
Abstract
We report autoradiographic measurements of the in vivo uptake of [3H]nimodipine during the nonischemic depolarization of cortical spreading depression (CSD) in rat brain. [3H]Nimodipine uptake in brain was determined regionally in rats undergoing CSD (n = 8) and was significantly increased in cortex (14 ± 7%) and hippocampus (10 ± 6%) on the stimulated side relative to the contralateral hemisphere when compared with the same measurements in a control group (n = 8). A similar measurement using the physiologically inert radiotracer [14C]iodoantipyrine to control for potential effects of CSD on radioligand distribution showed a minimal increase (2.4 ± 0.7%) of radiotracer uptake in cortex after CSD. This increase was significantly less than that observed in the [3H]nimodipine uptake studies. We hypothesize that increased in vivo [3H]nimodipine uptake in CSD identifies regions of depolarization and thus infers activation of the L-type voltage sensitive calcium channels.
Cortical spreading depression (CSD) is a reversible wave of depressed electrocortical activity and direct current (DC) depolarization that propagates across the cerebral cortex at a characteristic rate of 3 mm/min (Leao, 1944). The brief period of brain tissue depolarization is associated with massive shifts of K+ out of cells and Na+, Cl–, Ca2+ and water into cells (see review by Somjen et al., 1992). Excitatory neurotransmission and calcium channel activation may be factors in CSD. Cortical spreading depression is reduced or completely suppressed with N-methyl-
We have previously shown increased in vivo binding of the 1,4-dihydropyridine L-type VSCC antagonist [3H]nimodipine to the L-type VSCC in acutely ischemic brain by using quantitative autoradiography (Hakim and Hogan, 1991; Hogan et al., 1991). We proposed that increased in vivo uptake of [3H]nimodipine into ischemic brain was a consequence of the greater affinity of the L-type VSCC in depolarized cell membranes to bind 1,4-dihydropyridine VSCC antagonists (Kokubun et al., 1986; Triggle and Rampe, 1989) thus inferring activation of this channel. If this hypothesis is correct in vivo uptake of [3H]nimodipine should increase during the nonischemic depolarization associated with CSD.
METHODS
Adult male Sprague Dawley rats were fasted overnight before study. Halothane anesthesia (4% induction, 0.5% maintenance) was maintained throughout surgery and during recurrent CSD.
[3H]Nimodipine binding during cortical spreading depression
Mean arterial blood pressure and arterial blood gases were monitored in spontaneously breathing rats. Body temperature was maintained between 36 and 37°C. Burr holes were placed stereotactically over the right occipital and right frontal cortices. A microdialysis probe was inserted 2 mm into the right occipital cortex. To record CSD, a 13 μm-diameter, platinum wire electrode was inserted 2 mm into the right frontal cortex and connected to a DC amplifier (Gould Instruments Inc., Valley View, OH, U.S.A.). In 8 rats CSD was induced by infusion of 3 mol/L KCl solution through the microdialysis probe at a starting rate of 2 μL/min. Control animals (n = 8) were infused with normal saline at the same rate. After 60 minutes of recurrent CSD, 200 μCi of [3H]nimodipine (specific activity 130 Ci/mmol, NEN-Dupont, Boston, MA, U.S.A.) in 600 μL of carrier (BAY e9736 placebo, Miles Pharmaceuticals, Etobicoke, Ontario, Canada) were infused intravenously over 3 minutes at the start of the next recorded DC potential shift. In control studies the radiotracer was infused after 60 minutes of normal saline perfusion of the microdialysis probe. After 15 minutes of radiotracer circulation plasma, [3H]nimodipine activity was measured, the rat was decapitated, and the brain was immediately removed and frozen at −70°C.
