
Abstract
The potential of nitric oxide (NO) to influence positively or negatively the outcome of mechanically induced focal cerebral ischemia is still controversial. Recent evidence suggests that NO of vascular origin, whether synthesized from exogenously administered L-arginine (L-Arg) or from NO donor compounds, is beneficial but that of neuronal origin is not. However, the therapeutic potential of NO to ameliorate stroke induced by arterial thrombosis has not been reported. We assessed the therapeutic effect of L-Arg administration in spontaneously hypertensive rats (SHR) subjected to permanent photothrombotic occlusion of the distal middle cerebral artery (dMCA). The ipsilateral carotid artery was left unligated to enhance L-Arg delivery into the putative penumbral region. Local CBF (LCBF) was assessed at 30 min by the [14C]iodoantipyrine technique (n = 9), while histological infarct volumes and index of peripheral ischemic cell change were determined at 3 days (n = 7). Rats (n = 9) given 300 mg/kg L-Arg at 18 and 3 h before photothrombotic dMCA occlusion and at 5 min afterward displayed no significant differences in LCBF compared with animals (n = 8) injected with water (the carrier vehicle) and similarly irradiated. Infarct volumes were also similar, being 37.0 ± 9.7 mm3 (SD) in the vehicle-treated and 49.1 ± 17.2 mm3 (SD) in the L-Arg-treated groups (both n = 7), as were assessments of ischemic neuronal density in the penumbra. In contrast, L-Arg administered intravenously in a dose of 300 mg/kg to nonischemic SHR (n = 5) increased cortical CBF by ∼75% during a 70-min observation period. We conclude that thrombotic processes superimposed upon cerebral ischemia may facilitate tissue reactions that offset the potentially beneficial effect of L-Arg, and this caveat must be considered when proposing L-Arg for clinical treatment of focal thrombotic stroke.
Keywords
The synthesis of nitric oxide (NO) in the setting of focal ischemia, and its implications for tissue recovery or deterioration, have recently been subjected to much scrutiny, most of it controversial. In animals administered nonspecific NO synthase inhibitors while being subjected to mechanically induced permanent focal cerebral ischemia, infarct volumes have been reported to increase (Yamamoto et al., 1992; Zhang and Iadecola, 1993; Kuluz et al., 1993), decrease (Nowicki et al., 1991; Buisson et al., 1992; Nagafuji et al., 1992; Ashwal et al., 1993; Nishikawa et al, 1993), or not change significantly (Dawson et al., 1992) compared with animals administered vehicle. The basis for these discrepant reports is likely the complex involvement of NO in fundamental biochemical processes (Dalkara and Moskowitz, 1994). For example, therapeutic rationales for NO synthase inhibition include suppression of the potentially damaging peroxynitrite free radical (Beckman, 1991) or of N-methyl-D-aspartate-mediated cytotoxicity (Choi, 1993; Iadecola et al., 1994). On the other hand, NO synthesized in the vascular compartment is critical for maintenance of vascular function. NO can prevent platelet aggregation or disaggregate forming thrombi (Radomski et al., 1990; Radomski and Moncada, 1993) and also has been identified as the endothelium-derived relaxing factor, which through its vasodilator property is responsible for maintenance of vascular tone (Faraci and Brian, 1994). The substrate for NO synthase is L-arginine (L-Arg), and several enzyme isoforms have been found in different tissues, suggesting that NO has different functions according to the location of its synthesis (Forstermann et al., 1991; Marietta, 1993).
Mitigation of infarct development is currently recognized to involve treatment of the penumbra, the bioenergetically unstable region of diminished flow surrounding the infarct core (Hossmann, 1988), which can be incorporated into the evolving infarct if nothing is done to impede its metabolic degeneration (Back et al., 1995). Penumbral salvage might reasonably be based on enhancement of local CBF (LCBF) via activated collateral channels. In a recent test of this hypothesis, the vasodilator property of NO was augmented by means of L-Arg administration to spontaneously hypertensive rats (SHRs). This resulted in enhanced blood flow in the penumbral zone of the middle cerebral artery (MCA) territory following MCA occlusion (MCAO) (Morikawa et al., 1992a) and likely facilitated the reductions in infarct volume observed previously in SHRs (Morikawa et al., 1992b, 1994). In related work by others, administration of NO donor compounds was found also to reduce infarct volume and improve blood flow to marginally perfused tissues in SHRs (Zhang and Iadecola, 1993; 1994; Zhang et al., 1994). Subsequently, normal SV129 mice and their mutant knockout (Kn) counterparts deficient in neuronal NO synthase were subjected to MCAO by the suture technique (Huang et al., 1994). The results exhibited an interesting and vital complementarity: Compared with normals, the mutant Kn mice displayed decreased infarct volume and less severe neurological deficits, but infarct volume was increased in Kn mice treated with the endothelial NO synthase inhibitor nitro-L-arginine. These observations strongly indicate that, in the context of focal ischemia, the toxic effects of neuronal NO must be either mitigated by direct inhibition of neuronal NO synthase or dominated by vascular NO via collateral flow enhancement.
