
Abstract
Cerebrovascular damage leading to subsequent reductions in local cerebral blood flow (lCBF) may represent an important secondary injury mechanism following traumatic brain injury (TBI). We determined whether patterns of 111indium-labeled platelet accumulation were spatially related to alterations in lCBF determined autoradiographically 30 min after TBI. Sprague–Dawley rats (n = 8), anesthetized with halothane and maintained on a 70:30 (vol/vol) mixture of nitrous oxide/oxygen and 0.5% halothane, underwent parasagittal fluid percussion brain injury (1.7–2.2 atm). 111Indiumtropolone–labeled platelets were injected 30 min prior to TBI while [l4C]-iodoantipyrine was infused 30 min after trauma. Sham-operated animals (n = 7) underwent similar surgical procedures but were not injured. In autoradiographic images of the indium-labeled platelets, focal sites of platelet accumulation within the traumatized hemisphere were restricted to the pial surface (five of eight rats), the external capsule underlying the lateral parietal cortex (five of eight rats), and within cerebrospinal fluid (CSF) compartments (six of eight rats). In contrast, mild-to-moderate reductions in lCBF, not restricted to sites of platelet accumulation, were seen throughout the traumatized hemisphere. Flow reductions were most severe in coronal sections underlying the impact site. For example, within the lateral parietal cortex and hippocampus, lCBF was significantly reduced [p < 0.01; analysis of variance (ANOVA)] from 1.71 ± 0.34 (mean ± SD) and 0.78 ± 0.12 ml/g/min, respectively, versus 0.72 ± 0.17 and 0.41 ± 0.06 ml/g/min within the traumatized hemisphere. Significant flow reductions were also seen in remote cortical and subcortical areas, including the right frontal cortex and striatum. These results indicate that focal platelet accumulation and widespread hemodynamic depression are both early consequences of TBI. Therapeutic strategies directed at these early microvascular consequences of TBI may be neuroprotective by attenuating secondary ischemic processes.
Brain trauma is known to lead to early cerebrovascular abnormalities, including increased blood–brain barrier (BBB) permeability, endothelial lesions, autoregulatory abnormalities, as well as microvascular obstruction (Povlishock et al., 1978; Lewelt et al., 1980; Wei et al., 1980; Hekmatpanah and Hekmatpanah, 1985; Jiang et al., 1992; Tanno et al., 1992; Dietrich et al., 1994b). Vascular perturbations following traumatic brain injury (TBI) would be expected to alter normal brain perfusion and possibly lead to secondary cerebral ischemia. A major mechanism of secondary brain damage after TBI is suggested to be decreased local cerebral blood flow (lCBF) (Adams et al., 1982; Graham et al., 1978). Indeed, clinical data are available that demonstrate the occurrence of early ischemia after TBI (Marion et al., 1991; Bouma and Muizelaar, 1992; Obrist et al., 1984; Robertson et al., 1992; Steiger et al., 1994).
The model of fluid percussion (FP) brain injury has been used to experimentally investigate the early hemodynamic consequences of brain injury (Yuan et al., 1988; Le welt et al., 1980; DeWitt et al., 1986; Ishige et al., 1987; Unterberg et al., 1988; Yamakami and McIntosh, 1989; Muir et al., 1992). Using radiolabeled microspheres, Yamakami and McIntosh (1989) reported an initial global suppression of lCBF after lateral FP brain injury in rats with persistent focal reductions at the trauma site. In that study, alterations in lCBF recovered within 4 h after TBI. Using hydrogen clearance techniques in cats, Lewelt and colleagues (1980) reported only minimal reductions in CBF within the caudate nucleus at one hour after midline FP injury. In other FP studies, no significant early decreases in CBF have been reported (DeWitt et al., 1986; Unterberg et al., 1988).
Using a parasagittal FP injury approach, early evidence of venous thrombosis and acute BBB breakdown has recently been reported in rats (Dietrich et al., 1994b). In that study, ultrastructural evidence for endothelial damage, microvascular stasis, and arteriolar vasoconstriction were reported. Based on these morphological findings, one might predict that this injury model would result in perfusion deficits and depressed lCBF. In this regard, widespread his-topathological damage, consistent with ischemic processes, has been documented recently in this FP model (Dietrich et al., 1994a).
Indium-labeled platelet autoradiography is a powerful approach with which to visualize regional patterns of platelet accumulation (Dewanjee et al., 1983; Dietrich et al., 1993). For example, in a model of photochemically induced thromboembolic stroke, this methodology has proven useful in documenting early patterns of acute platelet accumulation after carotid artery thrombosis (Dietrich et al., 1993). Since indium has a relatively short half-life (2.8 days), this tracer can also be combined with other radioisotopes in a single study. Thus, regional patterns of indium-labeled platelet deposition, in combination with [14C]-iodoantipyrine for the assessment of lCBF, can be visualized together in the same animal. The major aim of the present study was to document regional patterns of platelet accumulation after TBI and to correlate sites of platelet accumulation with early alterations in lCBF.