[14C]Iodoantipyrine uptake during cortical spreading depression
[3H]Nimodipine freely crosses the blood brain barrier (Van den Kerckhoff and Drewes, 1985) and its initial clearance may be influenced by cerebral blood flow (CBF) changes during CSD. To assess for this possible effect, the uptake of the physiological inert and blood brain barrier permeable radiotracer [14C]Iodoantipyrine (Sakurada et al., 1978) was measured. Probe placements were identical to the [3H]nimodipine studies. After 60 minutes of either recurrent CSD (n = 6) or saline perfusion (n = 6) 30 μCi of [14C]Iodoantipyrine (specific activity 57 mCi/mmol, Amersham International PLC, England) in 600 μl of normal saline were infused over 3 minutes at the start of the next recorded CSD. After 15 minutes of radiotracer circulation plasma [14C]Iodoantipyrine activity was measured, the rat was decapitated, and the brain was rapidly removed and frozen at −70°C. To better control arterial P
Autoradiography
Horizontal cryostat brain sections 20-μm thick were obtained every 140 μm and apposed to tritium sensitive film (Hyperfilm, Amersham, Oakville, Ontario, Canada) for [3H]nimodipine studies or Kodak SB-5 film (Picker International, Brampton, Ontario, Canada) for [14C]iodoantipyrine studies. Tritium autoradiographs were calibrated using tissue paste standards prepared in the laboratory from forebrain homogenates. Microscale standards (Amersham International PLC, England) were used to calibrate the 14C autoradiographs. Autoradiographs were digitized (Imaging Research Inc., St. Catharines, Ontario, Canada) and regional radiotracer concentrations determined using a standardized region of interest template at five horizontal levels (Fig. 1). The observer was blinded to the treatment status of the rats. Previously determined metabolite corrections (Hogan et al., 1991) were applied and [3H]nimodipine volume of distribution calculated as the ratio of tissue [3H]nimodipine over plasma [3H]nimodipine concentration (Lassen and Perl, 1979).
