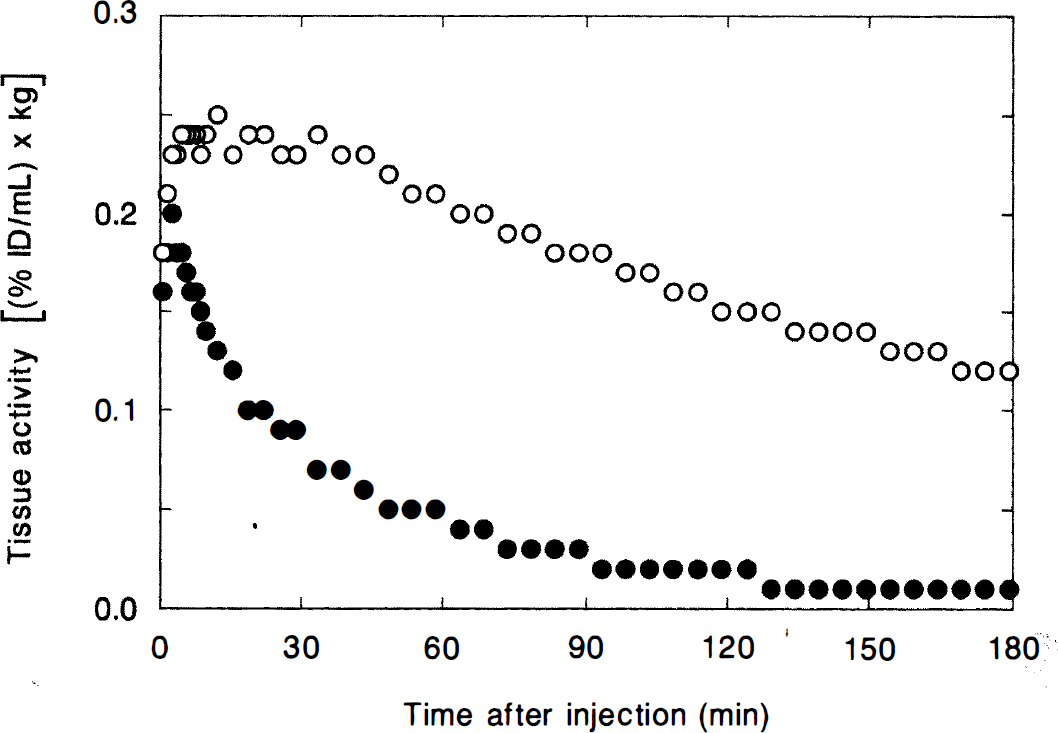
Tissue-activity curves for [18F]NMB as determined by PET in a control study. ○, striatum; •, cerebellum. Data was acquired with the PETT VI scanner.
The 953B scanner, with its greater resolution, gave even better discrimination between D2-like receptor-rich striatum and D2-like receptor-poor cerebellum. With use of this instrument, the striatum/cerebellum radioactivity ratios attained by [18F]NMB were 2.4 at 10 minutes and 35 at 3 hours. Differences in scanner resolution and sensitivity underlie the discrepancies between the control tissue-activity curves in Figs. 4, 5 and 6 (PETT VI) and the corresponding control curves in Figs. 7 and 8 (953B). Note the different scales for tissue activity in the two groups of figures.
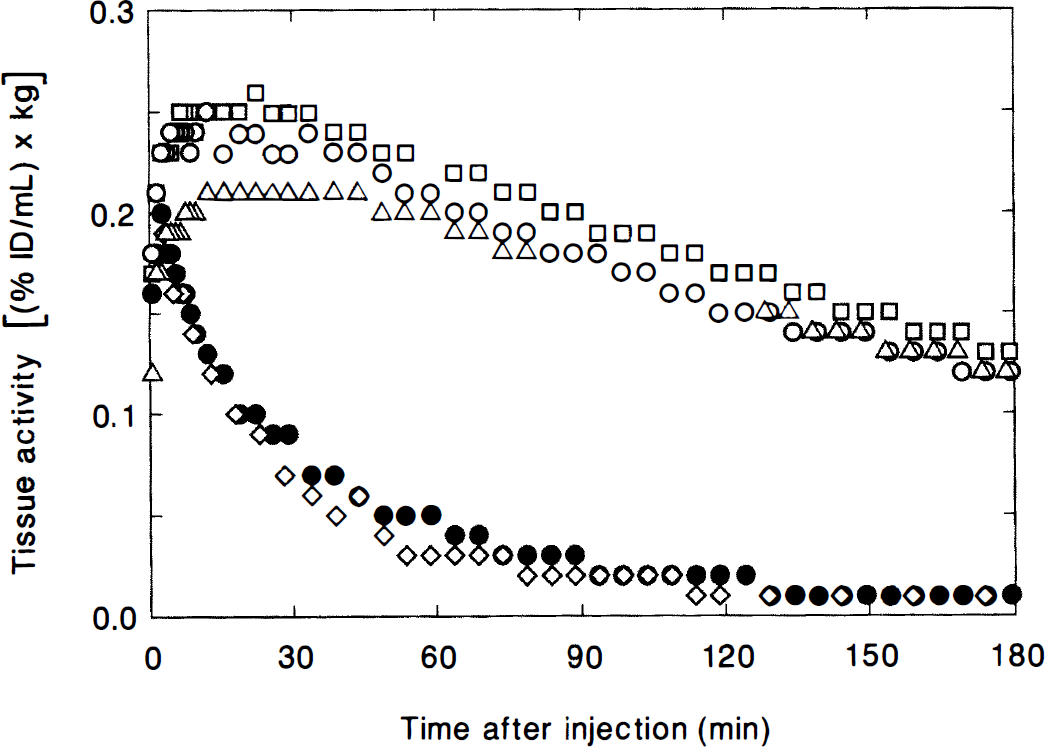
Localization in vivo of [18F]NMB after pretreatment with unlabeled receptor-specific antagonists. ○, control striatum; •, control cerebellum; ⋄, striatum (eticlopride pretreatment); □, striatum (SCH 23390 pretreatment); ▵, striatum (ketanserin pretreatment). Data was acquired with the PETT VI scanner.