Such correlations, gleaned via models of mechanically induced MCAO in rodents, have been very helpful in providing a simple basis for consolidating previous disparate reports of NO function. However, clinical stroke is often complicated by thrombosis, and this condition could not be addressed in the previous studies. In an effort to simulate vascular mechanisms believed to be activated during stroke, we have developed rodent models of photochemically induced thrombotic stroke. These models thus facilitate study of the interaction between thrombotic processes and simultaneously induced focal cerebral ischemia. In this connection, ultra-structural indexes of tissue injury were monitored following reversible MCAO induced by photochemical or mechanical means. Tissue exposed to thrombosis revealed much more severe changes than those exposed to mechanically induced ischemia, including marked vacuolization of the neuropil, swollen astrocytic processes, and the frequent appearance of dark shrunken neurons in regions well perfused by the microcirculation (Dietrich et al., 1989). Such signs of irreversible tissue deterioration demonstrate that thrombosis combined with ischemia greatly exacerbates the expression of reperfusion injury, probably owing to the secretion of platelet-derived factors such as serotonin (Wester et al., 1992), which damage microvascular integrity in downstream regions (Dietrich et al., 1988a, b ). Given these complications, we wished to determine whether L-Arg could still provide tissue protection to some degree in a thrombotic/ischemic environment, where the benefits of L-Arg-induced collateral activation might be compromised by enhanced vascular leakage and parenchymal exposure to toxic factors.
MATERIALS AND METHODS
Male SHRs (n = 31), weighing 270–345 g, were injected with L-Arg 300 mg/kg i.p. 18–20 and 3 h before distal MCAO (dMCAO) and 5 min after dMCAO (similar to Morikawa et al., 1992a). The last dose of L-Arg was administered intravenously. Animals prepared for histopathological analysis were given an additional dose of L-Arg 300 mg/kg i.p. 2 h after dMCAO. Animals were randomized for either LCBF (n = 17) or histopathological analysis (n = 14). Comparison animals underwent all experimental procedures, but were given equivalent volumes of sterile water in a similar manner. Cortical CBF and common carotid arterial flow were measured in another group of SHRs (n = 7) in which focal ischemia was not induced, but instead the animals were subjected to intravenous infusion of 300 mg/kg L-Arg over a 10-min period (as in Morikawa et al., 1994).
General preparation for dMCAO
Anesthesia was induced with 4% halothane and maintained with a 70:30 mixture of N2O/O2 and 0.5% halothane; animals were mechanically ventilated after muscle paralysis with pancuronium bromide 0.35 mg/kg i.v., followed by 0.1 mg/kg every 30 min for maintenance. Femoral venous and arterial catheters (PE-50) were inserted for fluid administration, blood gas determination, and blood pressure monitoring. Rectal and right temporalis muscle temperature were maintained at 37.0 and 36.0°C, respectively, by thermostatically regulated servo-controlled heating devices.
The right dMCA was exposed (Markgraf et al., 1993), and the beam of a 562-nm argon-pumped dye laser operating at 20 mW (model CR599; Coherent, Palo Alto, CA, U.S.A.) was diffracted by a Ronchi transmission grating into an 11-mW zero-order beam and two first-order 4.5-mW beams (total power 20 mW). The photosensitizing dye rose bengal (15 mg/ml) was administered intravenously at a dose of 20 mg/kg over a 90-s period, and irradiation of the dMCA (dMCAO) was begun with the zero-order beam placed most proximally and the first-order beams placed more distally at branch points (shown in Watson et al., 1995). Four minutes of irradiation was sufficient to occlude the dMCA at the three selected points, by means of platelet thrombi formed in response to the photochemically damaged endothelium. Similar procedures were used previously (Yao et al., 1993), but in the present study the common carotid arteries were not ligated to allow unimpeded access of drugs to the ischemic penumbral region via the collateral circulation.
Measurement of local and cortical CBF
LCBF was measured autoradiographically at 30 min following the induction of focal ischemia by the [14C]iodoantipyrine method (Sakurada et al., 1978) according to the procedures of Dietrich et al. (1991). The study was terminated by decapitation; brains were removed in <1.5 min and frozen in liquid nitrogen after prehardening them by suspension over liquid nitrogen vapor. Brains were processed for sectioning in a cryostat and were then exposed, together with calibrated [14C]-methylmethacrylate standards, to Kodak Hyperfilm Beta-max film for 10 days and digitally imaged according to Zhao et al. (1995).
In the SHR group administered L-Arg in the absence of focal ischemia, cortical CBF and common carotid arterial flow were measured simultaneously by a laser-Doppler flow probe (model BPM2; Vasamedics) placed over the right cerebral cortex and a 1-mm-diameter ultrasonic perivascular flowmeter (model T206; Transonic Systems), respectively, over a 70-min period (see Morikawa et al., 1994) with recordings made every 10 min.
Construction and analysis of averaged three-dimensional autoradiographic data sets
The procedure for reconstructing three-dimensional (3D) autoradiographic images is based on the disparity analysis image alignment algorithm whose theory and validation have been recently published in detail (Zhao et al., 1993, 1995). The subserial coronal sections of each brain studied for LCBF were first computer aligned by disparity analysis. Next, corresponding coronal sections from individual brains were placed in register with one another using a common coronal reference level (bregma +0.7 mm) (Paxinos and Watson, 1982). To permit correlations with functional anatomic regions, coronal outlines taken from the atlas of Zilles (1985) provided the “template,” into whose contours corresponding autoradiographic sections of each brain were mapped at each coronal level by means of an averaging procedure similar to that described already. By this process, an averaged 3D data set, together with a data set representing the SD of the measurement, were calculated and displayed on a video screen. Comparisons between the LCBF data sets for the vehicle- and L-Arg-treated animals groups were undertaken on these averaged and SD data sets. Data analysis was carried out on the neocortex. A digitized brain atlas (based upon Zilles, 1985) was used to overlay the autoradiographic sections and thereby to identify the neocortex at coronal levels ranging from bregma +2.2 mm to bregma −8.8 mm. This procedure allowed neocortical regions extracted from the average and SD image data sets to be segmented into 16 sectors in each hemisphere. These sectors were defined on the basis of radiating lines separated by 12.5°, originating at the center of each section. The mean and SD values for the ith section were calculated by the following formulas:
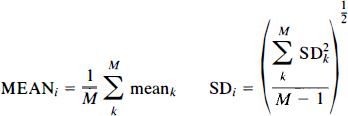
where mean k is the mean value for the kth pixel in the ith sector, SD k is the SD value for the kth pixel in the ith sector, and M is the total number of pixels in the ith sector. The mean value for the ith sector is thus the averaged mean for all pixels of the sector. The SD for each sector was calculated using the same formula as was used to calculate the SD for the corresponding sector from the SD image data set but with a mean value of zero. A t test was used to compare corresponding sectors of the two experimental groups.
