To investigate the role of superoxide in the toxicity of nitric oxide (NO), we examined the effect of nitric oxide synthase (NOS) inhibition on brain infarction in transgenic mice overexpressing CuZn-superoxide dismutase (SOD-1). Male SOD-transgenic mice and nontransgenic littermates (30–35 g) were subjected to 60 min of middle cerebral artery occlusion followed by 24 h of reperfusion. Either NG-nitro-l-arginine methyl ester (l-NAME; 3 mg/kg), a mixed neuronal and endothelial NOS inhibitor, or 7-nitroindazole (7-NI; 25 mg/kg), a selective neuronal NOS inhibitor, was administered intraperitoneally 5 min after the onset of ischemia. At 24 h of reperfusion, the mice were decapitated and the infarct volume was evaluated in each group. In the nontransgenic mice, l-NAME significantly increased the infarct volume as compared with the vehicle, while 7-NI significantly decreased it. In the SOD-transgenic mice, l-NAME-treated animals showed a significantly larger infarct volume than vehicle-treated ones, whereas there were no significant differences between 7-NI- and vehicle-treated mice. Our findings suggest that selective inhibition of neuronal NOS ameliorates ischemic brain injury and that both neuronal and endothelial NOS inhibition may result in the deterioration of ischemic injury due to vasoconstriction of the brain. Since l-NAME increased infarct volume even in SOD-transgenic mice, the protective effect of SOD could result from the vasodilation by increased endothelial NO as well as the reduction of neuronal injury due to less production of peroxynitrite compared to wild-type mice. Moreover, the neurotoxic role of NO might not be dependent on NO itself, but the reaction with superoxide to form peroxynitrite, because of no additive effects of SOD and a neuronal NOS inhibitor.
Despite intensive research efforts, the role of nitric oxide (NO) in the pathogenesis of ischemic brain injury still remains unclear and is controversial. NO has been proposed as playing either a neurotoxic or a neuroprotective role after cerebral ischemia (Dawson et al., 1992; Choi, 1993; Iadecola et al., 1994). One possible mechanism underlying the neurotoxic role of NO is its rapid reaction with superoxide radical (O2−) forming peroxynitrite (ONOO−), which is a strong oxidant in its own right and can form a species with the reactivity of hydroxyl radical (OH) during decomposition (Beckman et al., 1990). Previous reports demonstrated that nitric oxide synthase (NOS) inhibitors attenuate N-methyl-d-aspartate excitotoxicity in rat hippocampal slices (Izumi et al., 1992) and afford protection from glutamate-mediated neurotoxicity in primary cortical cultures (Dawson et al., 1991). Also in in vivo experiments, inhibition of NOS reduces infarct volume and ischemic neuronal damage following focal cerebral ischemia (Nowicki et al., 1991; Buisson et al., 1992; Nagafuji et al., 1992; Nishikawa et al., 1994). On the other hand, NO is believed to be a potent cerebrovasodilator (Palmer et al., 1988) and an inhibitor of platelet aggregation (Radomski et al., 1987), suggesting that NO may increase CBF in the ischemic territory. Some reports show that NOS inhibitors increase infarct size (Yamamoto et al., 1992; Kuluz et al., 1993) and that NO donors reduce brain damage in focal ischemia (Morikawa et al., 1994; Zhang et al., 1994). Therefore, the precise role of NO in cerebral ischemia remains to be established. In the present study, we used transgenic mice, which overexpress CuZn-superoxide dismutase (SOD-1) activity in the brain and other organs (Epstein et al., 1987), to investigate the involvement of superoxide in the toxic role of NO in focal cerebral ischemia.