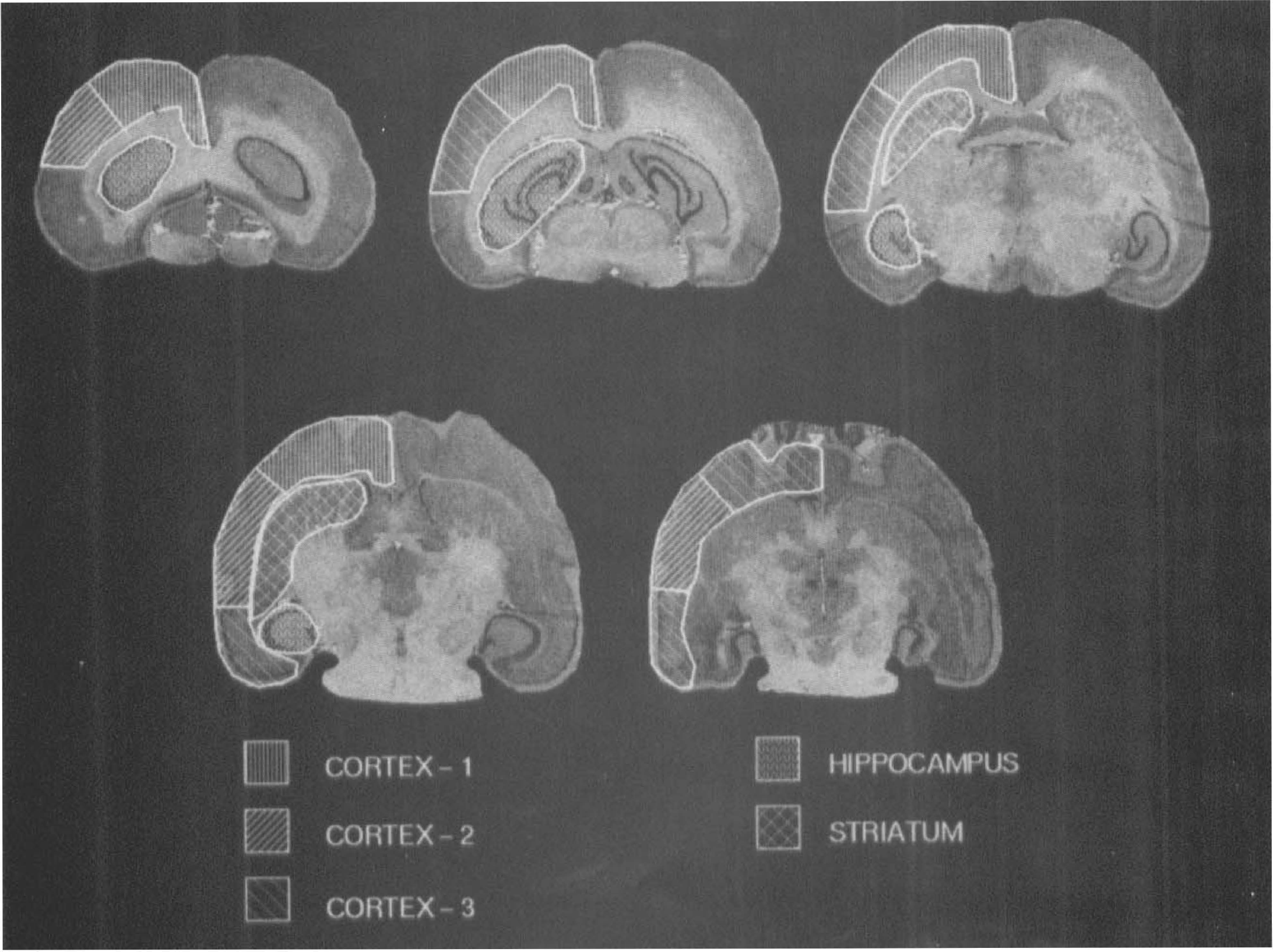
Region of interest template used to analyze the autoradiographic binding data in one cerebral hemisphere. The five groups of regions of interest which form the volumes of interest, generated by anatomical and cluster analysis criteria, are identified by the hatched markings.
Data analysis and statistics
Physiological data comparisons were performed using independent t tests. The ratio of the volume of distribution of [3H]nimodipine in the stimulated hemisphere over that in the contralateral hemisphere was calculated for each region of interest. Striatal and hippocampal regions of interest data were grouped and K-means cluster analysis (Hartigan and Wong, 1979) was used to subdivide the cortical regions of interest into three groups. All subsequent analysis used data averaged over these five volumes of interest (Fig. 1). The Mann-Whitney U test (BMDP Statistical Software, Inc., Los Angeles, CA, U.S.A.) with Bonferonni correction was used to assess differences between the CSD and control groups.
RESULTS
Physiologic measurements
Mean arterial blood pressure and arterial blood gases did not differ between the CSD and control groups in the [3H]nimodipine and [14C]iodoantipyrine studies. P
Cortical spreading depression
Recurrent DC potential shifts with initial negative deflection of 1 to 5 mV of 30 to 60 seconds duration consistent with CSD depolarization (Somjen et al., 1992; McLachlan, 1992) were recorded from the frontal cortex. In the [3H]nimodipine CSD group, 8.0 ± 2.6 DC depolarizations were recorded whereas in the [14C]iodoantipyrine CSD group, 9.0 ± 2.2 DC depolarizations were observed. In the [3H]nimodipine studies only one DC depolarization was observed after [3H]nimodipine infusion whereas in the [14C]iodoantipyrine studies six DC depolarizations were observed in five rats after radiotracer infusion. One DC depolarization was observed in the [3H]nimodipine control group.