Morphological methods
Routine light microscopic analysis was carried out on animals 3 days following dMCAO. The procedures for perfusion-fixation, processing, cutting, and staining of brain sections as well as computerized quantitation of infarct volume have been given previously (Markgraf et al., 1993). To compare infarct volumes derived from histological sections to the stereotaxically based LCBF distribution (see later), compensation must be made for shrinkage artifact during histopathological processing. In a previous study (Wester et al., 1995), correction factors of 1.28 and 1.18 were derived to compensate for areal (coronal section) and linear (anteroposterior) shrinkage, respectively. Stereotaxically corrected volumes are derived by multiplying the histological volumes by the combined correction factor of 1.51 (the product of the linear and areal factors).
To determine whether L-Arg administration might affect the severity of incomplete infarction, characterized by sporadic neuronal necrosis in the penumbra (Markgraf et al., 1993), the number of eosinophilic neurons was quantitated in the paramedian cortex at bregma level + 0.7 mm and in the dorsolateral and lateral cortex at bregma level −4.1 mm (×10 magnification). These counts were compared with the number of normal neurons in the corresponding contralateral areas.
RESULTS
Physiological variables
Physiological variables in the vehicle- and L-Arg-treated SHRs are shown in Table 1. Mean arterial blood pressure averaged 150–160 mm Hg in both groups. Arterial blood gases were in the normal range, and there were no significant intergroup differences.
Physiological variables from vehicle and L-arginine-treated rats