MATERIALS AND METHODS
Heterozygous transgenic mice of strain TgHS/SF-218–3 carrying human CuZn-SOD genes with a threefold increase in CuZn-SOD enzymatic activity were derived from the founder stock described by Epstein et al. (1987) and genetically characterized by Shi et al. (1994) and were bred on a CD-I mouse background. SOD-transgenic mice were identified by qualitative demonstration of human CuZn-SOD using nondenaturing gel electrophoresis followed by nitroblue tetrazolium staining (Epstein et al., 1987). There were no observable phenotypic differences between SOD-transgenic mice and nontransgenic normal littermates.
Male SOD-transgenic mice and nontransgenic littermates (30–35 g) were subjected to middle cerebral artery (MCA) occlusion and reperfusion according to the method described previously (Kamii et al., 1994). Briefly, mice were anesthetized with chloral hydrate (350 mg/kg i.p.) and xylazine (4 mg/kg i.p.). The rectal temperature of the animals was maintained at 37°C by a Homeothermic Blanket Control Unit (Harvard Apparatus, Natick, MA, U.S.A.). The left femoral artery was cannulated for MABP, Pao2, Paco2, and pH after MCA occlusion. The left common carotid artery was exposed, and the external carotid artery and its branches were isolated and coagulated. A 5–0 monofilament nylon suture, blunted at the tip, was introduced into the internal carotid artery through the external carotid artery stump up to the anterior cerebral artery. After 60 min of proximal MCA occlusion, blood flow was restored by removal of the nylon suture. The mice were decapitated at 24 h of reperfusion and their brains were rapidly removed, frozen, and stored at −70°C. Brain sections (20 μm) were cut at 500-μm intervals on a cryostat at −20°C and stained with cresyl violet to evaluate the infarcted area using a videodigitizer system (Swanson et al., 1990). The infarct volume was calculated by taking the sum of the infarcted area of different brain sections times the interval between the sections (500 μm).