MATERIALS AND METHODS
Surgical preparation
These experiments were conducted on 15 fasted male Sprague-Dawley rats weighing between 250 and 300 g. Eight rats underwent TBI and seven rats served as sham-operated controls. Rats were initially anesthetized with 3% halothane, 30% oxygen, and a balance of nitrous oxide. Tracheal intubation was performed and the rats were placed in a stereotaxic frame with a 4.8 mm craniotomy made over the right parietal cortex (3.8 mm posterior to bregma and 2.5 mm lateral to bregma) (Zilles, 1985). A plastic injury tube was next placed and anchored as previously described (Dietrich et al., 1994a). The next day, a FP device was used to produce experimental TBI (Dixon et al., 1987). Intubated anesthetized rats (70% nitrous oxide, 1.5% halothane, and 30% oxygen) were traumatized by moderate head injury ranging from 1.7–2.2 atm. Brain temperature was monitored indirectly with a thermistor probe inserted into the left temporalis muscle (Jiang et al., 1991). Temporalis muscle and rectal temperatures were maintained at 37°C before and after TBI. In sham-operated controls, all surgical steps were conducted but rats were not traumatized.
Platelet labeling with [111In]-tropolone
[111In]-labeled platelet procedures have been previously described in detail (Dewanjee et al., 1983; Dietrich et al., 1993). For each study, a total of 30 ml of blood was withdrawn from the femoral arteries of two donor rats (250–400 g) for platelet labeling. Blood was then centrifuged through several steps and the platelet pellet resuspended in 1 ml of acid citrate dextrose solution. Next, 25 μg of tropolone was added to 200 μCi of [111In] chloride in a centrifuge tube and vortexed. Resuspended platelets were then added to the [111In]-tropolone and allowed to incubate for 30 min at room temperature. For each rat, 1 ml fraction of [111In]-labeled platelets was withdrawn into a 10-ml syringe. The remaining fraction of labeled platelets was saved to determine harvesting efficiency by counting platelets with a Coulter counter. Radioactivity of labeled platelets and washings was measured with a gamma counter. Labeled platelets were next injected into experimental rats and allowed to circulate for 30 min prior to TBI or sham procedures.
Local CBF studies
lCBF was determined using [14C]-iodoantipyrine autoradiographic procedures (Sakurada et al., 1978). At 30 min after TBI, 20 μCi of the radioactive tracer, [14C]-iodoantipyrine (specific activity 40 μCi/mmol; New England Nuclear), dissolved in 1 ml of isotonic saline, was infused intravenously at a constant rate over 45 s via a Harvard infusion pump. Blood samples were obtained from the tip of a freely flowing femoral artery catheter at 2-s intervals. Studies were terminated by decapitation of the animals; brains were removed, frozen over liquid nitrogen, and immediately sectioned as previously described (Dietrich et al., 1991).
Autoradiographic procedures
For platelet imaging, frozen sections were exposed for 5 days. This relatively short exposure has been shown, in preliminary studies, to visualize primarily labeled platelet aggregates without significant background contamination from the 14C radiotracer. Regional patterns of labeled platelet accumulation were assessed by documenting the locations of platelet aggregates. Following initial exposure, sections were refrigerated for 25 days to allow the indium label to decay. Following this period, sections were used again for the visualization and quantitation of lCBF. Regional densitometry of ipsilateral and contralateral hemispheres was performed by means of an automated digitizing densitometer (Optronics) interfaced with a microVAX minicomputer image processor (Grinnell Systems). Brain regions associated with the impact site, as well as more remote regions, were given special attention.
Statistical analysis
Statistical analyses data are expressed as means ± standard deviation (SD). Repeated measure of analysis of variance (ANOVA) was used to compare levels of CBF with respect to side-to-side changes. One-way ANOVA was used to compare levels of local cerebral blood flow (lCBF) with respect to control and traumatized rats.
RESULTS
Physiological data
Control animals exhibited stable arterial blood pressures and normal physiological variables before TBI and during the CBF study. Physiological data from sham-operated controls and TBI rats were all within normal limits. In all TBI rats, arterial blood pressure increased immediately after trauma and remained elevated for ∼1–2 min. Peak blood pressure averaged 198 ± 37 mm Hg (mean ± SD).