[3H]Nimodipine binding
Figure 2 shows autoradiographs of in vivo uptake of [3H]nimodipine into brain in four horizontal sections (corresponding to the first four levels in Fig. 1) from a selected study in the recurrent CSD group. The stimulated hemisphere is on the left side of these images. [3H]Nimodipine uptake was increased in both cortex and hippocampus of the CSD-stimulated hemisphere relative to the contralateral hemisphere. No change was observed in striatum. The narrow margin of intense [3H]nimodipine uptake around the microdialysis probe was excluded from analysis. The ratio of [3H]nimodipine uptake in the stimulated hemisphere over the contralateral hemisphere for each volume of interest is summarized in Table 1. This ratio was increased in cortex 1 by 14% (p = 0.019), in cortex 2 by 11% (p = 0.031), and in the hippocampus by 10% (p = 0.016) in the CSD group compared with the control group. In the control group, the average cortical [3H]nimodipine volume of distribution was 104 ± 20 mL/100 g and was 112 ± 17 mL/100 g in the striatum and 125 ± 23 mL/100 g in the hippocampus. Volumes of distribution in the contralateral hemisphere did not differ between control and CSD studies.
Ratio of [3H]nimodipine uptake in volumes of interest
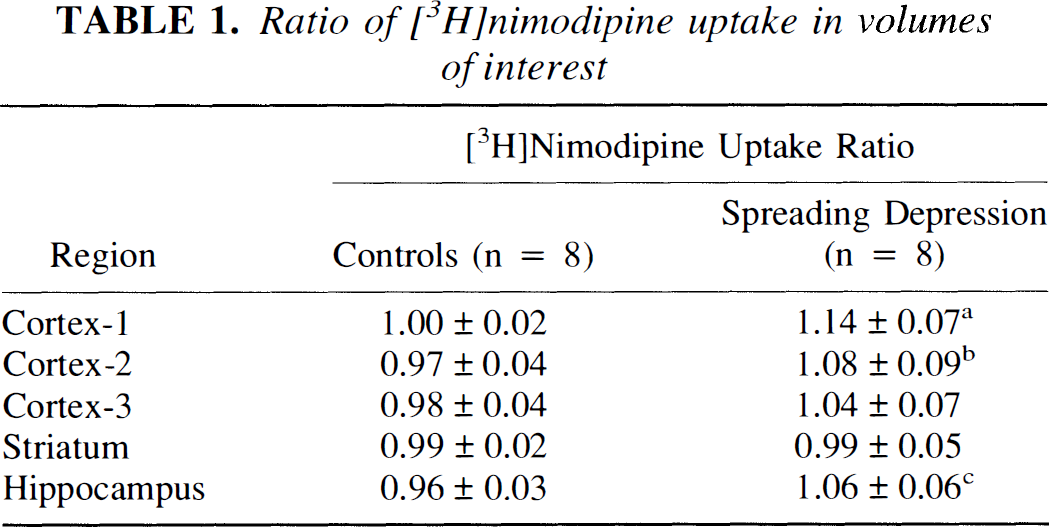
Ratio of [3H]nimodipine volumes of distribution in volumes of interest in the hemisphere with probe implantation over the corresponding volume of interest in the contralateral hemisphere for both control and cortical spreading depression groups. Differs from controls at
p = 0.019
p = 0.031
p = 0.016 (Mann-Whitney U [two tailed] with Bonferonni correction for multiple comparison).