Two kinds of NOS inhibitors were used in the present study. One was NG-nitro-l-arginine methyl ester (l-NAME; 3 mg/kg), a mixed neuronal and endothelial NOS inhibitor, and the other was 7-nitroindazole (7-NI; 25 mg/kg), a selective neuronal NOS inhibitor (Babbedge et al., 1993). Either l-NAME or saline was administered intraperitoneally 5 min after the onset of ischemia. Moreover, either 7-NI or arachis oil was given in another experiment. The saline and arachis oil were used as vehicles for l-NAME and 7-NI, respectively. These compounds or vehicles were administered in SOD-transgenic and nontransgenic mice (n = 10 per group).
Changes in MABP, blood gases, and infarct volume were evaluated by repeated-measures analysis of variance and by Student's t test.
RESULTS
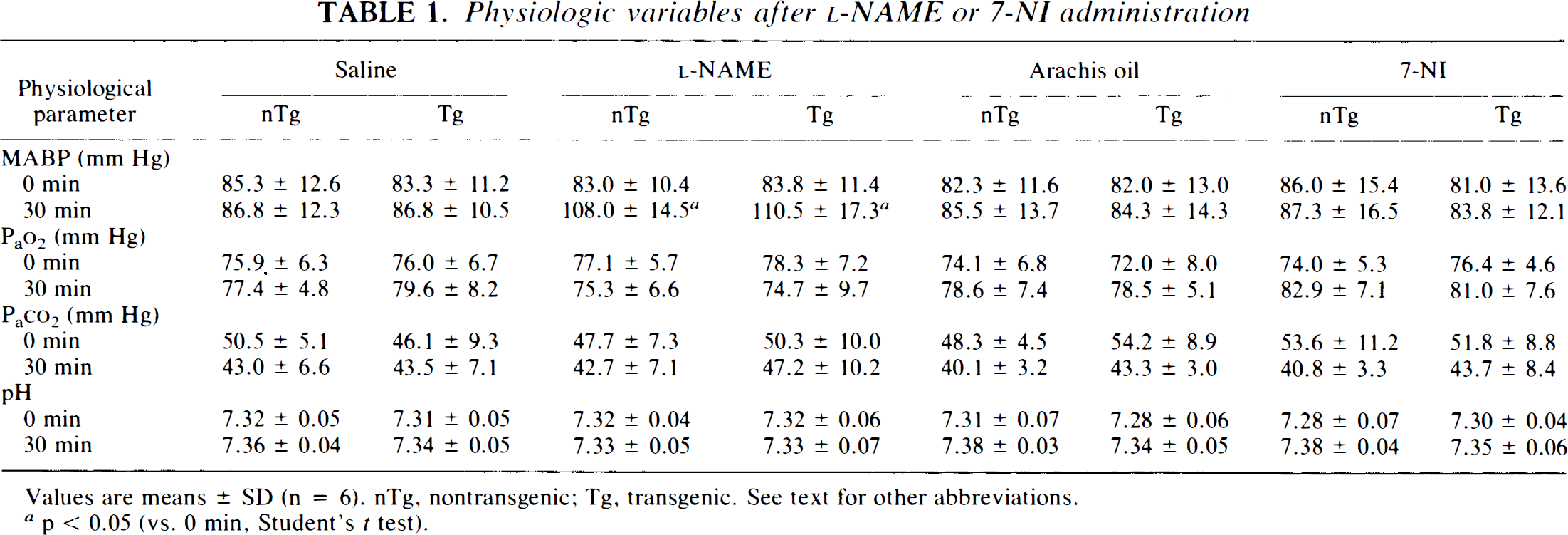
All mice survived the 60 min of transient MCA occlusion. Table 1 shows that l-NAME significantly increased MABP at 30 min after the intraperitoneal injection (at 35 min after the onset of ischemia) in both nontransgenic and SOD-transgenic mice, while 7-NI did not change it. There were no significant differences in blood gas measurements and pH between the drug- and the vehicle-treated animals.
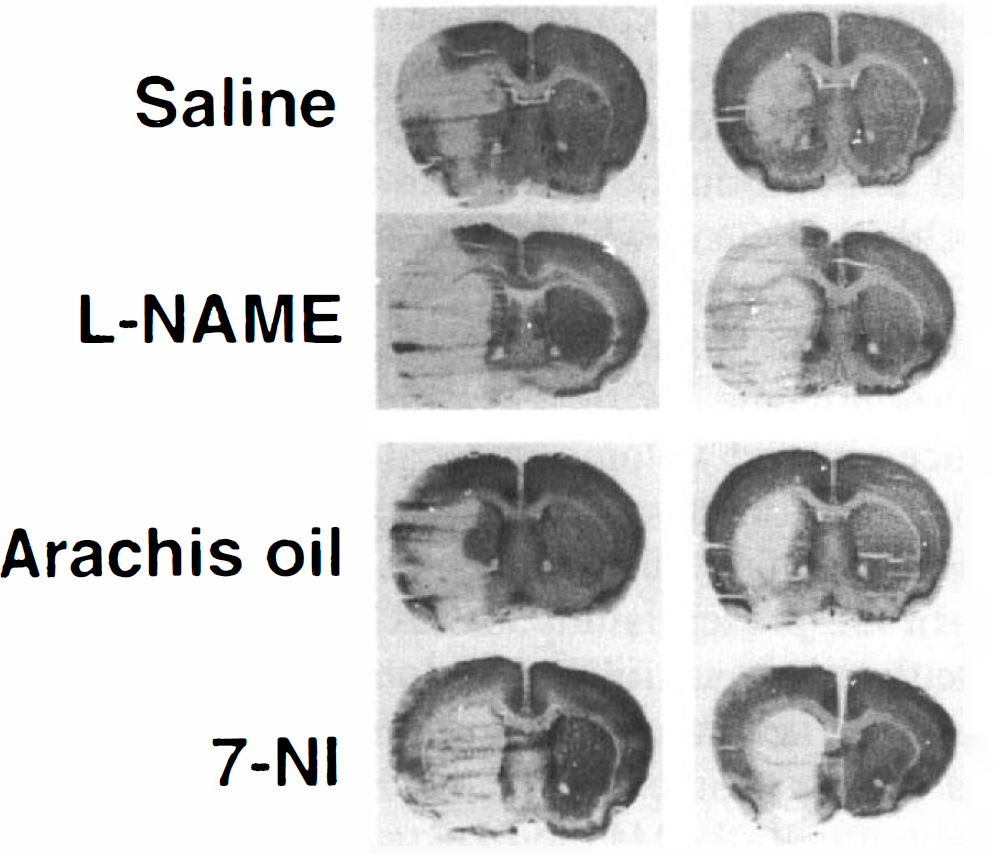
Figure 1 demonstrates representative photographs of brain sections at 24 h after 60 min of MCA occlusion in each group. In both nontransgenic mice and SOD-transgenic mice, l-NAME increased infarcted area as compared with saline. In contrast, 7-NI treatment resulted in decreased infarcted area in nontransgenic mice, while in SOD-transgenic mice, there was no reduction in infarction after 7-NI treatment. SOD-transgenic mice showed smaller infarcted areas than nontransgenic mice in both saline- and arachis oil–treated groups.