Platelet findings
Since the initial exposure for the visualization of indium-labeled platelets occurred over a relatively brief (5-day) period, a low background of [14C]-radioactivity was seen in all animals (Fig. 1 and 2). In sham-operated control rats, no evidence of abnormal indium-labeled platelet accumulation was seen. In contrast, focal sites of platelet accumulation, represented as dense spots of increased grain density, were seen in the right hemisphere of all traumatized rats. Regional patterns of labeled platelet aggregates are summarized in Table 1. As indicated, the majority of rats displayed labeled platelets within the subarachnoid space overlying the cortical convexities, the external capsule underlying the lateral parietal cortex, and those cerebrospinal fluid compartments separating the brain stem from the overlying cerebrum (Fig. 1 and 2). In some cases, labeled platelets appeared to be restricted to individual pial vessels. Platelet accumulation was most common overlying the external capsule beneath the lateral parietal and occipital cortices. However, platelet aggregates were not seen in remote regions, including the frontal cortex, striatum, cerebellum or brain stem.
Regional patterns of indium-labeled platelet aggregates after TBI

+, indium-labeled platelets detected; −, no indium-labeled platelets detected. Labeled platelets were not observed in the sham-operated series or in the contralateral (left) hemispheres of traumatized rats.
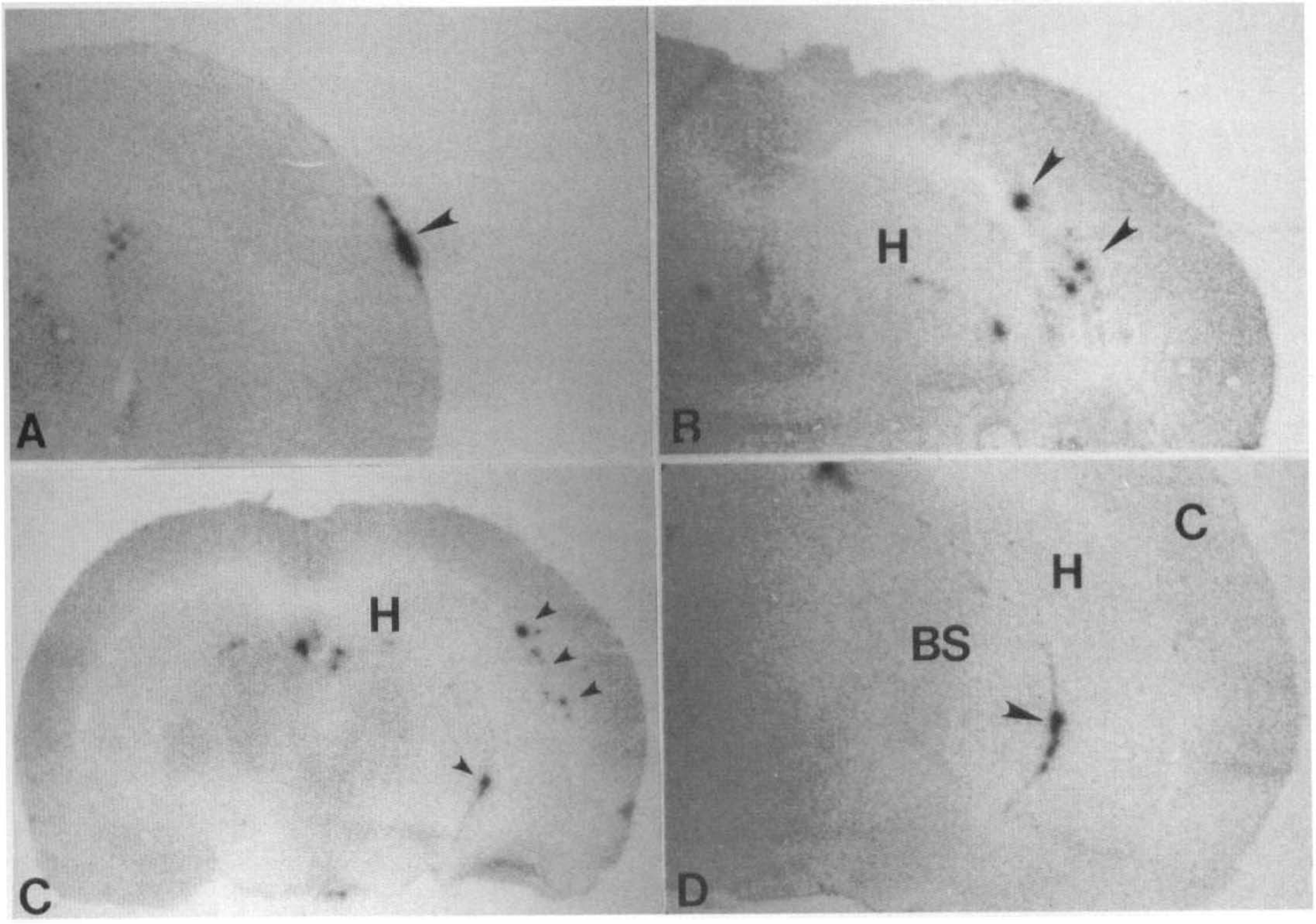
First exposure showing 111indium-labeled platelet accumulation within traumatized hemisphere.
