
Abstract
There is evidence of an intrinsic renin–angiotensin system in the brain. The goal of the study was to determine whether stimulation of endogenous angiotensin production by applying renin to the brain surface has an effect on pial arteriolar caliber and CBF. Pial vessel diameters were measured through a closed cranial window in anesthetized rabbits. Percent changes of blood flow in the cortical area under the cranial window were simultaneously measured by laser-Doppler flowmetry. Topical application of 0.01–0.1 U/ml renin induced maximum dilation of 18.9 ± 4% (mean ± SD) of pial arterioles within 2 min. Arteriolar calibers thereafter decreased slowly. Flow gradually increased to peak at 38 ± 15% 50 min after renin application. Angiotensin I levels in jugular blood, as measured by radioimmunoassay, increased to a peak 40 min after topical renin application. Angiotensin II levels in jugular blood and both angiotensin I and II levels in blood samples from the femoral artery did not change. Diameter and flow changes were inhibited by intravenous pretreatment with the converting enzyme blocker captopril (10 mg/kg body wt i.v.). Captopril did not affect the vasodilation and flow increase in response to hypercapnia. Topically applied Captopril (10−5 M) blocked renin-induced arteriolar dilation. We conclude that renin increases pial arteriolar diameters and cortical blood flow in the rabbit brain. Stimulation of angiotensin production is likely to be a mediator of this response.
Although debated for many years (Reid, 1977; Ganten, 1978), the existence of a renin-angiotensin system in the brain has now been established. True renin in the brain has been confirmed by its in vivo activity at neutral pH, its inhibition by specific renin antibodies, and the finding of renin mRNA (Saavedra, 1992). Immunohistochemistry revealed that renin is located mainly in neurons and that renin-containing cells are associated with small vessels in the brain (Inagami et al., 1980). The only substrate of renin, angiotensinogen, is abundant in extracellular brain and CSF, accounting for 2–3% of total CSF protein (Lynch and Peach, 1991). The intimate association of angiotensinogen-staining astrocytes with brain microvessels (Imboden et al., 1987) led investigators to suggest that the angiotensinogen found in astrocytes represents a component of a system involved in local control of CBF (Wright and Harding, 1992).
The major effector of the renin-catalyzed pathway, angiotensin II, can produce vasodilation (Toda and Miyazaki, 1981; Haberl et al., 1990; Tamaki et al., 1992) as well as vasoconstriction (Wei et al., 1978a; Whalley and Wahl, 1988) in the cerebral microcirculation. There may be degradation products of angiotensin II, such as angiotensin III and the hexapeptide angiotensin-(3–8), which also have an effect on brain arterioles (Haberl et al., 1991). It has been speculated that the effect of angiotensins on cerebral vessels depends on the species and on the size of the arteries under consideration (Hajdu et al., 1993). CBF studies with intravenous or intraarterial administration of angiotensin II have yielded controversial results. CBF did not change in humans (Olesen, 1972), slightly decreased in cats (Faraci et al., 1988), and increased in rabbits (Tamaki et al., 1992) and rats (Hajdu et al., 1993), when blood pressure was held constant. These studies did not address the question of whether angiotensin produced within the brain may alter CBF. The goal of the present study was to determine whether stimulation of endogenous angiotensin production by topical application of renin to the brain surface has an effect on brain microvessels and CBF.
METHODS
Experiments were carried out in 23 white male New Zealand rabbits (weight 2.9–3.6 kg). The experimental protocol was approved by the German government (Regierung von Oberbayern). The rabbits were anesthetized with sodium pentobarbital (25 mg/kg i.v.) and a mixture of urethane (560 mg/kg s.c.) and α-chloralose (38 mg/kg s.c.). Supplemental doses of pentobarbital were administered as required to maintain anesthesia. The animals were tracheotomized and mechanically ventilated with a positive pressure ventilator. End-expiratory CO2 was continuously monitored with an infrared CO2 analyzer (Heyer GmbH, Bad Ems, Germany) and was maintained at a level of ∼30 mm Hg throughout each experiment by adjusting ventilator rate and volume. Arterial blood pressure was measured by a pressure transducer (Gould, Cleveland, OH, U.S.A.) connected to a cannula that was inserted into the right femoral artery. Arterial blood samples (0.1 ml) were periodically drawn in a glass capillary and analyzed for arterial Pco2, Po2, and pH with a blood gas analyzer (AVL, Bad Homburg, Germany). The animals were kept at a constant temperature of 37.5°C by a rectal thermometer-controlled heating pad.