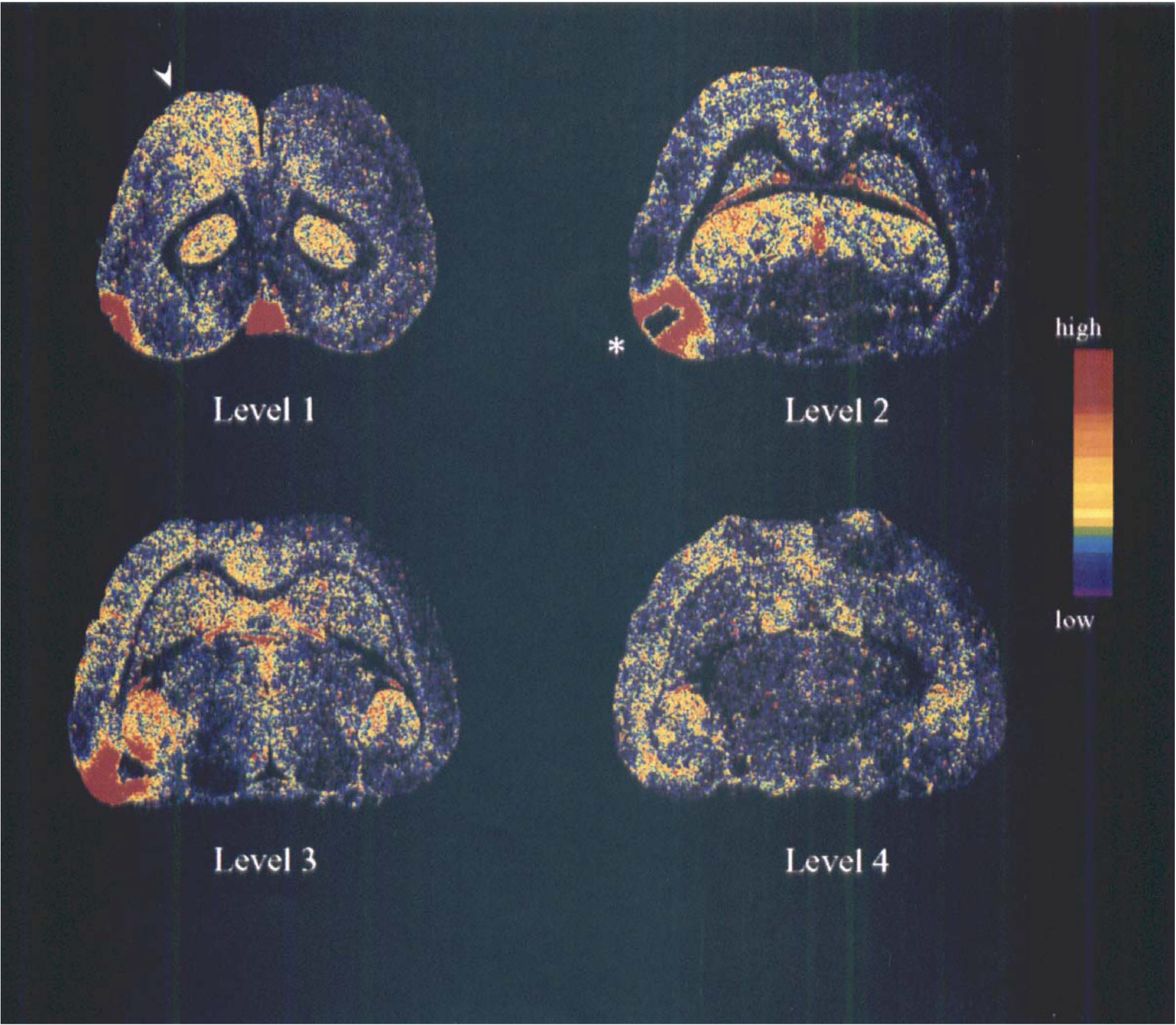
Autoradiographs of four horizontal brain sections from a selected study with recurrent CSD showing in vivo [3H]nimodipine uptake. The stimulated hemisphere is on the left side of the sections. The figure has been contrast enhanced to show the increase in [3H]nimodipine uptake in the frontal cortex ipsilateral to the recurrent CSD. Increased activity was also evident in the ipsilateral hippocampus but no change in uptake occurred in striatum. The location of the microdialysis probe and surrounding narrow band of depolarization is indicated by the asterisk. The arrow by the top left section indicates the location of the recording electrode. The horizontal sections correspond to the first four levels in Fig. 1. The pseudocolor scale has lower activities in blue and higher activities in yellow and red.
[14C]Iodoantipyrine uptake
Ratios of [14C]iodoantipyrine uptake in the stimulated hemisphere over the contralateral hemisphere for each volume of interest ranged from 1.005 ± 0.015 to 1.035 ± 0.027 in the control group and from 1.031 ± 0.006 to 1.065 ± 0.042 in the CSD group. This ratio was only increased in cortex 1 by 2.4% (p = 0.032) in the CSD group compared with the control group. This increased uptake of [14C]iodoantipyrine within cortex 1 was significantly less than the increase observed for [3H]nimodipine uptake within the same region (p = 0.02).