Double-label autoradiographic visualization of 111indium-labeled platelets and [14C]iodoantipyrine ICBF.
Local CBF findings
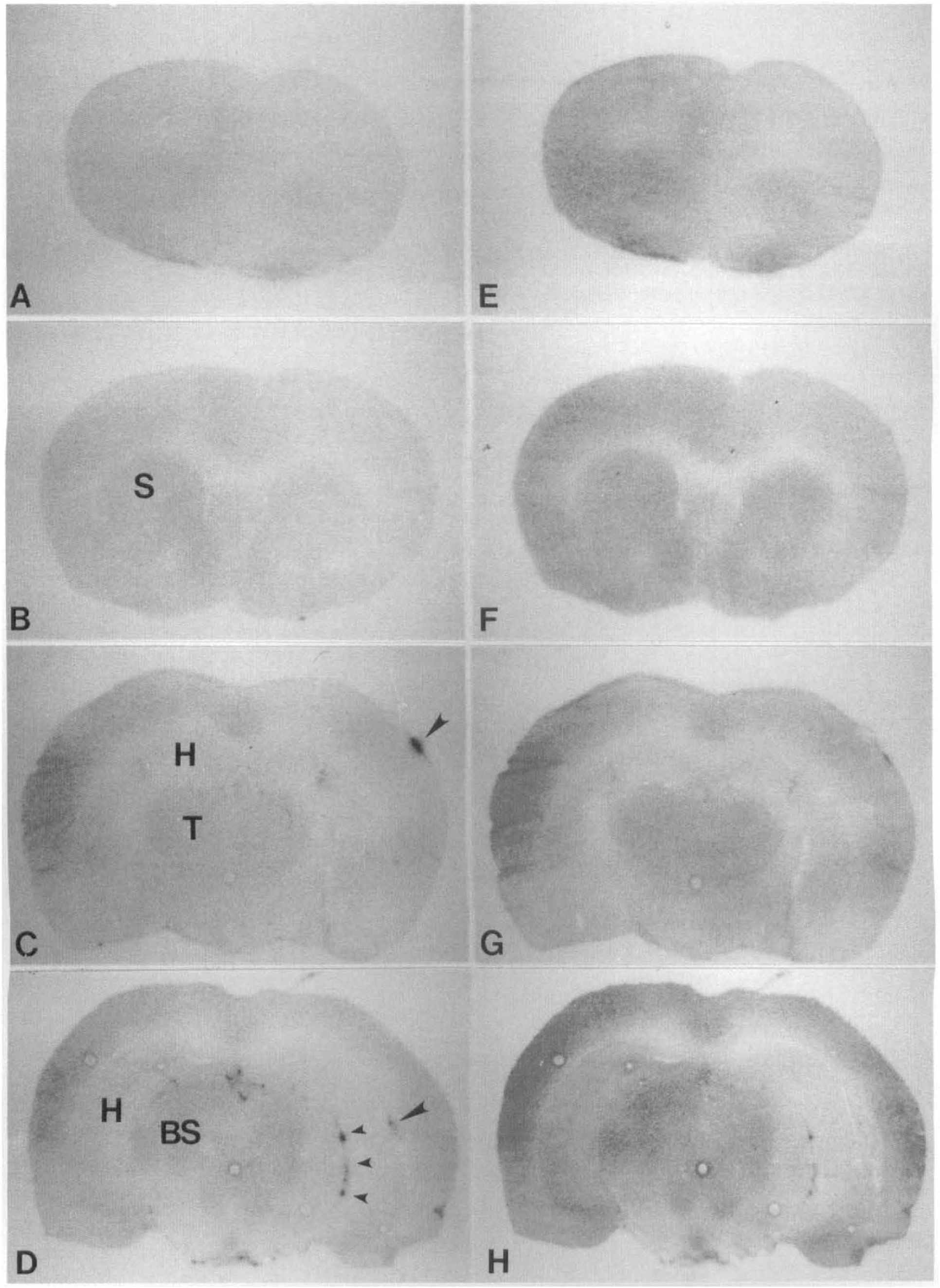
In sham-operated control animals, local levels of CBF were within normal ranges and no side-to-side differences were recorded (Tables 2 and 3). In contrast to the focal nature of the platelet response to TBI, moderate reductions in lCBF were seen throughout the right traumatized hemisphere (Tables 2 and 3, Fig. 2 and 3). Flow reductions were most consistent at the impact site, with reductions in lCBF ranging from 40 to 53% of control (Table 2). For example, cortical CBF directly underlying the site of impact was 0.63 ± 0.17 ml/g/min (mean ± SD) compared to 1.64 ± 0.59 ml/g/min in sham-operated specimens. Within the parietal cortex lateral to the impact site, rCBF was reduced to 0.72 ± 0.26 ml/g/min compared to control values of 1.71 ± 0.34 ml/g/min. Significant reductions were also documented within cortical and subcortical areas remote from the impact site (Table 3). Within the right frontal cortex, lCBF was reduced from 1.64 ± 0.59 ml/g/min in control rats to 0.79 ± 0.22 ml/g/min after TBI. A similar reduction was seen in the right striatum wherein lCBF was reduced after TBI to 61% of control. No significant lCBF changes were detected within the cerebellum or brain stem. In the second exposure for lCBF determination, pale images of the indium platelet response appeared to persist. This response may be the result of medium and low energy conversion and Auger electrons that are generated during the decay of 111In (Jonsson et al., 1992).