A closed cranial window was implanted on the midline of the skull as previously described in detail (Levasseur et al., 1975). Removal of the dura on each side of the sinus allowed observation of the pial vessels and the brain surface on both cerebral hemispheres. Two pial arterioles were studied in each animal. Artificial CSF and test solutions were topically applied by superfusion through an inlet–outlet port facility on opposite sides of the window. A third outlet was connected to a pressure transducer for continuous measurement of intracranial pressure. The outflow of the window was set at a fixed height to maintain an intracranial pressure of 5 mm Hg throughout the experiment. The plastic tubing connected to the three openings and the space under the window (∼0.3 ml in volume) were filled with artificial CSF (Levasseur et al., 1975).
Pial arterioles ranging in diameter from 38 to 100 μm and adjacent venules were observed with a trinocular microscope (Leitz GmbH, Munich, Germany). The microscopic field of the window was illuminated with a 100-W halogen lamp and a fiber optic ring probe fixed around the objective of the microscope. The field visualized through the microscope was recorded using a low light level video camera (Panasonic WV 1550/G) mounted on the photo tube of the microscope, a video recorder (Panasonic AG 6200-EG), and a video monitor (Barco CD 233, Kortrijk, Belgium). Total magnification on the video monitor was x80. Pial arteriolar diameters were measured with an image analysis system (Stemmer Elektronik GmbH, Munich, Germany).
Laser-Doppler flowmetry was used to measure blood flow on the brain surface visualized through the cranial window (Haberl et al., 1989). A detector probe of the LD flowmeter, 0.84 mm in diameter, was inserted through an additional opening of the cranial window and fixed in place with dental acrylic. The probe was placed over a cortex area without any large diameter vessels. The probe was kept in a fixed position 1–2 mm above the cortical surface throughout the experiment after a clear and constant flow signal was obtained. A model BPM 403 A instrument (Vasamedics, St. Paul, MN, U.S.A.) was used. The average flow output (time constant 1 s), MABP, intracranial pressure, and end-expiratory COz were recorded continuously with a personal computer running ASYST data acquisition software (Macmillian Software, New York, NY, U.S.A.).
To stimulate the local renin-angiotensin system in the brain, 0.01 and 0.1 U/ml renin in 1 ml CSF were topically applied to the brain surface in a cumulative manner. Renin (lyophilized powder from porcine kidney) was obtained from Sigma Chemicals (Deisenhofen, Germany). One unit renin was defined as liberating 100 μg angiotensin I from angiotensinogen per hour at pH 6.0 at 37°C. Renin was dissolved directly in artificial CSF. Renin was applied as soon as stable readings of diameter and flow were made following repeated topical application of 1 ml mock CSF. Flow was recorded continuously before and after renin application. Diameter readings were made after 2 min and then at 5-min intervals for 60 min after renin application (n = 8).
To investigate whether the responses to renin were mediated by generation of angiotensin, topical application of renin was repeated in a separate group of rabbits pretreated with the converting enzyme (CE) inhibitor Captopril (n = 6). Captopril was administered intravenously 25 min before renin application. Systemic instead of topical application of Captopril was used to obtain uniform enzyme inhibition in all vascular segments possibly contributing to the flow response, rather than local blockade in the area of the cranial window. Captopril (Heyden GmbH, Munich, Germany) was given at a dose of 10 mg/kg body wt, which in previous studies was shown to inhibit brain CE activity (Unger et al., 1988). To determine the effect of Captopril on other vasodilatory stimuli, the response to hypercapnia (ventilation with 5% CO2 in room air) before and after Captopril administration was tested.