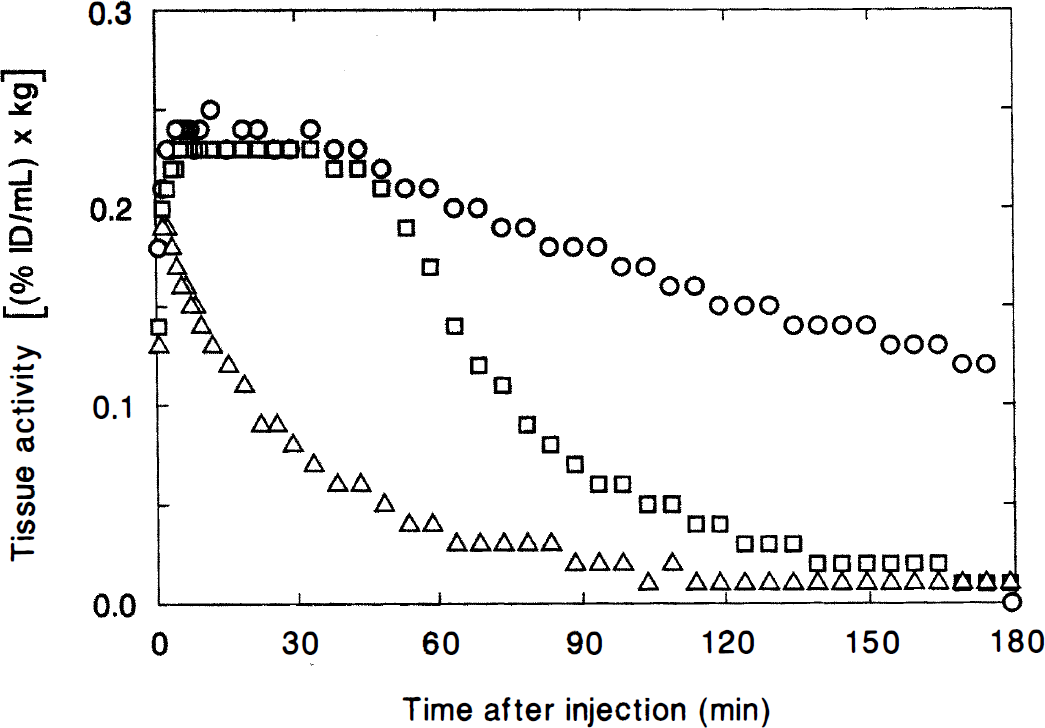
Reversibility of [18F]NMB binding to D2 receptors within baboon striatum. ○, striatum (control); ▵, striatum (unlabeled eticlopride 4 mg/kg, intravenously, 6 minutes before [18F]NMB); □, striatum (unlabeled eticlopride 4 mg/kg, intravenously, 45 minutes after [18F]NMB). Data was acquired with the PETT VI scanner.
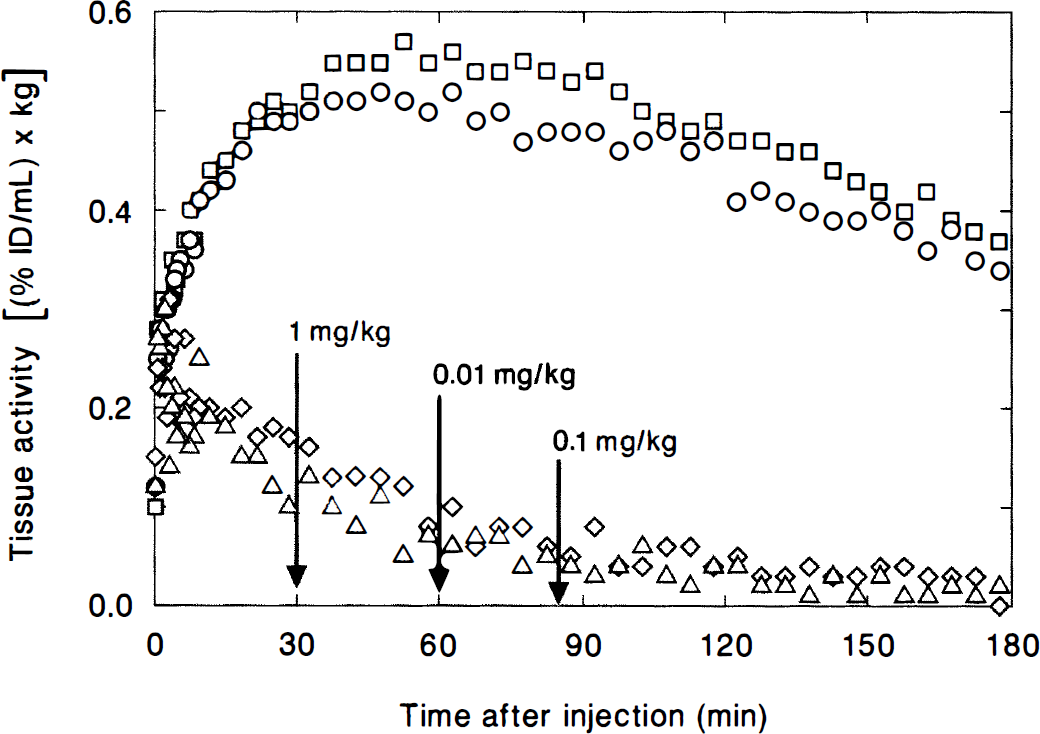
d-Amphetamine administration and in vivo localization of [18F]NMB. □, striatum and ⋄, cerebellum (d-amphetamine 0.01 mg/kg, intravenously, 60 minutes after [18F]NMB; d-amphetamine 0.1 mg/kg, intravenously, 85 minutes after [18F]NMB). ○, striatum and ▵, cerebellum (d-amphetamine 1.0 mg/kg, intravenously, 30 minutes after [18F]NMB). Data was acquired with the 953B scanner.