Local cerebral blood flow in structures at coronal levels underlying impact site
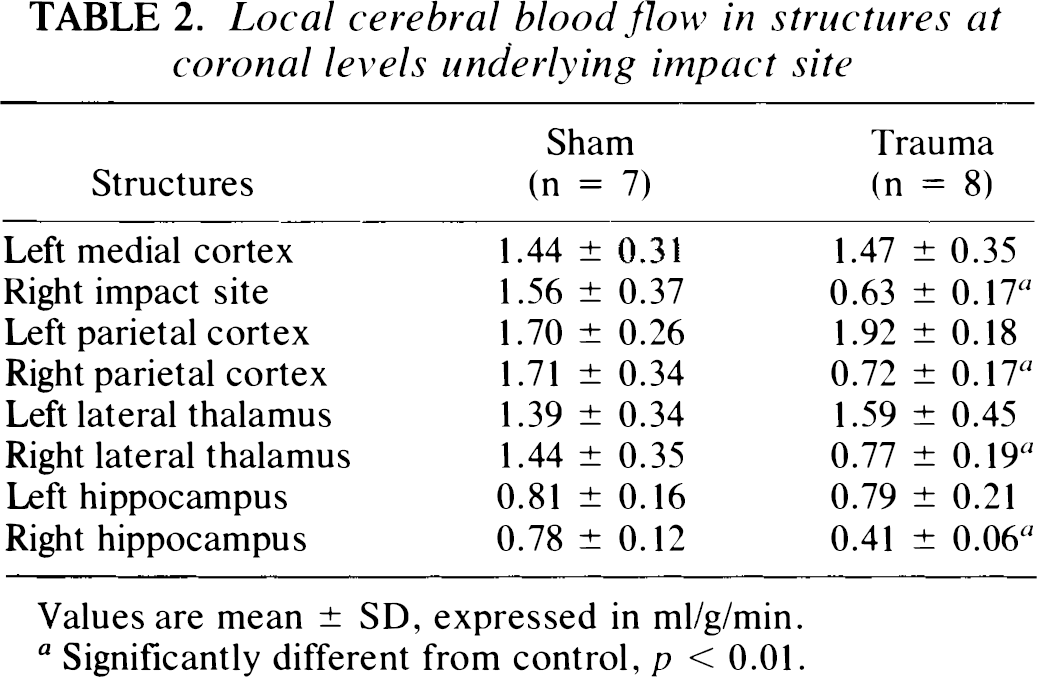
Values are mean ± SD, expressed in ml/g/min.
Significantly different from control, p < 0.01.