Representative photographs of brain sections at 24 h after 60 min of MCA occlusion in nontransgenic (nTg) and SOD-transgenic (Tg) mice. Either l-NAME or saline was administered intraperitoneally 5 min after the onset of ischemia. Moreover, either 7-NI or arachis oil was given in another experiment. The saline and arachis oil were used as vehicles for l-NAME and 7-NI, respectively. The brain sections were cut on a cryostat and stained with cresyl violet. In both nontransgenic and SOD-transgenic mice, l-NAME increased the infarcted area as compared with saline. In contrast, 7-NI treatment resulted in a decreased infarcted area in nontransgenic mice, while in SOD-transgenic mice, there was no reduction in infarction after 7-NI treatment. SOD-transgenic mice showed smaller infarcted areas than nontransgenic mice in both saline- and arachis oil–treated groups. See text for abbreviations.
Physiologic variables after l-NAME or 7-NI administration
Values are means ± SD (n = 6). nTg, nontransgenic; Tg, transgenic. See text for other abbreviations.
p < 0.05 (vs. 0 min, Student's t test).
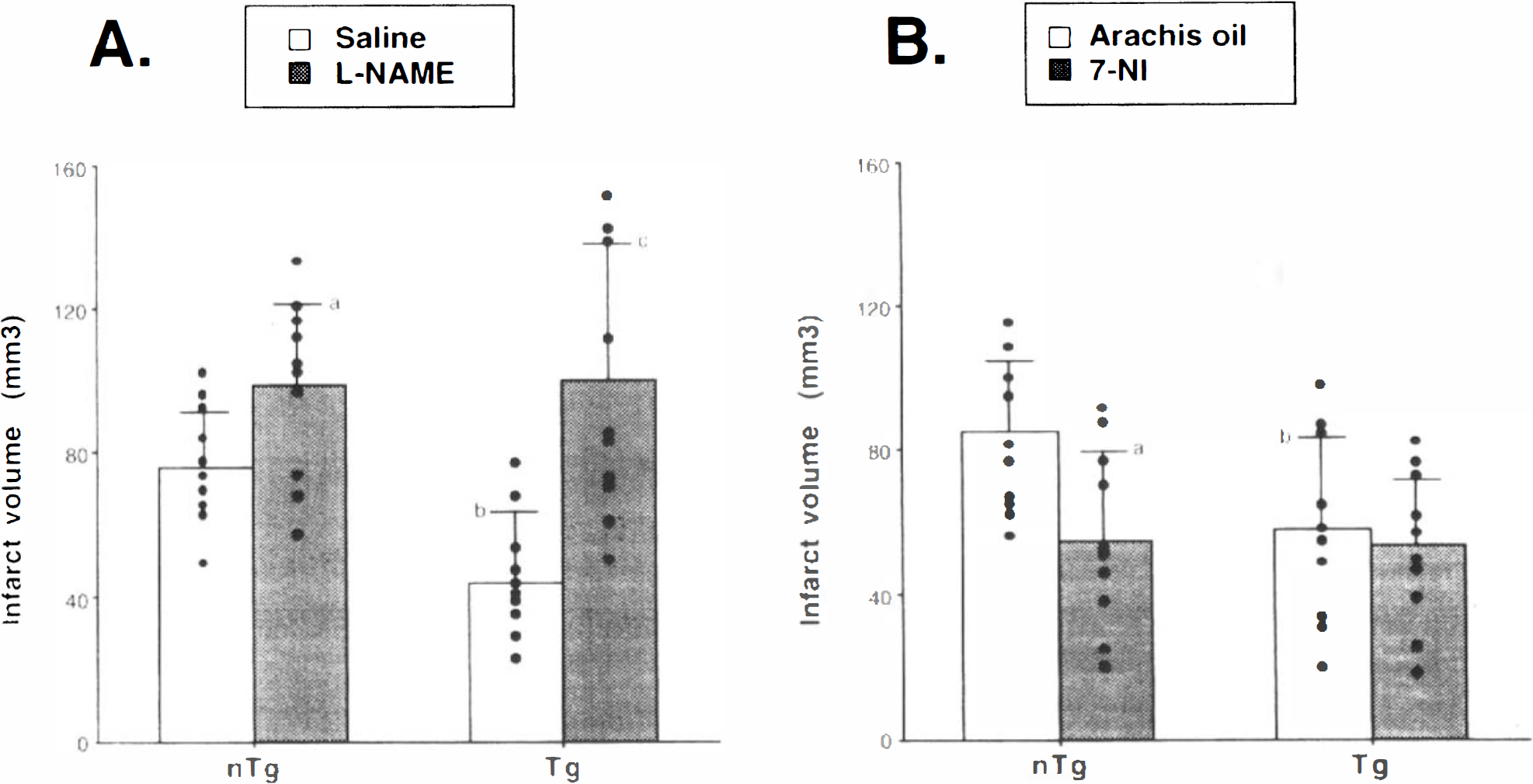
Figure 2 shows the infarct volume in each group (n = 10 per group). In Fig. 2A, l-NAME treatment significantly increased infarct volume when compared with saline treatment in nontransgenic mice (98.7 ± 22.6 vs. 75.2 ± 15.3 mm3; p < 0.05; mean ± SD). Also, in SOD-transgenic