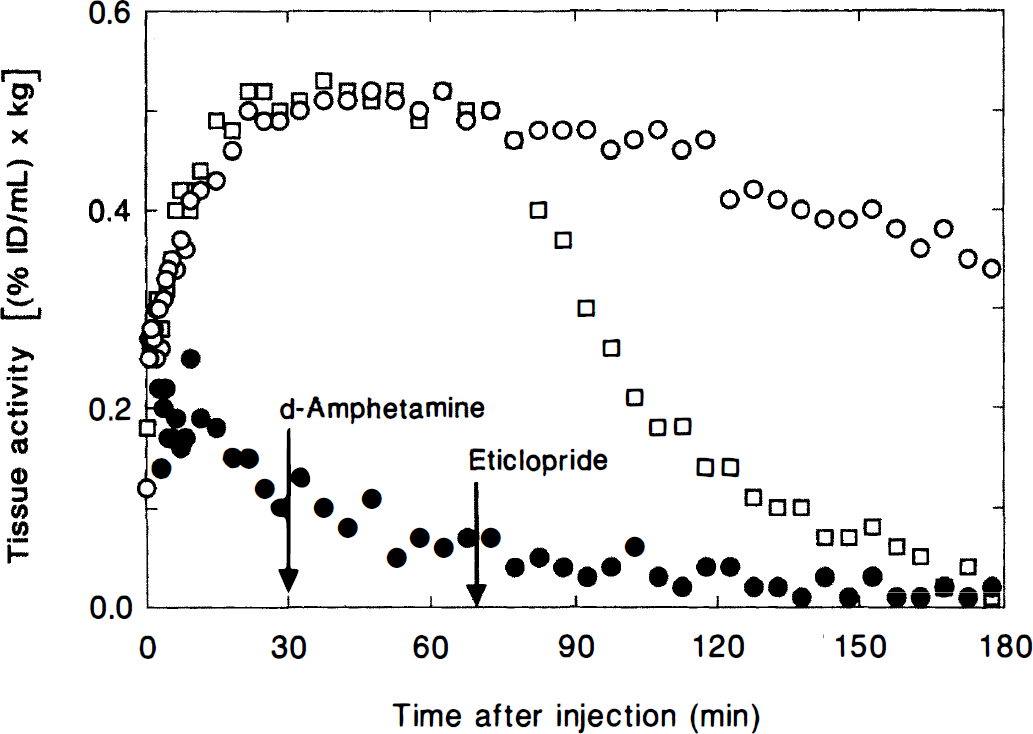
Noninternalization and trapping of [18F]NMB following in vivo D2-like receptor binding in baboon striatum. □, d-Amphetamine (1.0 mg/kg, intravenously) administered 30 minutes after [18F]NMB and eticlopride (4.0 mg/kg, intravenously) administered 70 minutes after [18F]NMB. ○, striatum (control); •, cerebellum (control). Data was acquired with the 953B scanner.
Receptor specificity studies
Fig. 5 illustrates the [18F]NMB tracer kinetics when the animal was pretreated with unlabeled receptor antagonists that bind to specific receptor sites. The tissue-activity curves are shown for the striatum and cerebellum in the control case. Striatal tissue-activity curves are also shown when unlabeled D2-like antagonist eticlopride (4 mg/kg intravenously), S2 antagonist ketanserin (0.6 mg/kg, intravenously), or D1-like antagonist SCH 23390 (1 mg/kg, intravenously) were injected into the animal 60 minutes, 30 minutes, and 60 minutes before radioligand administration, respectively. Note that selective localization of [18F]NMB was abolished when the D2-like antagonist was given; accumulation of the tracer in striatum was not significantly different from the nonspecific binding of the tracer in control cerebellum. In contrast, when the S2 and D1-like antagonists were given, radioactivity localization within the striatum did not deviate significantly from that in the striatum of the control study. There are some minor differences in the early parts of the tissue-activity curves in Fig. 5. These early data points are more influenced by ligand delivery, whereas later parts of the curve are not sensitive to radioligand delivery and almost entirely reflect radioligand binding. Note that there is little difference between the curves at these later times.