Values are mean ± SD. See text for abbreviations.
Local and regional CBF
LCBF values in vehicle- and L-Arg-treated rats are shown in Table 2. Cortical structures were read at five different coronal levels. The zone of highest-grade ischemia in this model lay in the dorsolateral cortex at bregma level −4 mm, where LCBF fell to mean values as low as 0.14 ml g−1 min−1. At other coronal levels, mean LCBF values in the dorsolateral and lateral cortices lay within the penumbral or suprapenumbral range (as defined in a previous study in normotensive Sprague–Dawley rats) (Back et al., 1995). Comparisons of LCBF values in vehicle and L-Arg rats by unpaired t tests revealed no significant differences for any structure. Representative autoradiographic LCBF images from the two groups are shown in Fig. 1.
Local CBF in vehicle and L-arginine-treated rats
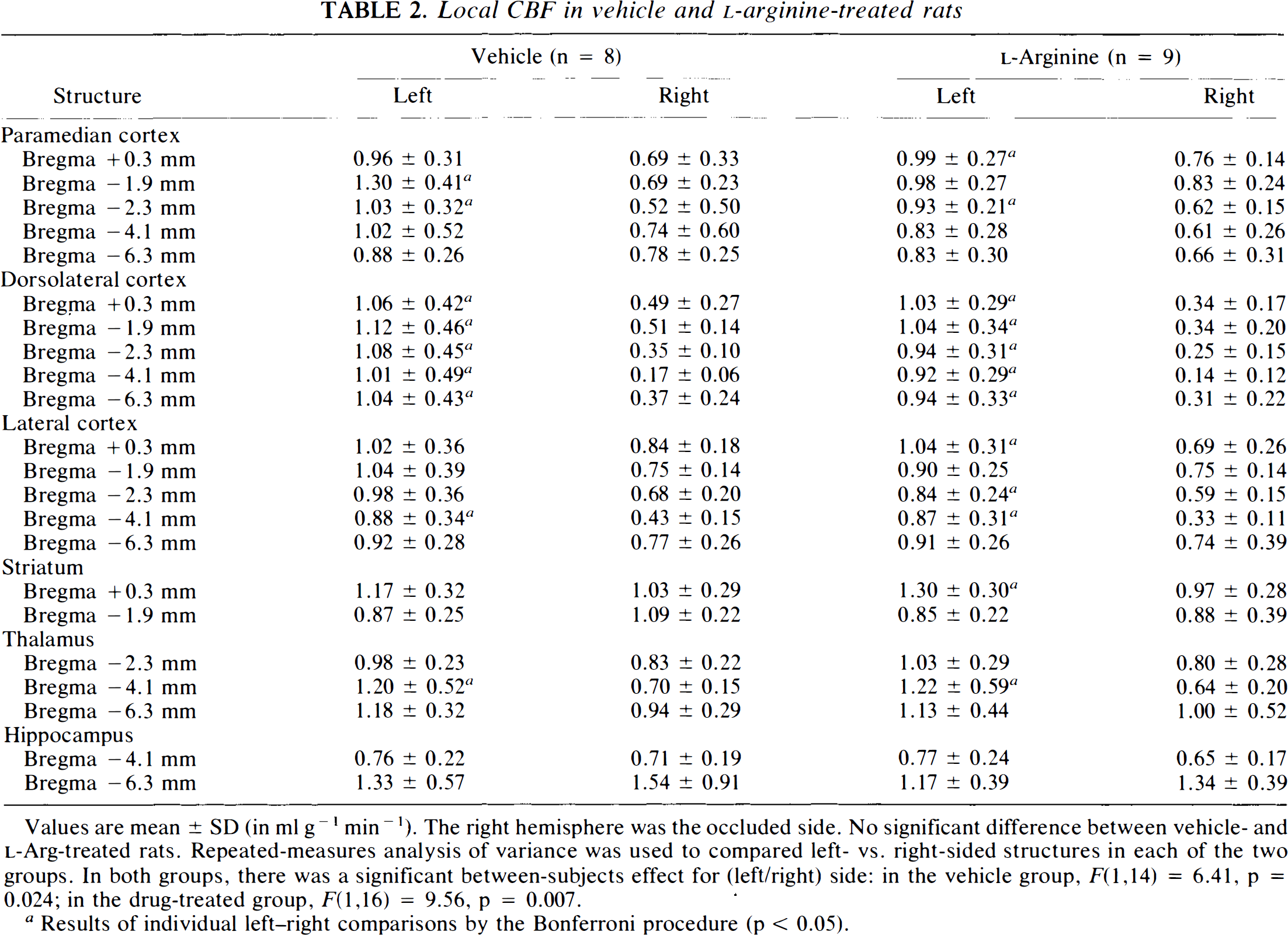
Values are mean ± SD (in ml g−1 min−1). The right hemisphere was the occluded side. No significant difference between vehicle- and L-Arg-treated rats. Repeated-measures analysis of variance was used to compared left- vs. right-sided structures in each of the two groups. In both groups, there was a significant between-subjects effect for (left/right) side: in the vehicle group, F(1,14) = 6.41, p = 0.024; in the drug-treated group, F(1,16) = 9.56, p = 0.007.
Results of individual left-right comparisons by the Bonferroni procedure (p < 0.05).
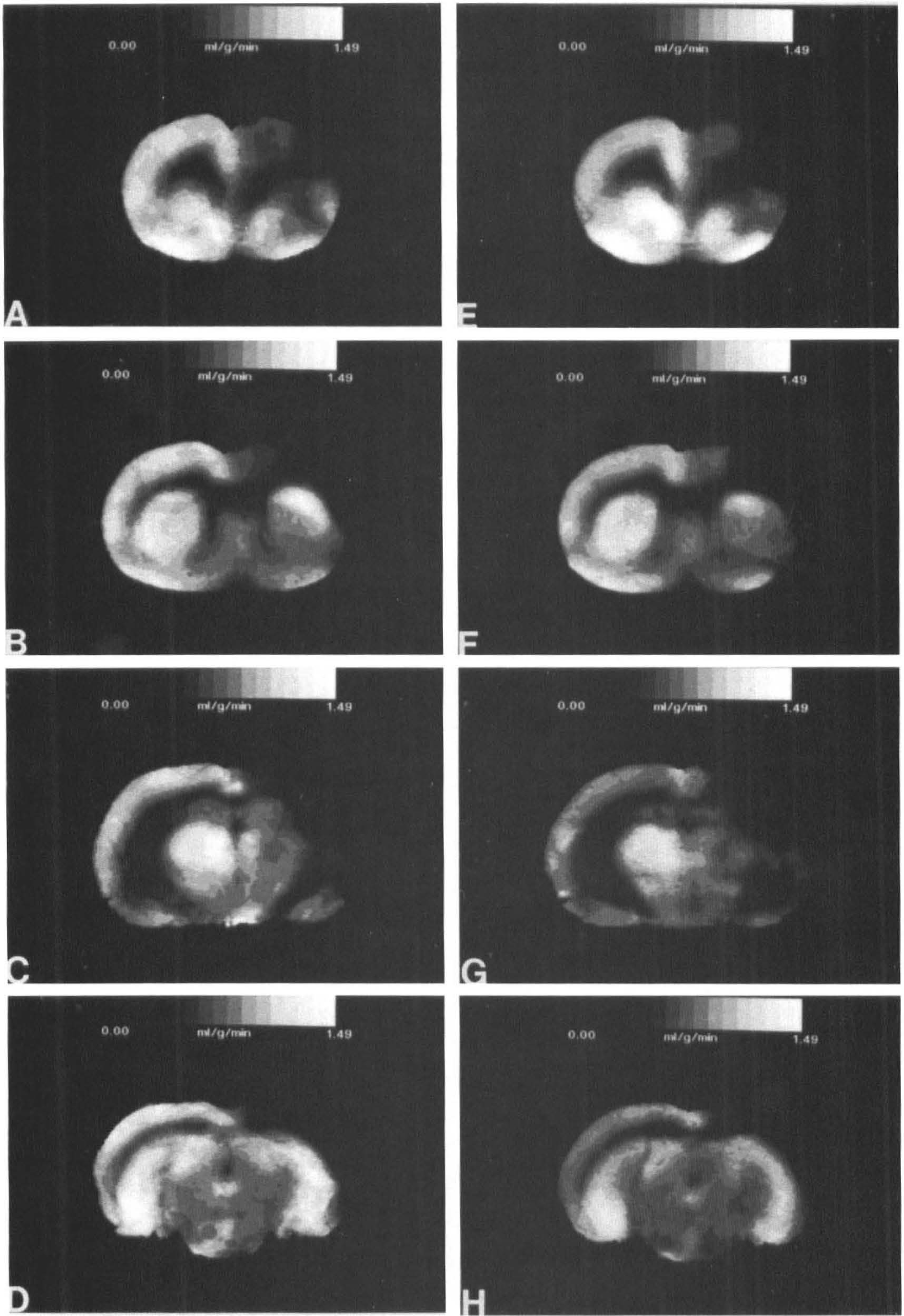
Computer-generated averaged autoradiographic image data sets for local CBF (LCBF) at four different coronal levels (bregma +2.2, +0.3, −2.3, and −6.3 mm).