To provide evidence of whether renin stimulates the release of angiotensins, angiotensin I and angiotensin II levels were measured in the venous outflow blood of the brain in a separate group of four rabbits before and at 2, 5, 10, 40, and 60 min after topical application of 0.1 U/ml renin. Venous blood rather than the CSF from the cranial window was analyzed, because the volume of fluid from the cranial window was too small to perform reliable measurements and because repeated measurements at several time points after topical renin application were possible in this way. Since neuropeptides are reported to diffuse rapidly by a non-carrier-mediated mechanism from CSF into the blood (Passaro et al., 1982; Pardridge, 1983), angiotensin levels in the venous outflow blood were assumed to indirectly reflect angiotensin production in the brain. Arterial blood samples taken from the femoral artery in the same animals were analyzed for control. For cerebral blood withdrawal, a catheter was inserted into the internal jugular vein. Blood samples were drawn in plastic syringes that contained a mixture of 0.125 M ethylenediaminetetraacetate-Na4 and 0.025 M 1.10 o-phenan-throline to give 0.05 ml/ml blood. Blood was transferred into plastic tubes kept at 4°C, and plasma was prepared by centrifugation (6,000 g, 4°C, 3 min). The plasma was extracted and frozen at −20°C until analysis. The extraction procedure and the radioimmunoassay (RIA) for angiotensin I and II were performed as described by Hermann et al. (1990). Antisera to angiotensin I and angiotensin II were produced in white New Zealand rabbits, and 125I-labeled angiotensin I and angiotensin II were purchased from New England Nuclear (NEN Dupont, Germany). Assays were performed in 0.05 M Tris–HCl buffer (pH 7.2) with 0.1% bovine serum albumin using 0.1-ml samples. Both antibodies were used at a final dilution of 2:200,000 with a 50% intercept at 18 and 10 fmol of angiotensin I and angiotensin II. The detection limit for both assays was 1.0 