Displacement studies
The in vivo accumulation of [18F]NMB in the striatum is shown for three different experiments in Fig. 6. Control data is given, along with results from the study in which eticlopride (4 mg/kg, intravenously) was administered 60 minutes before the radioligand was injected. Also shown is the tissue-activity curve when eticlopride (4 mg/kg, intravenously) was administered 45 minutes after [18F]NMB was administered. In the latter experiment, the striatal concentration of radioactivity decreased to the same low level as when the animal was pretreated with the unlabeled D2-like antagonist.
Dopamine challenge studies
Fig. 7 illustrates the localization of [18F]NMB in the striatum and cerebellum for two separate studies. In one of these, d-amphetamine (1.0 mg/kg, intravenously) was administered 30 minutes after [18F]NMB was injected. The other study involved two intravenous administrations of d-amphetamine at dosages of 0.01 mg/kg and 0.1 mg/kg at 60 minutes and 85 minutes after [18F]NMB, respectively. For all dosages of the unlabeled drug, note the insignificant change in the striatal localization of [18F]NMB in vivo.
Radioligand internalization study
The effect of sequential administration of two separate drugs on the localization of [18F]NMB in the striatum and in the cerebellum is shown in Fig. 8. In this study, d-amphetamine (1.0 mg/kg, intravenously) was administered 30 minutes after injection of the radioligand, and a subsequent dose of eticlopride (4.0 mg/kg, intravenously) was administered 70 minutes after [18F]NMB. There was no change in striatal accumulation of the radioligand when the animal was treated with d-amphetamine. Despite the prior treatment with amphetamine, injection of eticlopride induced a rapid decrease in the striatal concentration of radioactivity.
Tracer kinetic modeling
The results of the application of the three-compartment nonsteady state method to control PET data for [18F[NMB acquired using the 953B scanner are given in Table 1. The free fraction in blood (f1) was 0.025 ± 0.001, which is lower than the corresponding values of 0.045 ± 0.011 for [18F]spiperone (Perlmutter et al., 1991b). The fit of the model to the observed tissue-activity curves were assessed by calculation of the average deviation of the calculated points from the observed points. For these data, the average deviation was 3.8%. The calculated f2 of 0.012 was identical to the 0.012 ± 0.002 value for [18F]spiperone (Perlmutter et al., 1991b), whereas the permeability surface area product for [18F]NMB was 0.43 ± 0.12, which is over seven times higher than the permeability-surface area product for 18F]spiperone (0.060 ± 0.014) (Perlmutter et al., 1991b). In some cases, there was a high correlation (>0.9) between estimates of the CFRC and the DRC, making it difficult to clearly distinguish these variables. The binding potential BP was therefore computed because it has a lower coefficient of variation and is thus a more conservative interpretation of the data. The BP for [18F]NMB (750 ± 88) is about 50% higher than the corresponding value for [18F]spiperone (473 ± 115) (Perlmutter et al., 1991b). Similar data analysis strategies were used for both types of studies, which which involved 3-hour data PET data collection for [18F]NMB and [18F]spiperone. Some of the differences between the estimates for [18F]NMB and those of [18F]spiperone can be explained by differences in imaging instrument capabilities. Data for [18F]NMB estimates were acquired with the higher-resolution Siemens 953B scanner, which has four times the axial sampling and nearly twice the trans-axial reconstructed resolution compared with PETT VI.
Three-compartment nonsteady-state analysis of [18F]NMB and [18F]spiperone receptor-binding in vivo
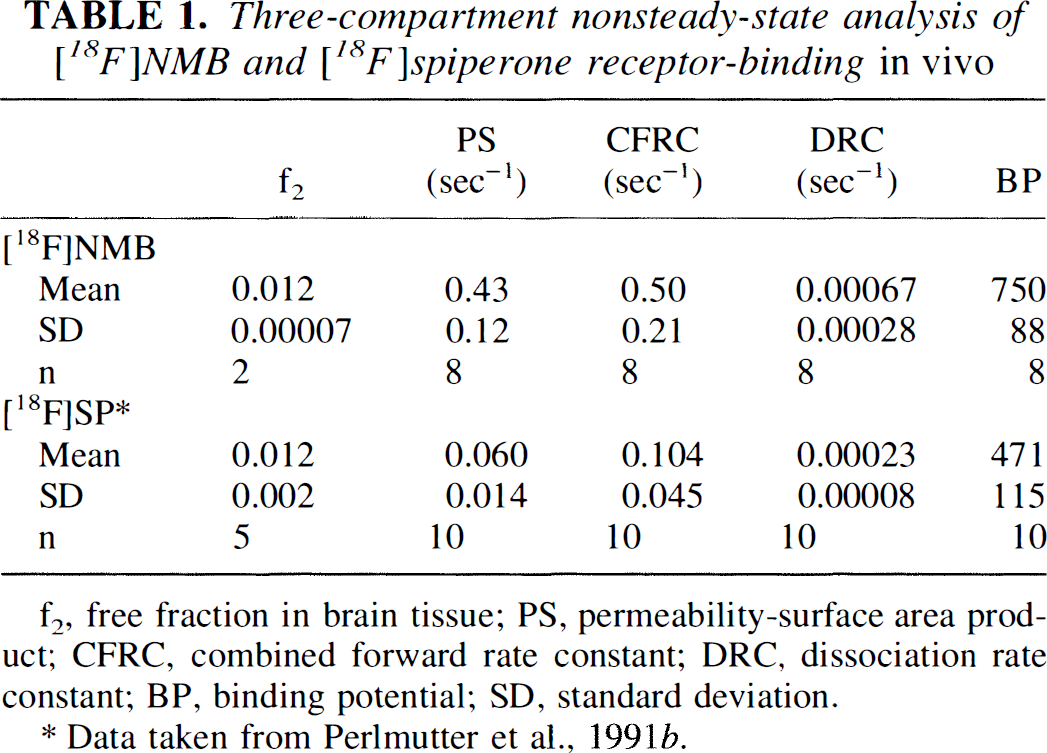
f2, free fraction in brain tissue; PS, permeability−surface area product; CFRC, combined forward rate constant; DRC, dissociation rate constant; BP, binding potential; SD, standard deviation.