Local cerebral blood flow in structures remote from impact site
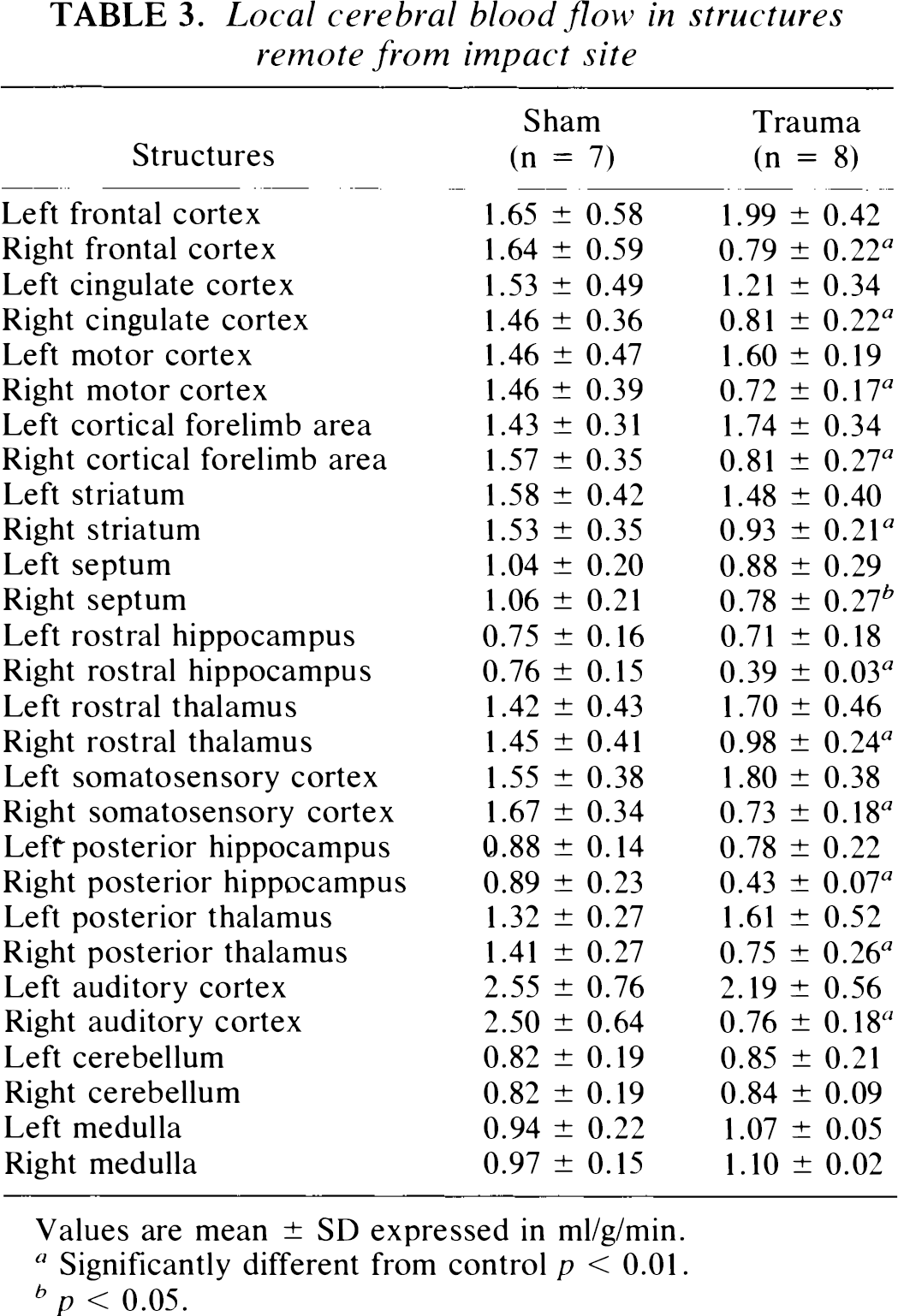
Values are mean ± SD expressed in ml/g/min.
Significantly different from control p < 0.01.
p < 0.05.
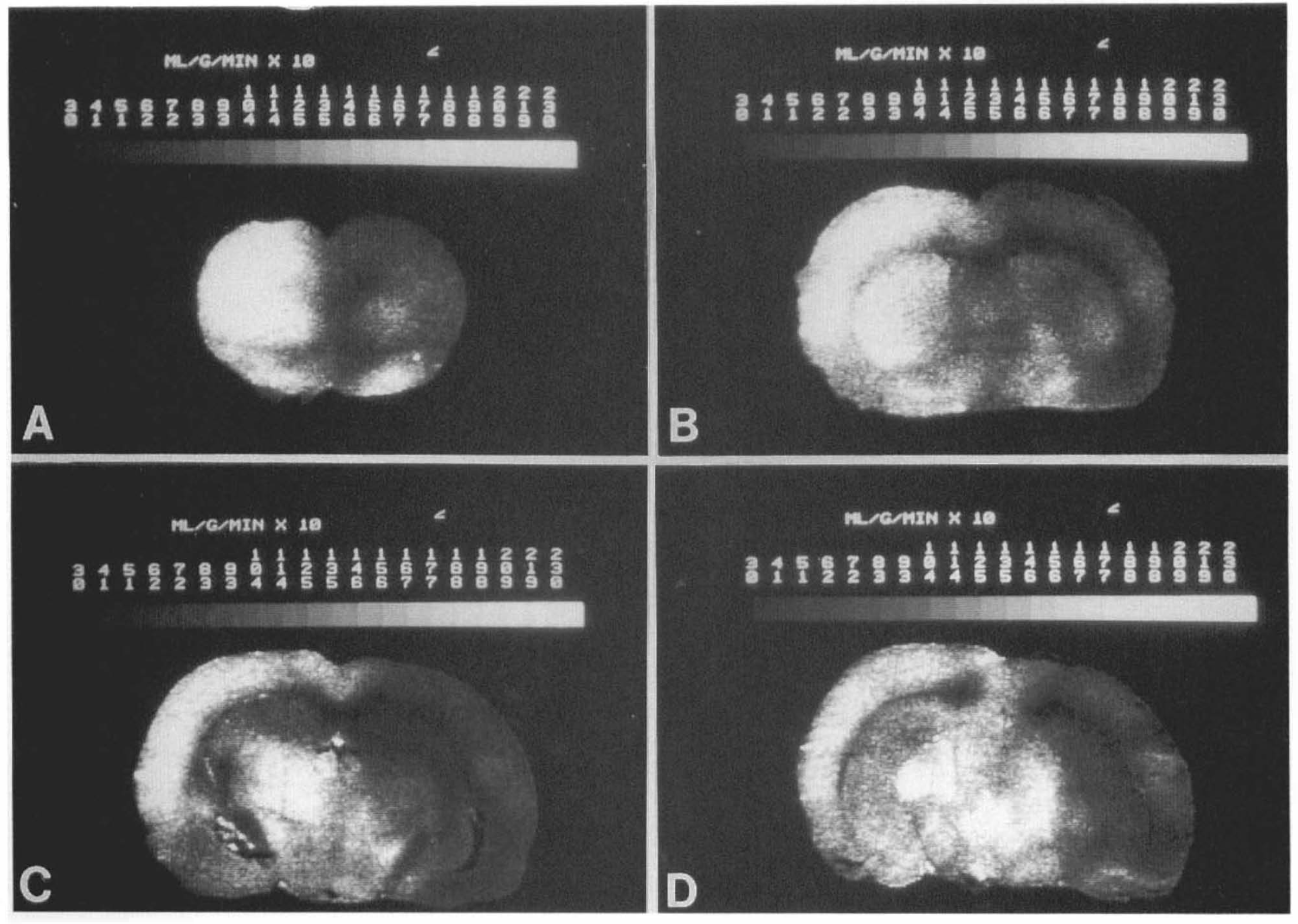
Computer-generated [14C]-iodoantipyrine autoradiographic images of rat brain 30 min after TBI. The gray scales represent levels of ICBF(ml/g/min × 100).
In the left, uninjured hemisphere of traumatized rats, lCBF tended to be mildly increased in cortical regions compared to that in sham-operated controls (Tables 2 and 3). Although no significant increases in lCBF were documented between individual structures, average CBF values for cortical flow (i.e., mean value from frontal, motor, somatosensory, parietal, and occipital cortices) between sham-operated controls and the left hemispheres of traumatized rats were significantly different (1.68 ± 0.39 versus 1.82 ± 0.24 ml/g/min, p < 0.05).
DISCUSSION
These results demonstrate that in this FP model of brain injury, focal patterns of early platelet accumulation after trauma are associated with widespread hemodynamic abnormalities. As previously discussed, some (but not all) models of FP injury have been shown to result in early decreases in lCBF. The present findings are the first autoradiographic data describing regional changes in CBF, using a FP trauma model that results in consistent histopathological damage. Thus, the opportunity to correlate these early hemodynamic abnormalities with regional patterns of histopathological vulnerability appears advantageous. In addition, the ability to correlate sites of early platelet accumulation with regionally specific hemodynamic abnormalities should allow cause and effect relationships to be addressed.
Using the present FP model, we recently reported platelet aggregates within the evolving hemorrhagic contusion using ultrastructural techniques at 1 h after TBI (Dietrich et al., 1994b). Interestingly, platelet aggregates were detected when immersion-fixation (but not perfusion-fixation) was employed. These morphological findings indicated that platelet aggregates could be dislodged by routine perfusion-fixation procedures. In the present study, autoradiographic visualization of indium platelets was used to verify these earlier morphological findings. Again, labeled platelets were documented at sites known to be most severely damaged in this brain injury model. For example, subarachnoid hemorrhage overlying the pial surface of the traumatized parietal cortex has been reported (Dietrich et al., 1994b). In the present study, luminal platelet accumulation was also observed within individual pial vessels in close proximity to the impact site. A second common site of labeled platelets was the external capsule underlying the lateral parietal cerebral cortex. This region has been shown to contain mechanically disrupted venules, and commonly evolves into a hemorrhagic contusion. A third site of platelet accumulation was within cerebrospinal fluid compartments containing cerebral vessels. Our previous ultrastructural study demonstrated this last site to undergo early horseradish peroxidase extravasation, most likely due to shearing stress (Dietrich et al., 1994b).