Infusion of 300 mg/kg L-Arg i.v. to SHRs under nonischemic conditions led to a sustained increase in cortical CBF to 174.9 ± 26.4% (expressed as the average of recordings over the period of 10–70 min following the end of the infusion period) with respect to a baseline of 100 ± 28.5%, obtained over a 30-min preinfusion period (n = 5). This measurement reached a significance level of p = 0.01 in comparison with the baseline cortical CBF by paired t test. On the other hand, blood flow in the carotid artery was not significantly altered by L-Arg administration (data not shown).
Threshold LCBF analysis
For this analysis, the effect of L-Arg or vehicle treatment on blood flow was assessed by determining the volume of cortical tissue displaying LCBF in a specified interval (incremental flow range) and arranging the resultant volumes histogrammatically as a function of LCBF interval. This analysis was accomplished by superimposing and aligning a standardized rat brain atlas (Zilles, 1985) on the averaged 3D image data sets of vehicle- and L-Arg-treated rats. This allowed the neocortical mantle of the left (contralateral) and right (ispilateral) hemispheres to be identified by computer. LCBF threshold analysis was then performed, from which the cortical volumes encompassing LCBF within specified limits could be determined. The results are shown in Fig. 2. The LCBF intervals are expressed as percentage intervals of the control level, accepting an LCBF value of 1.2 ml g−1 min−1 as control (Back et al., 1995). In the left hemisphere (Fig. 2A), the distribution of cortical volumes as a function of normalized LCBF interval was unimodal. Repeated-measures analysis of variance revealed no significant difference between the vehicle- had L-Arg-treated groups. In the right (ischemic) hemisphere (Fig. 2B), the threshold LCBF distribution was bimodal; a cortical volume of ∼40 mm3 and LCBF values below 10% of control, and a volume of ∼65 mm3 had LCBF values below 20% of control. Repeated-measures analysis of variance revealed no significant difference in LCBF distribution between vehicle- and L-Arg-treated rats for the right hemisphere.
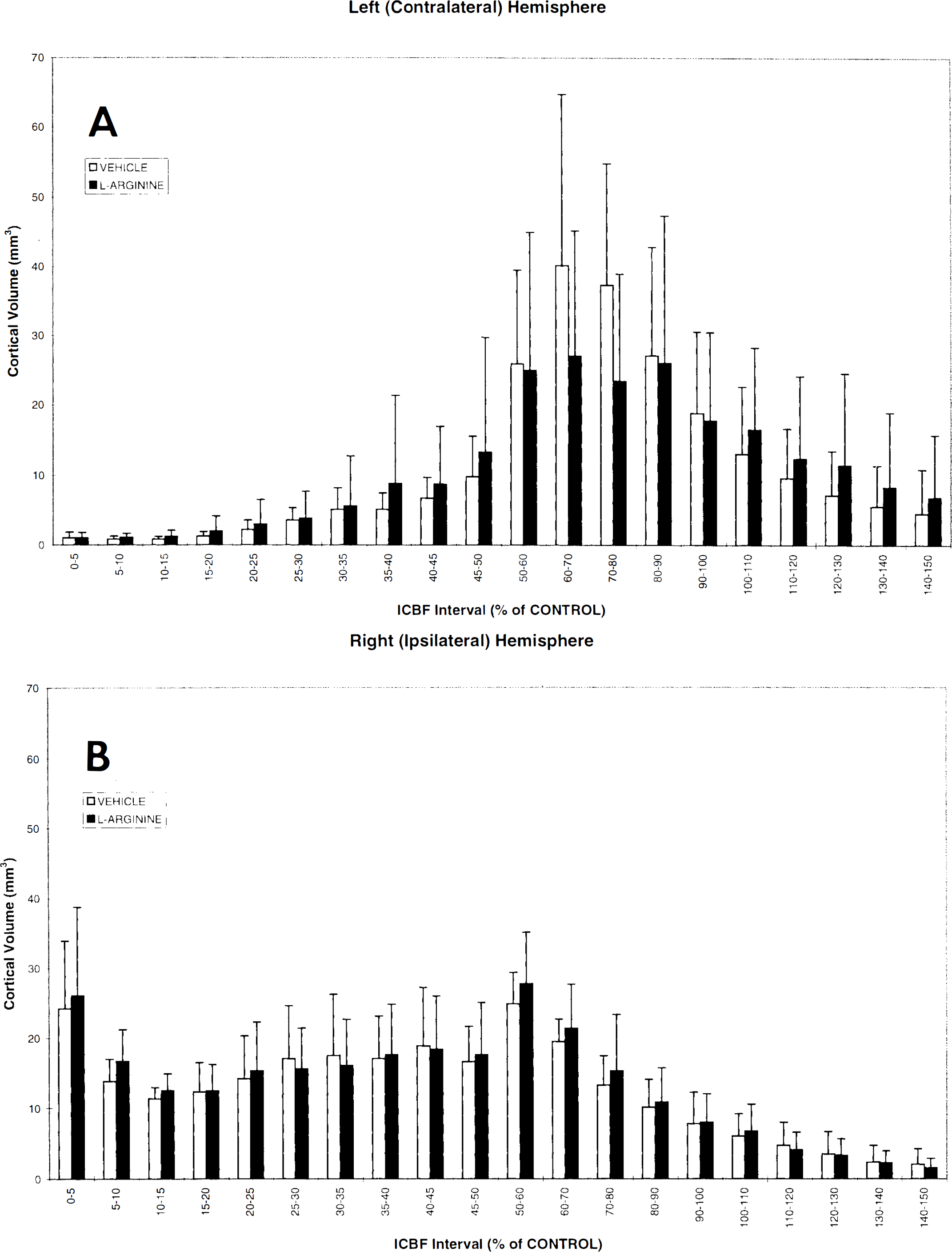
Histogram of cerebral cortical volumes displaying local CBF (LCBF) in a specified LCBF interval (incremental flow range), arranged in sequence of corresponding LCBF intervals, for left (
Figure 3 shows the 3D image data sets for (A) vehicle- and (B) L-Arg-treated rats, viewed from the ischemic (right) hemisphere. A percent difference image was computed by image subtraction and division of the primary data sets, according to 100 × (Arg — vehicle)/vehicle. Pseudocolor display of the percent difference data set revealed a coherent area of the right frontoparietal cortex in which a consistent difference between the vehicle and L-Arg groups was apparent (LCBF was lower in the L-Arg group); this is displayed in Fig. 3C–E.