Data taken from Perlmutter et al., 1991b.
DISCUSSION
Specificity of a PET radioligand for dopamine receptors in an important criterion for clinical use as a PET radiopharmaceutical. Receptor specificity is clearly necessary when PET is used for assessing changes in D2-like receptor activity with pathology, given the multitude of different neuroreceptors present in the brain. Specificity is also important when PET is used to evaluate psychiatric agents in drug challenge studies. Whereas binding to dopamine receptors is correlated with clinical potency of neuroleptics (Creese et al., 1976; Seeman et al., 1976; Seeman, 1992), binding to serotonin receptors is also a characteristic of atypical antipsychotics (Kapur et al., 1995; Protais et al., 1994; Schotte et al., 1993; Stockmeier et al., 1993). Discrimination of the role of each of these receptor systems in drug action and pathophysiology thus requires a PET radioligand that binds specifically to either dopaminergic receptors or to serotonin receptors.
The impetus of this research to develop D2-like specific radioligands is spurred by the potential versatility of such tracers for PET imaging of dopamine receptor changes associated with psychiatric and movement disorders, as well as during therapy of these processes. The clinical potency of antipsychotic drugs correlates with their affinity for binding to D2-like receptors (Creese et al., 1976; Seeman et al., 1976; Seeman, 1992). Moreover, the high Bmax of D2-like receptors in human striatum (13.3 to 14.7 pmol/g) (Hall et al., 1988) suggests that this receptor class is relevant to movement disorders such as Parkinson's Disease, dystonia, or Tourette's Syndrome. It is important to note that the D2-like family includes D2, D3, and D4-specific receptors that are presently better distinguished by mRNA expression for these proteins than by pharmacologically-specific probes. In baboon striatum, mRNA for D2-specific receptors are present in greater amount than D3 whereas D4 mRNA is not found (Todd et al, 1996). Because we have not defined the subtype specificity of [18F]NMB, the striatal measurements that we describe reflect a composite of predominantly D2-specific and D3-specific binding, although we cannot entirely exclude a contribution from D4-specific receptors as well. We do, however, distinguish clearly between D2-like and D1-like receptors with [18F]NMB.
Two classes of radioligands have been used in research studies worldwide for PET studies of D2-like receptor binding in human subjects. Positron-emitting analogs of spiperone and [11C]raclopride have been successfully used in clinical studies of D2-like receptor binding changes in healthy controls, as well as for investigation of pathological processes and the effects of centrally-acting drugs. [18F]Spiperone itself has been used for such clinical PET studies (Perlmutter et al., 1987; 1997), as well as carbon-11 (Wagner et al., 1983, 1984; Wong et al., 1984, 1986; Gjedde and Wong, 1987; Hägglund et al., 1987), and fluorine-18 (Arnett et al., 1986; Wienhard et al., 1990) labeled analogs of the ligand. [11C]Raclopride has proven to be useful for PET detection of D2-like receptor-binding changes in human subjects with neurological disorders (Rinne et al., 1990, 1993; Brooks et al., 1992; Sawle, et al., 1993), as well as for examination of drug effects in humans (Farde et al., 1988, 1992; Farde and von Bahr 1990; Farde, 1991; Dewey et al., 1992, 1993; Nordström et al., 1993).
Despite these advances, the radiopharmaceuticals currently used for PET study of D2-like receptor binding in humans suffer from deficiencies. The spiperone analogs listed above require long imaging protocols for assessment binding parameters, and have poor D2-like selectivity as they also bind avidly to serotonin receptors in vivo (Frost et al., 1987; Coenen et al., 1988; Perlmutter, et al., 1991b). However, [11C]Raclopride has relatively low binding affinity for D2-like receptors which may contribute to competitive displacement by endogenous dopamine under nonequilibrium conditions. Whatever the mechanism, displacement of a radioligand by endogenous dopamine from a radioligand-receptor complex complicates tracer kinetic modeling, and makes interpretation of some experiments more difficult if alterations in endogenous dopamine occur. These suboptimal characteristics of clinically-used PET radioligands prompted us to investigate [18F]NMB as a potentially superior D2-like receptor-binding PET radiopharmaceutical.
[18F]NMB selectively localizes in vivo within regions of the brain that are highly innervated by dopaminergic neurons. As shown in Fig. 3, the emission data for the control experiment clearly demonstrate that the radioligand preferentially accumulates in the basal ganglia, with relatively modest localization in other cerebral tissues. The in vivo localization of [18F]NMB thus parallels the Bmax values for D2-like receptors in primate tissue (Köhler and Radesäter, 1986), and suggests that the D2-like receptor-binding selectivity of the radioligand is high and that nonspecific binding is low.