DISCUSSION
We have shown increased in vivo [3H]nimodipine uptake in the cortex and hippocampus ipsilateral to recurrent CSD in rat brain. Only a minimal increase in the cortex was observed with the physiologically inert radiotracer [14C]iodoantipyrine administered in an identical manner. We hypothesize that increased [3H]nimodipine uptake is a consequence of cell-membrane depolarization indicating activation of the L-type VSCC during CSD.
We have previously shown increased in vivo uptake of [3H]nimodipine in ischemic brain (Hakim and Hogan, 1991) with kinetics of in vivo binding consistent with saturable and specific binding to the L-type VSCC (Hogan et al., 1991). This increased uptake is rapidly reversible with restoration of CBF (Hogan and Hakim, 1992; Takizawa et al., 1994) and we concluded that in vivo [3H]nimodipine binding may be used to identify transient cell-membrane depolarization and thus infer activation of the L-type VSCC. The current study extends these observations to a model of nonischemic depolarization in which interpretation of binding changes is not confounded by severe CBF depression and histologic injury.
The 14% increase of [3H]nimodipine uptake during CSD is less than the 90% increase observed in irreversible focal cerebral ischemia (Hakim and Hogan, 1991). In ischemia, tissue depolarization is continuous whereas CSD produces an intense but transient depolarization. In vivo binding of [3H]nimodipine to the L-type VSCC may require up to 30 minutes to reach equilibrium (Hogan et al., 1991). Binding to the L-type VSCC is reversible with dissociation occurring over minutes in membrane fraction preparations (Glossmann and Ferry, 1985). The current experimental design was a compromise between attaining equilibrium of distribution of radiotracer between plasma and brain but preventing complete dissociation of [3H]nimodipine bound to the L-type VSCC after repolarization. Only a fraction of the available binding sites during depolarization may be expected to remain labelled at study termination and our observations will underestimate the degree of depolarization during CSD.
Cerebral blood flow may increase by 100% during the initial depolarization of CSD (Hansen et al., 1980) but subsequently CBF decreases to approximately 70% of control values (Lauritzen et al., 1982; Duckrow, 1991). The in vivo uptake of [14C]iodoantipyrine administered in an identical manner to [3H]nimodipine showed only a small increase in cortical uptake after CSD. This increase was significantly less than the more widespread increase observed with [3H]nimodipine despite a greater number of CSD depolarizations after radiotracer administration in the [14C]iodoantipyrine studies. We have previously observed that [3H]nimodipine distribution between brain and plasma was not impaired even with marked CBF reduction (Hogan et al 1991). Thus increased [3H]nimodipine uptake 15 minutes after CSD is not a consequence of CBF fluctuations. The blood volume in brain has been estimated to be 3.4 to 4.5 mL/100 g for the range of arterial P
In cerebral ischemia, CSD may be harmful by increasing the energy demands of tissue in the peri-infarct border zone (Mies et al., 1993). Ischemia-induced CSD may explain the small increase in [3H]nimodipine uptake previously reported (Hakim and Hogan, 1991) in the cingulate cortex adjacent to ischemic brain in a rat model of middle cerebral artery occlusion. It has also been observed that CSD may induce a delayed neuroprotection to subsequent ischemia (Kawahara et al 1994; Matsushima et al., 1996). Both CSD and calcium-channel activation will induce immediate early gene expression (Herrera and Robertson, 1990, Murphy et al., 1991) and may underlie the molecular changes observed in CSD. Thus, the phenomenon of CSD with its nonischemic activation of calcium channels may be a useful experimental tool to further probe the metabolic and molecular responses involved in neuronal survival in neurological disease.
Footnotes
Acknowledgment:
We thank Dr. Kazushi Matsushima for his technical assistance and Ms. Georgette Roy and Ms. Rose Moore their secretarial assistance.