fmol/tube. The cross-reaction of the angiotensin I antibody with angiotensin II, angiotensin III, and angiotensin-(3–8) was <0.1% and with other peptides <0.1%. The cross-reaction of the angiotensin II antibody with angiotensin III and angiotensin-(3–8) was 100%, with shorter angiotensin fragments <0.1%, and with other peptides <0.01% (Hermann et al., 1988).
Data are expressed as means ± SD. Diameters and flow values at different time points after renin application as well as angiotensin I levels in jugular venous blood were compared by multivariate analysis of variance. The responses to renin and renin in the presence of Captopril were compared by the nonparametric Mann-Whitney test. Values of p < 0.05 were considered statistically significant.
RESULTS
Topical application of 0.01 U/ml renin did not change pial arteriolar diameters or flow (data not shown). Subsequent cumulative application of 0.1 U/ml renin induced 18.9 ± 4% dilation of pial arterioles within 2 min (Fig. 1). Diameters slowly returned toward baseline. There was no change in pial venular diameters. Flow increased after topical renin application, but the increase was delayed. Maximal flow was attained at 50 min after renin application. MABP did not change significantly during the 60-min observation period (Fig. 1).
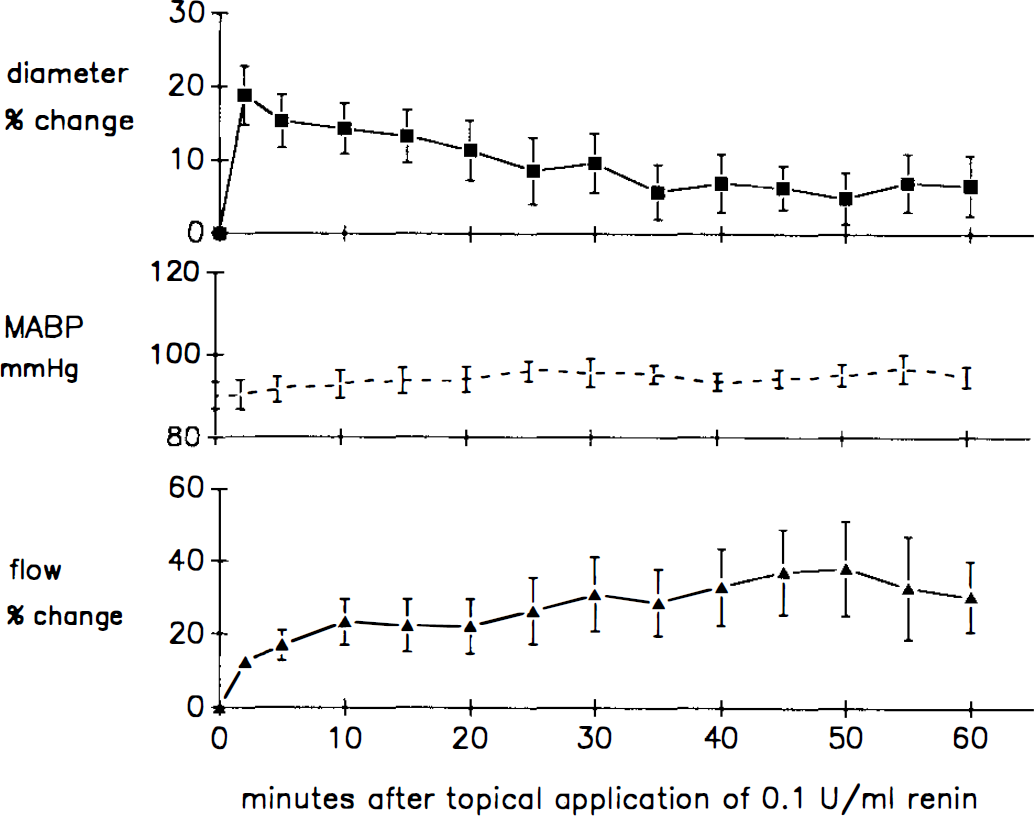
Effect of topical application of 0.1 U/ml renin on pial arteriolar diameter, MABP, and laser–Doppler flow. Data represent means ± SD of eight experiments. Baseline vessel diameter in the eight rabbits was 81 ± 5 μm. Multivariate analysis of variance indicated that diameters were significantly different from baseline 2–30 min after renin application (p < 0.05). Flow was different from baseline 20–60 min after renin application (p < 0.05). MABP did not change significantly.