Because D2-like receptor densities in primate brain are high in the basal ganglia and low in the cerebellum (Hall et al., 1988), comparison of the radioactivity concentrations in these two brain tissues indicates the relative receptor-specific to nonspecific localization of [18F]NMB. Fig. 4 illustrates the tissue-activity curves (normalized for animal body weight) of [18F]NMB in the striatum and cerebellum as a function of time after injection. The concentration of the tracer within the D2-like receptor-rich striatum reached peak levels within 10 to 15 minutes, and cleared slowly during the next 3 hour. Radioactivity in the receptor-poor cerebellum decreased rapidly as nonspecifically-bound radioligand cleared from nonspecific binding sites. After 15 minutes, the relative striatum/cerebellum radioactivity concentration was about 2.4; this ratio increased to approximately 35 during the 3-hour imaging interval (953B data). The rapid localization of the radioligand in D2-like receptor-rich tissues, together with the relatively fast clearance of nonspecifically-bound [18F]NMB is a significant advantage over [18F]spiperone (Perlmutter et al., 1991b). The improved image contrast attained early after tracer injection not only implies the utility of 20-minute half-lived 11C as a radiolabel for NMB, but is also important when 18F is used as a label because clinical imaging procedures can be substantially shortened without loss of receptor-binding information.
The low percentage of brain activity that is nonspecifically-bound may be caused, in part, by the lack of radiometabolites that cross the blood-brain barrier. The distribution of total blood radioactivity between [18F]NMB or radiometabolites is shown as a function of time after injection in Fig. 2. The radioligand is rapidly metabolized in vivo to polar metabolites only. Based on the metabolism of benperidol (Soudijn et al., 1967), these polar metabolites are most likely p-[18F]fluorohippuric acid and p-[18F]fluorobenzoic acid. The polarity of these radiometabolites may prevent their partitioning across the blood-brain barrier, which minimizes the nonspecifically-bound signal in PET images of the brain.
Receptor-blocking studies show [18F]NMB localized in vivo specifically on D2-like receptors. Fig. 5 illustrates the great extent to which [18F]NMB localization within the basal ganglia was hindered when D2-like receptors were saturated with the unlabeled D2-like receptor antagonist eticlopride. The striatal enrichment characteristic of the control study was absent when the radiotracer was administered after injection of unlabeled eticlopride. Nonspecific localization of the radioligand in the cerebellum was not different from control when the animal was pretreated with eticlopride. Because eticlopride binds specifically to D2-like receptors (Hall et al., 1985), these results are further evidence of the high selectivity of [18F]NMB in binding to D2-like receptors in vivo. Nonreceptor-based mechanisms were not responsible for this tracer behavior because rCBF, rCBV, tracer free fraction, and the peripheral metabolism of [18F]NMB were not substantially changed by eticlopride, and changes in these factors would only alter the early points on the tissue-activity curve. The selective localization of [18F]NMB within the striatum can thus be attributed specifically to binding of the radioligand to D2-like receptors in vivo. This contrasts with [18F]spiperone, in which only 68 to 73% of the uptake in basal ganglia was blocked by eticlopride; 27 to 35% of striatal localization of this tracer was caused by binding to S2 receptors and was thus blocked only by the serotonergic antagonist ketanserin (Perlmutter et al., 1991b).
Receptor-binding selectivity of [18F]NMB is further indicated by the lack of effect of non-D2-like receptor antagonists on the in vivo localization of the radioligand. As shown in Fig. 5, when receptor-saturating doses of the D1-like antagonist SCH 23390 or the S2 antagonist ketanserin were administered to the animal before the radioligand, there was no significant change in the selective accumulation of [18F]NMB within the striatum compared to control. Tissue-activity curves for the radioligand in the cerebellum were unaltered in these interventional studies compared with the control results. These data indicate that the radioligand binds to neither dopaminergic D1-like sites nor serotonergic S2 receptors in vivo, and is thus a more specific D2-like receptor ligand for PET than [18F]spiperone (Perlmutter et al., 1991b).
For applications of PET in pharmacological studies, a useful characteristic is reversibility of the radioligand-receptor interaction. In vivo of centrally-acting drugs requires the displacement of specifically-bound radioligand as a measure of the potency and kinetics with which these drugs bind to targeted receptors of the brain (Frost and Wagner, 1990). As shown in Fig. 6, [18F]NMB was competitively displaced from its binding sites by injection of unlabeled eticlopride into the animal 45 minutes after tracer injection. This administration time was based on the time of peak striatal uptake of the radioligand. The displacement of [18F]NMB was rapid, clearance from D2-like sites occurring within minutes of injection of the displacing drug. Note that the radioactivity in the striatum was displaced to the same low level as in the striatum of the eticlopride pretreatment study or the cerebellum of the control case. This suggests that all of the [18F]NMB that binds to D2-like receptors does so in a reversible manner because there is an insignificant component of nondisplaced radioligand. The reversible binding of this radioligand may be applied in pharmacological evaluation of drugs that interact with brain D2-like receptors because PET can be used to measure of the effect of the unlabeled drugs on the in vivo binding of [18F]NMB. This characteristic of [18F]NMB is a major advantage over radiolabeled spiperone, which binds in a less reversible manner to D2-like receptors in vivo (Perlmutter et al., 1991b), and is thus unsuitable for these applications.