mice, l-NAME-treated animals demonstrated a significantly larger infarct volume than saline-treated ones (100.8 ± 38.2 vs. 44.2 ± 19.5 mm3; p < 0.01). In the saline-treated group, SOD-transgenic mice showed significantly smaller infarct volume than nontransgenic mice (44.2 ± 19.5 vs. 75.2 ± 15.3 mm3; p < 0.01). In contrast, as shown in Fig. 2B, 7-NI administration significantly decreased infarct volume as compared with arachis oil administration in nontransgenic mice (54.2 ± 25.1 vs. 85.6 ± 19.4 mm3; p < 0.05), whereas there were no significant differences between 7-NI- and arachis oil–treated animals in SOD-transgenic mice (53.9 ± 18.3 vs. 57.8 ± 25.3 mm3). Also in the arachis oil–treated group, SOD-transgenic mice showed significantly smaller infarct volume than nontransgenic mice (57.8 ± 25.3 vs. 85.6 ± 19.4 mm3; p < 0.05).
The infarct volume in each group (n = 10 per group) at 24 h after 60 min of MCA occlusion in nontransgenic (n I g) and SOD-transgenic (Tg) mice. The infarct volume was calculated by taking the sum of the infarcted area of different brain sections times the interval between the sections (500 μm). A:ap < 0.05 compared with saline group, Student's t-test. bp < 0.01 compared with nTg saline group. cp < 0.01 compared with saline group. B:ap < 0.05 compared with arachis oil treatment in nTg mice. bp < 0.05 compared with arachis oil treatment in non-Tg mice. See text for abbreviations.