Intravenous pretreatment with 10 mg/kg Captopril inhibited pial arteriolar dilation and the increase of flow induced by topical application of 0.1 U/ml renin (Fig. 2). Pretreatment with Captopril decreased MABP from 92 ± 4 mm Hg to a new stable baseline of 67 ± 6.5 mm Hg within 20 min, associated with a 13.9 ± 3.7% autoregulatory dilation of the pial arterioles and no change in flow. Dilation in response to systemic hypercapnia before intravenous Captopril was 16.5 ± 2.3% and 20 min after Captopril pretreatment was 15.6 ± 2.1% for ΔPco2 of 19.2 ± 2 and 18.9 ± 3 mm Hg, respectively (n = 4). To provide further evidence that angiotensin inhibition rather than the autoregulatory decrease in transmural pressure by intravenously applied Captopril blocks renin-induced dilation, the response of pial arterioles to 0.1 U/ml renin was studied after topical application of 10−5 M Captopril in three additional rabbits. Topically applied Captopril did not change pial arteriolar diameters or MABP. Topical application of renin in the presence of 10−5 M Captopril had no effect on pial arterioles (+1.2 ± 1%, six arterioles in three rabbits).
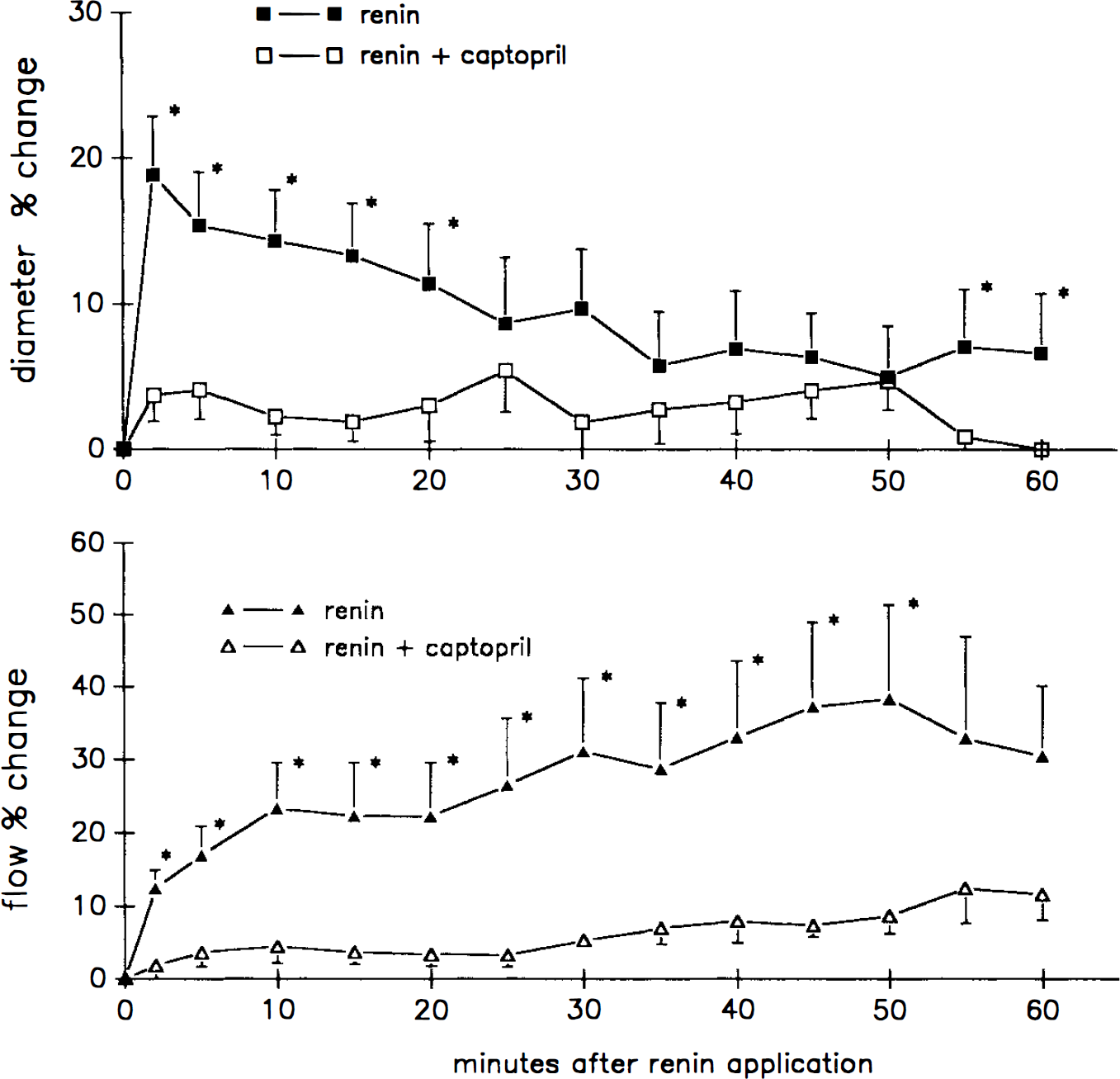
Inhibition of the diameter and flow response to 0.1 U/ml renin by intravenous pretreatment with 10 mg/kg i.v. Captopril. Diameter values at all time points and flow values 2–50 min after renin application were significantly different from the responses to renin in the absence of Captopril (Mann-Whitney test, *p < 0.05).
Angiotensin I levels, as measured by RIA in the internal jugular blood, increased from 2 min to a peak value at 40 min after topical renin application (Fig. 3). Angiotensin II levels in the internal jugular blood did not change from baseline throughout the observation period. Angiotensin I levels (410 ± 48 pg/ml) and angiotensin II levels (50 ± 26 pg/ml) in samples taken from the femoral artery did not change after topical renin application.
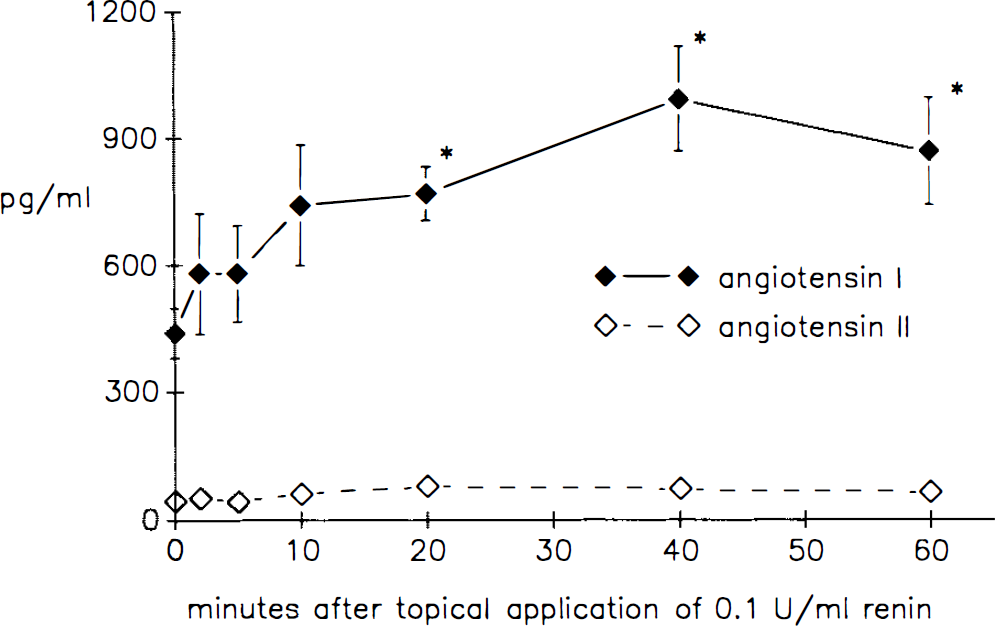
Angiotensin I and angiotensin II levels in jugular venous blood before (time 0) and 2, 5, 10, 20, 40, and 60 min after topical renin application. “Significantly different from the zero value (multivariate analysis of variance, p < 0.05).”