The displacement of [18F]NMB by eticlopride also provides an opportunity to assess the performance of the tracer kinetic method. Eticlopride produces a rapid decrease in striatal radioactivity that primarily represents the competition between the unlabeled eticlopride and [18F]NMB for specific binding sites. Specific binding sites become available as [18F]NMB dissociates from the radioligand-receptor complex. Under normal circumstances. i.e., in the absence of a large excess of eticlopride, [18F]NMB would rebind to the receptor sites. In the presence of excess eticlopride, there is opportunity for competition for specific binding sites, and the decrease in striatal tissue activity represents primarily the dissociation rate of the radioligand. Although there is probably a small amount of rebinding of [18F]NMB under these competitive conditions, this is likely to be small because of the excess amounts of unlabeled eticlopride. Fitting an exponential to the displacement curve permits estimation of the half-life of the dissociation (about 20 minutes) and subsequent calculation of a dissociation rate constant of 0.00057 sec−1. This result compares favorably with the value of 0.00067 ± 0.00028 sec−1 (Table 1), which is the mean estimate of the dissociation rate constant as determined using our tracer kinetic model and parameter estimation. The close correspondence between the two dissociation rates provides additional validation of our tracer kinetic model applied to [18F]NMB.
Like [18F]NMB, [11C]raclopride also binds specifically and reversibly to D2-like receptors in vivo (Farde et al., 1985). However, [11C]raclopride is susceptible to displacement by endogenous dopamine. If the purpose of the study is to measure changes in endogenous dopamine, this characteristic is an advantage (Dewey et al., 1992; Volkow et al., 1994). For other PET applications, susceptibility to displacement by dopamine is a limitation of [11C]raclopride, as the data analysis must include correction for the effects of endogenous dopamine. The localization of [18F]NMB in the striatum and cerebellum for two dopamine challenge studies is shown in Fig. 7. In one of these studies, d-amphetamine (1.0 mg/kg, intravenously) was administered 30 minutes after radioligand injection. The other study involved the intravenous injection of d-amphetamine at 0.01 and 0.1 mg/kg at 60 and 85 minutes after [18F]NMB, respectively. For all three experiments, there was no decrease in D2-like receptor binding by the radioligand. Note that d-amphetamine when administered intravenously at a dose of 1 mg/kg has been shown to increase striatal dopamine levels from a baseline of 5 fmol/µL to approximately 100 fmol/µL within 40 minutes (Moghaddam et al., 1993). Because this dose of d-amphetamine has been shown to dramatically decrease striatal localization of [11C]raclopride (Dewey et al., 1993), our results indicate the substantially greater resistance of [18F]NMB to displacement by endogenous dopamine compared to the benzamide radioligand.
The most apparent explanation for this difference in receptor binding in vivo is the higher dopamine receptor-binding affinity of 18F]NMB compared to [11C]raclopride. Under equilibrium conditions, displacement of a radioligand by dopamine would depend only on the concentration and affinity of dopamine for D2 receptors, and not depend on the affinity of the radioligand. This situation, however, does not apply to the nonsteady state conditions of our studies, just as it does not apply to [18F]spiperone. An alternative explanation for the differences in displacement by dopamine may be a substantial difference in the dopamine receptor subtype affinities of the two radioligands.
Another potential explanation for the resistance of the [18F]NMB-receptor interaction to displacement by the neurotransmitter is that the radioligand-receptor complex is internalized and trapped after it binds to D2-like receptors. The internalized radioligand-receptor complex would thus no longer be located near D2-like receptors on neuronal cell surfaces, and would be less accessible for displacement by extracellular dopamine. This does not seem a likely mechanism, however. [11C]Raclopride can be displaced by amphetamine-induced changes in dopamine, which suggests that the radioligand-receptor complex is still exposed to synaptic dopamine at the cell surface. Because dopamine is a charged molecule, it penetrates postsynaptic cell membranes poorly and is therefore not expected to rapidly displace a radioligand from an internalized radioligand-receptor complex. Nevertheless, it has been suggested that internalization of the D2-like ligand spiperone is promoted by dopaminergic agonists (Chugani, 1988). Hypothetically, the lack of effect of d-amphetamine on the striatal localization of [18F]NMB may simply be a net effect whereby any decrease in striatal activity from displacement of receptor-bound radioligand by dopamine is offset by an increase in striatal activity induced by agonist-mediated internalization and subsequent trapping.
To evaluate this hypothesis, we examined the effect of sequential d-amphetamine and eticlopride treatment on the tissue-activity curves for [18F]NMB. These data are illustrated in Fig. 8. Similar to the results given in Fig. 7, d-amphetamine (1.0 mg/kg, intravenously) injected 30 minutes after [18F]NMB did not alter the in vivo localization of the radioligand within D2-like receptor-rich striatum of this animal. However, when eticlopride (4.0 mg/kg, intravenously) was injected 70 minutes after [18F]NMB (40 minutes after d-amphetamine), there was rapid clearance of radioactivity from the striatum. Like the results in Fig. 6, the radioactivity concentration in the striatum began to decrease within minutes of giving the unlabeled D2-like antagonists, and eventually was indistinguishable from that in the cerebellum.