DISCUSSION
In the present study, we used two kinds of NOS inhibitors: l-NAME and 7-NI. l-NAME is one of the most widely used NOS inhibitors. This compound inhibits the activity of both neuronal and endothelial NOS, the enzyme that synthesizes NO from l-arginine (Moncada et al., 1991). In contrast, 7-NI is a selective inhibitor of neuronal NOS in vitro and in vivo with potency similar to or greater than that of l-NAME (Babbedge et al., 1993). Moore et al. (1993) reported that 7-NI (25 mg/kg, the same dose as in our study) decreased mouse cerebellar NOS activity measured 15 min thereafter by >55%, while a higher dose of l-NAME (50 mg/kg) produced only 46% inhibition of this enzyme. Moreover, 7-NI (25 mg/kg) did not increase MABP over the 45-min experimental period, whereas the result with l-NAME was a prolonged increase in MABP in a dose-dependent manner (Rees et al., 1990). Although NOS activity was not measured in the present study, the change in MABP after the injection of l-NAME or 7-NI was consistent with previous results.
Our recent studies have shown that infarct size in SOD-transgenic mice is significantly smaller than in nontransgenic mice following focal cerebral ischemia (Kinouchi et al., 1991; Yang et al., 1994). Also, in the present study, a significant reduction in infarction at 24 h of reperfusion after 60 min of MCA occlusion in both saline- and arachis oil–treated groups was demonstrated in SOD-transgenic mice. Although the focal ischemic model used in the present study is different from those in our previous studies, the finding that infarct volume in SOD-transgenic mice was significantly reduced in the present study supports the notion that oxygen free radicals, superoxide in particular, play an important role in the pathogenesis of focal cerebral ischemia and that the preexisting high level of SOD-1 offers cerebral protection against ischemic brain injury (Chan, 1994).
The findings that 7-NI administration significantly decreased infarct volume as compared with arachis oil administration in nontransgenic mice are similar to results published in a recent report (Yoshida et al., 1994). In this report, 7-NI decreased cerebral infarction by ˜25% at 24 h after proximal MCA occlusion in rats, suggesting that enzymatic products of neuronal NOS promote ischemic brain injury. Furthermore, it has been reported that infarct volume significantly decreased at 24 and 72 h after MCA occlusion in mutant mice deficient in neuronal NOS activity (Huang et al., 1994). The mutant mice had regional CBF reductions comparable with those of the control groups after MCA occlusion. However, administration of l-NAME, an inhibitor of both endothelial and neuronal NOS, produced larger infarcts in the mutant group. Therefore, the authors conclude that neuronal NO production appears to exacerbate acute ischemic injury, whereas vascular NO plays a protective role after MCA occlusion. This hypothesis is supported by the observation that administration of l-arginine or NO donors increases CBF and reduces infarct volume in focal cerebral ischemia in the rat (Morikawa et al., 1994; Zhang and Iadecola, 1994; Zhang et al., 1994).
In the present study, l-NAME (3 mg/kg) significantly increased infarct volume as compared with saline in nontransgenic mice. Since l-NAME inhibits neuronal and endothelial NOS, the infarct volume after l-NAME administration may result from the sum of the protective effects by decreased production of neuronal NO and the deleterious effects by decrease in endothelial NO. Thus, the results in nontransgenic mice suggest that l-NAME, at least at a dosage of 3 mg/kg, caused deleterious effects over protective ones on the ischemic brain in our experiments. Moreover, the infarct volume after l-NAME administration in SOD-transgenic mice was comparable with that of the l-NAME group in nontransgenic mice, indicating that l-NAME abolished the protective effect of SOD. These findings may also lead to the idea that the deleterious effect by less production of endothelial NO is larger than the protective effect by lower levels of superoxide and hydroxyl radicals in SOD-transgenic mice. We believe that a selective endothelial NOS inhibitor, if it were in existence, could cause a larger infarct volume than l-NAME in nontransgenic mice while causing the same infarct volume as l-NAME in SOD-transgenic mice. On the other hand, the level of NO produced by endothelial NOS might be higher in SOD-transgenic mice than in nontransgenic mice, since superoxide can be rapidly scavenged to hydrogen peroxide, but can hardly be reacted with endothelial NO. Therefore, the protective effect of SOD may result from vasodilation by a higher level of endothelial NO as well as a decrease in superoxide and hydroxyl radicals.