DISCUSSION
The new finding of our study is that renin induces pial arteriolar dilation and a delayed CBF increase in the rabbit brain. Inhibition by the CE inhibitor Captopril is compatible with an angiotensin-mediated mechanism of the responses to renin.
Angiotensins have been shown in previous investigations to produce dilation of cerebral arterioles in rabbits (Haberl et al., 1991; Tamaki et al., 1992; Haberl, 1994) and rats (Haberl et al., 1990; Hajdu et al., 1993) and vasoconstriction in cats (Acar and Pickard, 1978; Wei et al., 1978b; Whalley and Wahl, 1988) and hamsters (Mayhan et al., 1986; Joyner et al., 1988). More consistent results were obtained with larger cerebral arteries of cats (Edvinsson et al., 1979; Mayhan et al., 1988), dogs (Manabe et al., 1989; Toda et al., 1990), rats (Näveri et al., 1994), and humans (Whalley et al., 1987; White and Robertson, 1987), where angiotensin II induced contraction in in vivo and in vitro models. Thus, the impression emerges that the response to angiotensin II depends on both the species and the size of the cerebral vessels under observation. CBF studies have shown an increase in response to intracarotid infusion of angiotensin II in rabbits and rats (Tamaki et al., 1992; Hajdu et al., 1993) and no change during intravenous infusion in cats (Faraci et al., 1988). Resistance data from these studies suggest that the sensitivity of dilating factors released by angiotensins increases with smaller size of the arterioles. While the CBF response after systemic application depends on the relative contribution of large and small arteries to total resistance, locally generated angiotensins may have more contact with small microvessels than with large arteries, thereby accounting for the considerable CBF response in the present study. The response of human cerebral arterioles to angiotensin II and the significance for CBF regulation in humans are not known.
The presence of renin in the brain has been established by a number of different approaches (Unger et al., 1988). Most importantly, renin mRNA has been quantified in brain tissue by special solution hybridization assays (Paul et al., 1988) and by the polymerase chain reaction method (Iwai and Inagami, 1992). Renin mRNA is present only in small amounts, and renin activity in the brain has been estimated in the range of 0.1–25 ng angiotensin I/h/ml (Husain et al., 1981), which would be less than the activity used in the present study. It is believed, however, that renin activity is distributed heterogeneously in the brain, with high expression confined to small regions that were not identified by the techniques currently available (Moffett et al., 1987; Paul et al., 1993). Levels that were shown to be vasoactive in the present study may be reached in these microdomains.
There was a striking difference in the temporal pattern of the pial arteriolar response to renin and the delay in the flow increase. The reason for this dissociation remains uncertain. One possibility is that unmeasured subsurface arterioles dilate later than the pial arterioles. As speculated in a previous study, the intraparenchymal arterioles may be the major determinants of resistance in the vascular bed assessed by laser-Doppler flowmetry (Haberl et al., 1989). Diffusion of renin into deeper layers of the cortex or indirect effects of renin, with release of dilator substances in a time-dependent manner, could be potential causes for such a delay. Regardless of the exact mechanism, however, the data indicate that release of renin in the brain may cause long-lasting changes of local CBF.