These data are a cogent illustration that [18F]NMB is not displaced from D2-like receptors by endogenous dopamine, does not undergo agonist-mediated internalization with trapping, but is completely displaced from its D2-like binding sites by unlabeled eticlopride. The radioligand-receptor complex may hypothetically undergo internalization, and the lipid-soluble eticlopride could theoretically displace the radioligand from the internalized complex as if the receptor were still located on the cell membrane. However, one would expect that there may be a decrease in the rate of displacement of striatal radioactivity if this were the case, and inspection of Figs. 7 and 8 does not reveal such a difference. This alone does not prove that there is no internalization without trapping because comparisons of these tissue activity curves may not be sufficiently sensitive to detect a distinction in a potential delay of displacement, by the highly lipophilic D2-like antagonist eticlopride, of an internally located versus externally located radioligand-receptor complex. More importantly, because d-amphetamine-induced increases in extracellular dopamine displace receptor-bound [11C]raclopride but not [18F]NMB, the radioligand-receptor complex must be exposed externally to synaptic dopamine. This implies that the distinction between the receptor-binding behavior of [11C]raclopride and [18F]NMB under these in vivo conditions is determined by differences in the receptor-binding affinity or the dopamine subtype specificity of the two radioligands.
A standard nonsteady state three-compartment model and parameter estimation technique was applied for quantification of binding of [18F]NMB. This approach has been implemented and validated for use with [18F]spiperone in humans and nonhuman primates (Perlmutter et al., 1986, 1987, 1989, 1991b). Although the variances of the parameter estimates is substantial, the model fit the data well. Because there is a close correlation between the estimate of the CFRC (the product of the association rate constant and the maximum number of available binding sites) and the DRC, we also report the estimates of the BP (CFRC divided by DRC). The estimates of BP were found to have much lower variance than either of the individual parameter estimates. The estimates for the BP of [18F]NMB also have a lower variance than similar estimates for [18F]spiperone (Perlmutter et al., 1989, 1991b), which is expected because the dissociation rate for [18F]NMB is faster and more dissociation events occur during the course of a 3-hour PET study. These results do not validate the binding measurements for [18F]NMB, but rather show the feasibility of fitting this model to the data.
The above data provide convincing evidence of the unique characteristics of [18F]NMB compared to the clinically-used PET D2-like ligands ([11C]raclopride or radiolabeled spiperone analogs). It is also appropriate that [18F]NMB be compared to recently-developed D2-like ligands that have shown similar characteristics in preclinical trials. Positron emission tomographic studies of primates show that [18F]benperidol shares the advantages of D2-like specificity and reversibility (Moerlein et al., 1995) with [18F]NMB. However, compared with the nonmethylated derivative, [18F]NMB has greater striatal accumulation and greater image contrast for D2-like receptor-rich tissues. With use of the 953B scanner, after 3 hours [18F]benperidol has a striatal accumulation of approximately 0.25 (%ID/mL) × kg and a striatum/cerebellum concentration ratio of about 10 (Moerlein et al., 1995). The corresponding values for [18F]NMB using the same scanner are a striatal accumulation of about 0.4 (%ID/mL) × kg, and a striatum/cerebellum ratio of 35 (Figs. 7 and 8).
There are also radioligands of the benzamide class that share advantageous characteristics with [18F]NMB. Like [18F]NMB, the epidipride derivatives [18F]FPS (Mathis et al., 1992) and [18F]FPrEPID (Kessler et al., 1991) have high selectivity for binding to D2-like receptors, bind to D2-like receptors in a reversible manner, and are completely (Mathis et al., 1992) or predominantly (Kessler et al., 1991) inert to displacement by endogenous dopamine. To date, application of these benzamide radioligands for PET studies of human subjects has not been reported.
It would be beneficial if a D2-like radioligand with characteristics of [18F]NMB or one of the epidipride derivatives be implemented for PET studies of subjects with Parkinson's Disease, schizophrenia, or dystonia. Although [18F]FPS has greater striatal localization and is less affected by endogenous dopamine than [18F]F-PrEPID (Mathis et al, 1992), there are no clear advantages of this radioligand over [18F]NMB. In the development of [18F]NMB as a radiopharmaceutical, it has already been assessed that the critical organ with regard to absorbed radiation dosimetry is the lower intestinal wall. Up to 8.5 mCi (315 MBq) of the tracer can be administered to human subjects (Moerlein et al., 1997), meeting requirements set by the United States Food and Drug Administration for institutional radioactive drug research committee approval (radiation absorbed dose limit of 3 rem per procedure to the lens, gonads, or blood-forming organs, and 5 rems to other organs or the whole body). Preliminary studies also indicate the utility of [18F]NMB for PET studies of D2-like receptor binding in human subjects (J.S. Perlmutter, unpublished results). We thus anticipate that the implementation of 18F]NMB as a clinically-used PET radiopharmaceutical to be relatively efficient compared to the investigational benzamides.
CONCLUSION
To summarize, [18F]NMB is a novel radioligand that has in vivo characteristics unavailable in any other PET tracer used for clinical study of cerebral D2-like receptors. Advantages of [18F]NMB over the spiperone series of PET tracers are that the radioligand binds specifically to D2-like sites, receptor binding is reversible, and that high striatum/cerebellum ratios are more rapidly achieved. The noteworthy advantage of [18F]NMB over [11C]raclopride is that its binding to D2-like receptors is resistant to displacement by synaptic dopamine. The radioligand is not internalized and trapped, which makes it amenable to in vivo evaluation of unlabeled drugs that act on central D2-like receptors. Furthermore, preliminary application of a 3-compartment model suggests that binding may be quantified using standard parameter estimation techniques. [18F]NMB is a unique D2-like radioligand with great promise for PET examination of human pathological conditions and for evaluation of pharmacotherapy in vivo.
Footnotes
Abbreviations used
Acknowledgements
We thank L. Lich and J. Carl for expert technical assistance with the animal studies, J. Hood and J. Giovanni for data processing, W. Margenau and J. Robison for radioisotope production, and K. Lechner for quality-control testing.