In SOD-transgenic mice, there were no significant differences in infarct volume between 7-NI-and arachis oil–treated animals. The lack of an additive effect of SOD and a neuronal NOS inhibitor leads us to believe that the neurotoxic role of NO may be dependent on its reaction with superoxide to form peroxynitrite. However, in the presence of a high level of SOD-1 activity, this reaction is limited. In addition, a smaller amount of superoxide can increase the level of NO by prolonging the half-life of NO itself in SOD-transgenic mice. Although NO itself may induce neuronal injury by the formation of NO-iron complexes with several enzymes necessary for DNA replication and mitochondrial energy production (Moncada et al., 1991), our observations suggest that the reaction of NO with superoxide is the most important determinant of NO-mediated neurotoxicity and the toxic role of NO itself might be negligible in neurons after focal cerebral ischemia.
In conclusion, NO plays an important role in the pathophysiology of focal cerebral ischemia. Vascular NO can reduce ischemic brain injury by increasing CBF, whereas neuronal NO may mediate neurotoxicity following brain ischemia mainly by its reaction with superoxide to generate peroxynitrite. These findings could contribute to a strategy for the treatment of cerebral ischemia.
Footnotes
Abbreviations used
Acknowledgment:
We thank Julie Weigel for her editorial assistance. This work was supported by NIH grants AG-08938, NS-14543, and NS-25372.
References
1.
BabbedgeRCBland-WardPAHartSLMoorePK (1993) Inhibition of rat cerebellar nitric oxide synthase by 7-nitro indazole and related substituted indazoles. Br J Pharmacol110:225–228.
2.
BeckmanJSBeckmanTWChenJMarshallPAFreemanBA (1990) Apparent hydroxyl radical production- by peroxynitrite: implication for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci USA87:1620–1624.
3.
BuissonAPlotkineMBouluRG (1992) The neuroprotective effect of a nitric oxide inhibitor in a rat model of focal cerebral ischaemia. Br J Pharmacol106:766–767.
4.
ChanPH (1994) Oxygen radicals in focal cerebral ischemia. Brain Pathol4:59–65.
5.
ChoiDW (1993) Nitric oxide: foe or friend to the injured brain?Proc Natl Acad Sci USA90:9741–9743.
6.
DawsonTMDawsonVLSnyderSH (1992) A novel neuronal messenger molecule in brain: the free radical, nitric oxide. Ann Neurol32:297–311.
EpsteinCJAvrahamKBLovettMSmithSElory-SteinOBryCGronerY (1987) Transgenic mice with increased CuZn-superoxide dismutase activity: animal model of dosage effects in Down syndrome. Proc Natl Acad Sci USA83:8044–8048.
9.
HuangZHuangPLPanahianNDalkaraTFishmanMCMoskowitzMA (1994) Effects of cerebral ischemia in mice deficient in neuronal nitric oxide synthase. Science265: 1883–1885.
IzumiYBenzAMCliffordDBZorumskiCF (1992) Nitric oxide inhibitors attenuate N-methyl-D-aspartate excitotoxicity in rat hippocampal slices. Neurosci Lett135:227–230.
12.