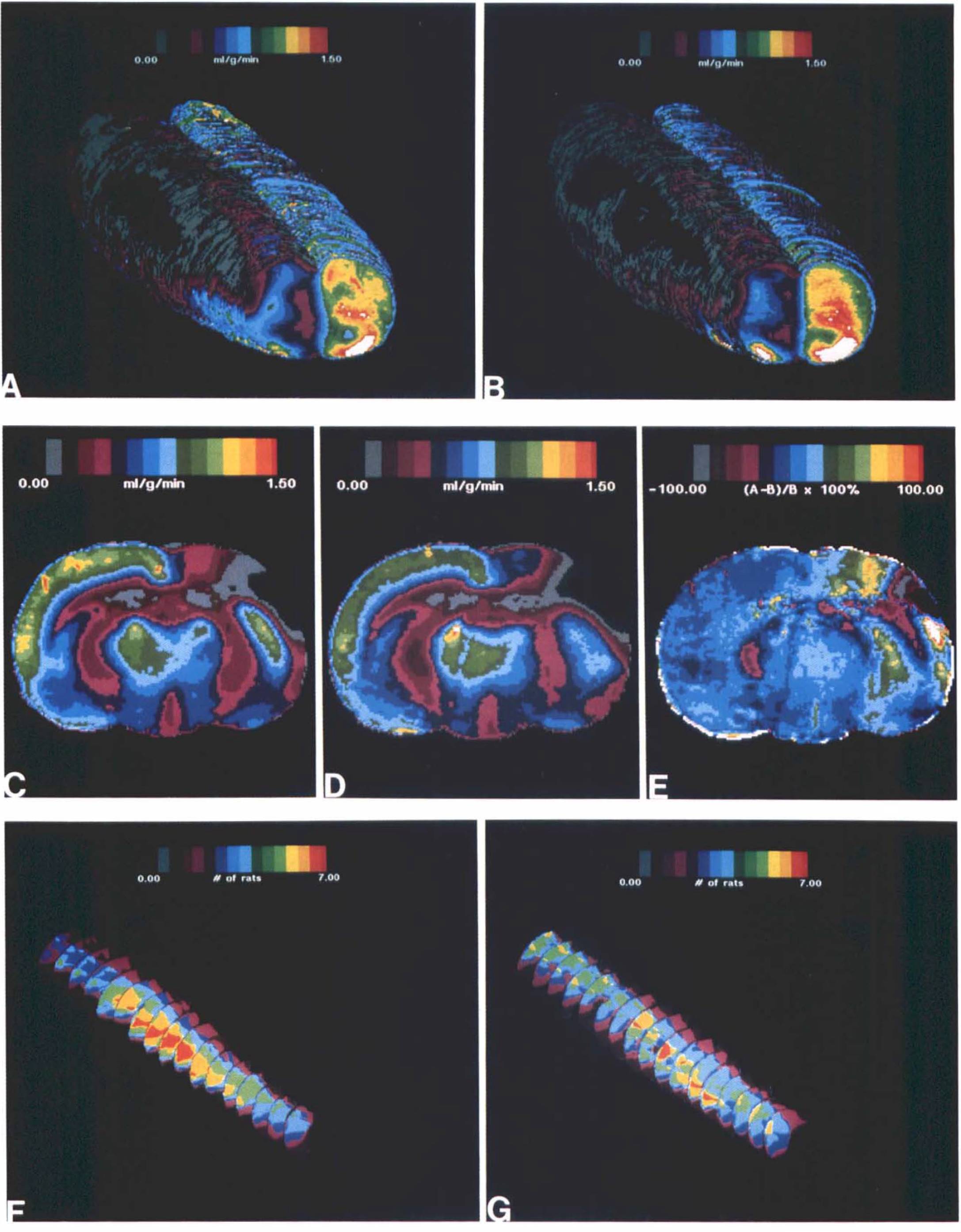
Differences between the vehicle and L-Arg groups were further analyzed at 30 coronal levels spanning a 3-mm anteroposterior extent through the zone of maximally apparent difference. At each coronal level, the neocortex was divided into 16 sectors, and t tests were conducted on each sector between the vehicle and L-Arg groups. This analysis revealed no intergroup differences for any sector of either the right or the left hemisphere.
Histopathology
Hematoxylin-and-eosin-stained sections in the vehicle and L-Arg groups demonstrated a zone of infarction comprising portions of the dorsolateral and lateral neocortex of the ischemic hemisphere and did not involve subcortical structures. The stereotaxically corrected infact volume averaged 37.0 ± 9.7 mm3 (SD) in the vehicle-treated group (n = 7) and 49.1 ± 17.2 mm3 (SD) in the L-Arg-treated group (n = 7). These values were not significantly different from one another.