Angiotensins are the most likely mediators of the responses to renin. The responses to renin were antagonized by the CE inhibitor Captopril. Captopril did not reduce dilatory responses of the pial arterioles in general, as shown by the preserved dilation to systemic hypercapnia. The possibility remains that vasoactive substances other than angiotensins are released by renin and CE. The only biologic action of renin, however, of which we are aware, is the cleavage of angiotensinogen with the release of angiotensin I.
Intravenous instead of topical application of Captopril was used to obtain widespread enzyme inhibition in the brain rather than localized inhibition in the vicinity of the cranial window chamber. We decided to use this protocol after the delayed flow response to renin, possibly involving remote intraparenchymal arterioles, was observed. One disadvantage of systemic application was the MABP response to Captopril, associated with autoregulatory dilation of the pial arterioles. The conclusion that CE inhibition rather than the autoregulatory change in vascular tone affects vasodilation to renin is supported by inhibition by topically applied Captopril, which had no systemic blood pressure effect. Controversy surrounds the question of how completely CE activity in the brain is blocked by intravenous Captopril. The majority of studies indicate that brain CE is inhibited after short-term Captopril treatment, although inhibition may not be uniform throughout the brain (Unger et al., 1988).
The generation of angiotensin I is supported by the finding that angiotensin I levels increase in internal jugular blood following topical renin application. Neuropeptides in CSF rapidly enter blood via absorption into the superior sagittal sinus at the arachnoid villi, with a half-time of 13 min following topical bolus injection in rabbits (Passaro et al., 1982; Pardridge, 1983). The measurement of jugular blood, therefore, may indirectly reflect the change in angiotensin levels in CSF. Alternatively, angiotensin I generation from circulating angiotensinogen may be catalyzed after renin diffusion into the blood. Angiotensinogen in plasma is relatively abundant [∼1 μM (Lynch and Peach, 1991)], and a direct effect of renin on circulating angiotensinogen may be excluded only by not finding an increase in angiotensin I in CSF. CSF studies were not done in the present study because reliable RIA determinations were not possible with the small volumes obtained from the cranial window chamber. The failure to find an increase of angiotensin II levels in jugular blood may be due to the fact that degradation of angiotensin II in circulating blood is exceedingly fast, with a half-life of 15–30 s (Johnston, 1990). Unlike the angiotensin II antibody, the angiotensin I antibody had low cross-reactivity with breakdown products such as angiotensin III and angiotensin-(3–8). Thus, the greater response of angiotensin I to renin administration may not be due to cross-reactivity of the angiotensin I antibody with shorter angiotensin fragments.
There is a striking coincidence of the increase in angiotensin I levels (Fig. 3) and the flow increase following renin application (Fig. 1). We considered the possibility that the flow response may have been caused by angiotensins acting from the luminal rather than the abluminal side. Intraarterially applied angiotensin II has been shown in previous studies to increase CBF in rabbits and rats (Tamaki et al., 1992; Hajdu et al., 1993). However, arterial levels of angiotensin I and II in samples taken from the femoral artery did not change following topical renin application. Angiotensin in cerebral venules and veins was not likely to have an effect on flow, because pial venular diameters did not change. We deem it more likely, therefore, that angiotensins produced within the CSF compartment may be the trigger of the caliber and flow changes seen in the present study.
The physiological relevance of our observation depends on how renin is released and regulated in the brain. Renin is present in high levels in nerve terminals and is released from brain slices by potassium-induced depolarization or by electrical stimulation (Saavedra, 1992). Angiotensinogen-staining astrocytes are in intimate contact with brain microvessels (Imboden et al., 1987). Renin-catalyzed angiotensin formation from angiotensinogen is believed to occur intracellularly in neurons and not in glial cells (Moffett et al., 1987; Saavedra, 1992). Particularly high concentrations of renin are found in certain brain areas such as the pituitary gland (Paul et al., 1993). Renin expression appears to be increased or decreased in spontaneously hypertensive rats, depending on the brain area studied (Iwai and Inagami, 1992; Paul et al., 1993). In view of the profound and long-lasting effects of topically applied renin on CBF in this study, it remains a challenge to find physiological and pathophysiological situations in which CBF may be regulated by the renin-angiotensin system.
Footnotes
Acknowledgment:
We thank Mrs. J. Gailer and Mrs. J. Benson for the help in the preparation of the manuscript. The study was supported by the Deutsche Forschungsgemeinschaft.