KamiiHKinouchiHSharpFRKoistinahoJEpsteinCJChanPH (1994) Prolonged expression of hsp70 mRNA following transient focal cerebral ischemia in transgenic mice overexpressing CuZn-superoxide dismutase. J Cereb Blood Flow Metab14:478–186.
13.
KinouchiHEpsteinCJMizuiTCarlsonEChenSFChanPH (1991) Attenuation of focal cerebral ischemic injury in transgenic mice overexpressing CuZn superoxide dismutase. Proc Natl Acad Sci USA88:11158–11162.
14.
KuluzJWPradoRJDietrichWDSchleienCLWatsonBD (1993) The effect of nitric oxide synthase inhibition on infarct volume after reversible focal cerebral ischemia in conscious rats. Stroke24:2023–2029.
15.
MoncadaSPalmerRMHiggsEA (1991) Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev43:109–142.
16.
MoorePKWallacePGaffenZHartSLBabbedgeRC (1993) Characterization of the novel nitric oxide synthase inhibitor 7-nitro indazole and related indazoles: antinociceptive and cardiovascular effects. Br J Pharmacol110:219–224.
17.
MorikawaEMoskowitzMAHuangZYoshidaTIrikuraKDalkaraT (1994) L-Arginine infusion promotes nitric oxide-dependent vasodilation, increases regional cerebral blood flow, and reduces infarction volume in the rat. Stroke25: 429–435.
18.
NagafujiTMatsuiTKoideTAsanoT (1992) Blockade of nitric oxide formation by N-omega-nitro-L-arginine mitigates ischemic brain edema and subsequent cerebral infarction in rats. Neurosci Lett147:159–162.
19.
NishikawaTKirschJRKoehlerRCMiyabeMTraystmanRJ (1994) Nitric oxide synthase inhibition reduces caudate injury following transient focal ischemia in cats. Stroke25: 877–885.
20.
NowickiJPDuvalDPoignetHScattonB (1991) Nitric oxide mediates neuronal death after focal cerebral ischemia in the mouse. Eur J Pharmacol204:339–340.
RadomskiMWPalmerRMJMoncadaS (1987) The role of nitric oxide and cGMP in platelet adhesion to vascular endothelium. Biochem Biophys Res Commun148:1482–1489.
23.
ReesDDPalmerRMJSchulzRHodsonHFMoncadaS (1990) Characterization of three inhibitors of endothelial nitric oxide synthase in vitro and in vivo. Br J Pharmacol101:746–752.
24.
ShiYPHuangEPCarlsonEJEpsteinCJ (1994) The mapping of transgenes by fluorescence in situ hybridization on G-band mouse chromosomes. Mamm Genome5:337–341.
25.
SwansonRAMortonMTWuGTDavidsonCSharpFR (1990) A semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metab10:290–293.
26.
YamamotoSGolanovEVBergerSBReisDJ (1992) Inhibition of nitric oxide synthesis increases focal ischemic infarction in rat. J Cereb Blood Flow Metab12:717–726.
27.
YangGChanPHChenJCarlsonEChenSFWeinsteinPEpsteinCJKamiiH (1994) Human copper-zinc superoxide dismutase transgenic mice are highly resistant to reperfusion injury after focal cerebral ischemia. Stroke25:165–170.
28.
YoshidaTLimmrothVIrikuraKMoskowitzMA (1994) The NOS inhibitor, 7-nitroindazole, decreases focal infarct volume but not the response to topical acetylcholine in pial vessels. J Cereb Blood Flow Metab14:924–929.
29.
ZhangFIadecolaC (1994) Reduction of focal cerebral ischemic damage by delayed treatment with nitric oxide donors. J Cereb Blood Flow Metab14:574–580.
30.
ZhangFWhiteJGIadecolaC (1994) Nitric oxide donors increase blood flow and reduce brain damage in focal ischemia: evidence that nitric oxide is beneficial in the early stages of cerebral ischemia. J Cereb Blood Flow Metab14: 217–226.