The observation that platelet accumulation occurs early in this model of TBI has various implications. First, platelet aggregates may present as a mechanical barrier to normal microvascular perfusion. In this regard, evidence for vascular stasis has been presented in the present model as well as in other models of TBI (Hekmatpanah and Hekmatpanah, 1985; Cortez et al., 1989; Dietrich et al., 1994b). However, our data clearly demonstrate regional flow reductions that are not spatially related to platelet foci and, thus, provide no direct evidence for causality.
Platelet-derived vasoactive substances released into the blood or surrounding brain regions following platelet activation may also potentially effect lCBF. Platelets are known to release a variety of vasoactive substances, including thromboxane A2 and serotonin, both of which may lead to vasoconstriction under pathological conditions (Ellis et al., 1977; Thompson et al., 1984). In this regard, DeWitt and colleagues (1988) reported elevated brain levels of thromboxane B2, the stable metabolite of thromboxane A2, 24 h after FP brain injury. The fact that platelet accumulation occurs in CSF spaces and is associated with sites of altered BBB permeability, indicates potential anatomical routes whereby platelet-derived blood-borne substances may gain access to remote or extravascular compartments.
In this model, moderate levels of hemodynamic depression were detected in both vulnerable and -nonvulnerable brain regions. For example, at 3 days after TBI, we previously detected necrotic neurons within layers V and VI of the parietal cortex (Dietrich et al., 1994a). In the present study, lCBF within the parietal cortex was reduced to only 42% of control. Similar reductions in flow were also observed in other vulnerable regions, including the hippocampus and lateral thalamus. Based on experimental studies, these CBF reductions alone do not appear to be sufficient to explain the neuronal damage seen at 3 days in these areas (Astrup et al., 1977). It is know that specific neuronal populations are extremely sensitive to secondary ischemia, induced following TBI (Jenkins et al., 1989). Based on the present data, multiple pathomechanisms, including reduced lCBF, would appear to be necessary to promote irreversible neuronal damage after TBI (Chan et al., 1984; Young, 1988; Katayama et al., 1990; Hayes et al., 1991; Hovda et al., 1992).
In addition to vulnerable regions, moderate reductions in lCBF were also seen in brain regions in which overt histopathological damage did not occur. Within the frontal cortex and striatum, for example, flow reductions on the average of 48 and 61%, respectively, were documented. In the normal brain, CBF and metabolism are normally coupled, and CBF is regulated by the metabolic demand of the tissue (Roy and Sherrington, 1890; Reivich, 1974). In this regard, alterations in local cerebral metabolic rate of glucose (lCMRglc) have been documented in models of FP brain injury (Hayes et al., 1988; Yoshino et al., 1991; Dietrich et al., 1994c). Yoshino and colleagues (1991) reported dynamic changes in glucose metabolism, including both immediate hypermetabolism followed by prolonged hypometabolism. Using the present FP model, we have reported moderately depressed levels of lCMRglc at 4 and 24 h after TBI within widespread cortical and subcortical areas (Dietrich et al., 1994c). Thus, some of the hemodynamic consequences of TBI may also represent secondary responses to depressed cerebral metabolism.
Mild increases in lCBF were documented throughout the noninjured cerebral cortex at 30 min after TBI. Using laser-doppler flowmetry, Muir and colleagues (1992) reported a transient hyperemic response within the contralateral hemisphere immediately after lateral FP injury followed by moderate flow reductions at 1 h. In head-injured patients, hyperemia is observed hours to days after injury and has' been associated with elevated intracranial hypertension (Fieschi et al., 1974; Obrist et al., 1983; Robertson et al., 1992). In our study, the acute hyperemic response may have resulted from widespread vasoconstriction of cerebral vessels within the traumatized hemisphere leading to a redistribution of blood flow to the contralateral hemisphere. Alternatively, these contralateral flow changes may reflect the vasculature's response to the initial traumatic and/or ischemic insult. Regardless of the exact mechanism underlying this contralateral effect, these data indicate that the contralateral hemisphere is acutely affected by parasagittal FP brain injury and, therefore, cannot be regarded as normal.
In summary, our results demonstrate evidence for early platelet accumulation and widespread hemodynamic alterations in a FP model that results in histopathological damage. Experiments using agents that decrease platelet number or block the actions of platelet-derived vasoactive substances are required to critically investigate whether abnormal platelet activation participates in the early hemodynamic consequences of TBI. These data also support previous work in the trauma field showing that hemodynamic abnormalities following trauma represent potentially important secondary injury mechanisms in TBI. Clarification of these ischemic processes and their specific roles in the pathophysiology of TBI should identify novel pathways that may be targeted for therapeutic intervention.
Footnotes
Acknowledgment:
This study was supported by USPHS Grants NS30291, NS05820, and NS 27127, as well as funds from the Titmus Foundation. The authors thank Yolanda Loor, Isabel Valdes, and Marcilia Halley for technical assistance and Helen Valkowitz for typing the manuscript.