Camera lucida drawings of infarct area at sequential coronal levels in the two animal groups were computer digitized, aligned, and displayed as frequency maps. These images are shown in Fig. 3F and G. Patterns of complete infarction in the vehicle and L-Arg groups were similar at their respective levels.
Ischemic neuronal counts in cortical areas bordering the core of infarction are shown in Table 3. In the histological locations selected, these measures were heterogeneously distributed among the animals studied. They were therefore expressed, for each of the vehicle- and L-Arg-treated groups, as the range of ischemic neuronal counts observed in each group as well as the percentage of ischemic neuronal counts with respect to the count of normal neurons in the same neuronal field on the contralateral side. The upper limits of these measures of incomplete infarction are substantial (∼20–30% of the neurons are dead). No significant differences could be detected between vehicle- and L-Arg-treated rats.
Cortical ischemic neuronal counts in vehicle- and L-arginine-treated rats
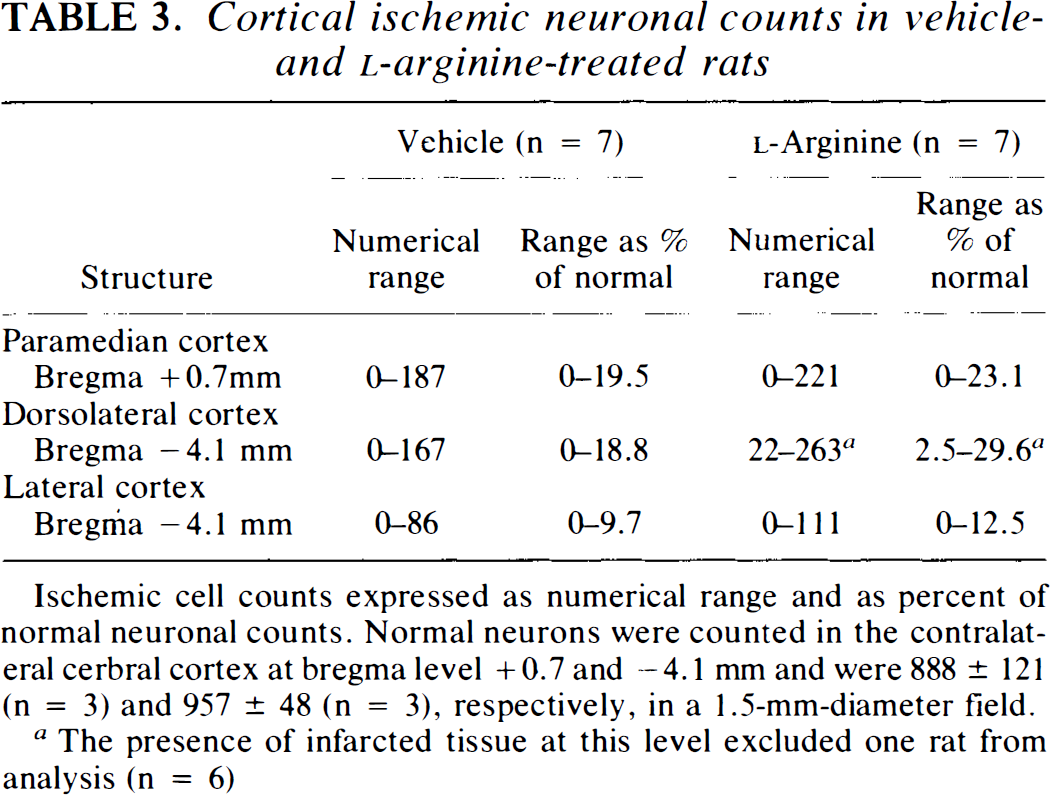
Ischemic cell counts expressed as numerical range and as percent of normal neuronal counts. Normal neurons were counted in the contralateral cerbral cortex at bregma level +0.7 and −4.1 mm and were 888 ±121 (n = 3) and 957 ± 48 (n = 3), respectively, in a 1.5-mm-diameter field.
The presence of infarcted tissue at this level excluded one rat from analysis (n = 6)
DISCUSSION
In these experiments, SHRs subjected to focal photothrombotic stroke of the MCA territory and treated with L-Arg did not differ from vehicle-treated rats, in terms of either LCBF pattern, infarct volume reduction, or ischemic neuronal counts in the region of incomplete infarction (Markgraf et al., 1993). These null results are intriguing in light of the recently emerging consensus that administration of L-Arg, and NO donor compounds as well, mitigates infarct development and acutely improves cerebral blood flow in SHRs (Morikawa et al., 1992b; Zhang and Iadecola, 1993, 1994; Zhang et al., 1994) and in normotensive Sprague–Dawley rats (Morikawa et al., 1994). Our observation that cortical flow increases over baseline by ∼75% in nonischemic SHRs given L-Arg is similar to the result observed previously in Sprague–Dawley rats (Morikawa et al., 1994) and suggests that the previously indicated potential therapeutic benefit of L-Arg was negated in the context of photothrombosis.
One possibility is that the photothrombotic insult, either directly at the site of irradiation or indirectly by toxic transportable factors, damages the endothelial NO synthase to the extent that intravascular L-Arg therapy is rendered ineffectual. A loss of vasodilator response to L-Arg in mouse brain arterioles at the sites of fluorescein-mediated photochemical injury was noted previously by Rosenblum et al. (1990), although downstream responsiveness was preserved. In this connection, Reidel et al. (1995) also observed destruction of vasodilator capacity to L-Arg following fluorescein-mediated photochemical injury to pial arterioles in normotensive WKY rats, but to a lesser degree in SHRs. Rose bengal–induced photothrombosis, however, is much more potent than fluorescein-mediated photodamage. The vasodilator capacity of L-Arg at the arterial irradiation site is evidently destroyed, as suggested by the severe vasoconstriction seen in irradiated arteries of all diameters (Watson et al., 1987); an accompanying feature is the intense deposition of platelets at overt breaches in the luminal surface (Watson et al., 1985, 1987). These effects are likely augmented by direct exposure of the underlying smooth muscle layer owing to areal shrinkage (contraction) of the endothelial cell layer following photochemical injury (Tseng et al., 1994) or exposure to thrombogenic factors (Nagy et al., 1995). Vascular damage in the MCA territory distal to the forming thrombus is also known to occur, as evidenced by enhanced permeability to horseradish peroxidase at sites downstream from the thrombus (Dietrich et al., 1988b). Such damage is presumed to result from transportable photoinduced thrombogenic factors, which may further lessen the probability of tissue recovery when produced in conjunction with a period of intervening ischemia (Dietrich et al., 1988b). These factors are capable of transforming tissue injury into an irreversible state from a potentially reversible one, thereby signaling the onset of reperfusion injury (Dietrich et al., 1989).
With further regard to the potential benefits of L-Arg therapy, it has been demonstrated that ischemia per se in the context of mechanically induced MCAO does not damage the response of the penumbral vasculature to L-Arg administration to SHRs, because L-Arg-stimulated increases in cortical CBF have been shown to precede the recovery of spontaneous electrical activity required for functional improvement (Dalkara et al., 1994). However, the concatenation of damaging effects induced by rose bengal–mediated photothrombosis in SHRs likely overwhelms any beneficial effect of L-Arg on collateral vascular patency and enhancement of peripheral blood flow, thus negating reductions in infarct volume. The combination of platelet secretions with the impaired vasodilator response of cerebral arterioles in SHRs is likely to accentuate local lesion severity further (Heistad et al., 1990).
Another explanation for the present results is that the photothrombotically induced enhancement of downstream vascular permeability may facilitate entry of L-Arg into the neuropil. If the neuronal form of NO synthase is thus activated, the resultant neuronal NO may lessen the possibility of recovery, because any enhancement of collateral flow by NO synthesized from L-Arg would tend to be counterbalanced by toxic effects (e.g., N-methyl-D-aspartate receptor overstimulation) accompanying intraparenchymal NO synthesis following L-Arg administration (Buisson et al., 1993). The effects of this conjecture are approximately inverse to those demonstrated previously in the Kn mice, in which infarct development was retarded by lack of neuronal NO synthesis and enhanced by inhibition of vascular NO synthase (Huang et al., 1994).
The infarct volumes observed in the present work are considerably smaller than those usually reported in SHRs after MCAO, owing to our exclusion of common carotid artery ligation. The latter was done to maximize the effectiveness of collateral channel activation in the region surrounding the infarct core and thus to enhance the possibility that a therapeutic agent could penetrate effectively at least into the penumbral zone. The completed infarct volumes at 3 days of the vehicle- and L-Arg-treated rats correspond approximately to the integrated CBF volume below the flow threshold corresponding to 15% of control (see Fig. 2B). This threshold of infarction is below that for SHRs subjected to ischemic insults yielding large infarct volumes (Jacewicz et al., 1992), presumably owing to the influence of unimpeded collateral channels in our model. The relatively constant (but diminished in magnitude) cortical volume distribution in the broad LCBF interval range between 15 and 50% of control implies the existence of an initial penumbra within that range. The persistence of a penumbra at 3 days is strongly suggested by the observation of ischemically compromised neurons in noninfarcted tissue adjacent to the infarct core (Table 3). This zone is nonetheless evidently amenable to treatment; in a similar model of photothrombotic occlusion of the distal MCA (augmented by mechanical occlusion of the common carotid arteries) in Wistar rats, we found previously that the glutamate antagonist MK-801 failed to reduce infarct core volume (Yao et al., 1993, 1994) but did reduce the volume of incompletely infarcted tissue containing ischemic neurons (Yao et al., 1994).
Despite the relatively small size of the infarct in the present work, reproducibility (coefficients of variation ∼25–30%) is similar to that resulting from coincident occlusion of the MCA and common carotid artery in SHRs. These features of small lesion volume with consistency comparable with the ligation models suggest an enhancement of lesion focality. Whether this smaller lesion, in the absence of thrombolysis, is amenable to potential flow-enhancing therapies other than L-Arg administration remains to be seen. The photochemically induced lesion, although reproducible, may be resistant to such therapies in an environment of intense platelet reactivity and concomitant release of substances toxic to blood–brain barrier integrity. Hence, at least some of the factors giving rise to incomplete infarction (ischemic neuronal injury) in the potentially treatable penumbral region in this photothrombotic stroke model are likely to be biochemically different from those ordinarily induced in mechanical models of MCAO. Finally, the existence of peripheral noninfarcted zones containing ischemic neurons at 3 days suggests that the thrombotically induced infarct is still evolving. These caveats should be considered before contemplating L-Arg therapy in humans experiencing stroke in evolution.
Footnotes
Acknowledgment:
This work was supported by NINDS grants NS23244, NS05820, and NS27127. We thank Susan Kraydieh for her diligence in preparing histological specimens, Isabel Garcia for her expert assistance during the LCBF experiments, and Yolanda Loor for precise tissue sectioning. Brant D. Watson is the recipient of a Jacob Javits Neuroscience Investigatorship (1992–1999).