
Abstract
The utility of microtubule-associated protein 2 (MAP2) immunostaining as a marker of acute focal ischemic injury was investigated. Permanent middle cerebral artery occlusion (MCAO) elicited a rapid reduction in MAP2 immunostaining that was visible 1 h post-MCAO and that increased in intensity and area encompassed over time. The ischemic lesion borders were well defined by loss of MAP2 immunostaining, but alterations in staining within the lesion were more heterogeneous. Lesion volume increased significantly from 1 to 4 h post-MCAO (from 63.8 ± 10.8 to 111.3 ± 19.0 mm3, mean ± SD). Thus, MAP2 immunostaining is a sensitive, quantifiable indicator of acute brain injury following focal ischemia.
Lesion volume in rodent models of focal cerebral ischemia may reach its maximum level within a relatively short time period. Thus, pathological processes initiated within the first few hours of an ischemic insult are likely to have a greater impact on outcome than later events. To investigate such acute pathological processes, it is necessary to have an indicator of ischemic injury that can reliably delineate the lesion at correspondingly early time points. Although conventional histological techniques such as triphenyltetrazolium chloride and hematoxylin and eosin (H&E) staining have been employed within 4 h of permanent focal ischemia (Tamura et al., 1981; Hatfield et al., 1991), their use is limited since triphenyltetrazolium chloride immersion staining may be unreliable for the quantification of acute ischemic damage (Hatfield et al., 1991), while H&E staining (at early time points) is difficult to interpret without extensive neuropathological expertise. As an alternative approach, ischemia-induced reduction in microtubule-associated protein 2 (MAP2) immunostaining has received considerable attention as a marker of early damage following transient global ischemia (Yanagihara et al., 1990; Tomioka and Yanagihara, 1992; Miyazawa et al., 1993), and therefore we decided to assess the utility of this technique as a marker of acute neuronal injury in the context of permanent focal ischemia.
MAP2 is one of a family of MAPs involved in maintaining the structural integrity of the neuronal cytoskeleton and modulating synaptic plasticity (Johnson and Jope, 1992). Under normal conditions, MAP2 is almost exclusively located in neurons and represents an effective somatodendritic label (De Camilli et al., 1984). While loss of MAP2 immunostaining has proven to be a highly sensitive marker of ischemic injury following transient global ischemia (Yanagihara et al., 1990; Miyazawa et al., 1993), its use has, until now, been predominantly restricted to a purely qualitative description of neuronal injury. Thus, the current study is novel in that it not only represents one of the first detailed attempts to use this technique for permanent focal ischemia, but it also exploits loss of MAP2 immunostaining to quantify lesion volume in the acute time period following onset of ischemia.
METHODS
Adult male (300–350 g) spontaneously hypertensive rats underwent permanent middle cerebral artery occlusion (MCAO) of 1-, 2-, or 4-h duration (n = 5 per group). Rats were anesthetized with halothane (5% for induction, 1–1.5% for maintenance) in nitrous oxide/oxygen (70:30) and artificially ventilated. Blood pressure, arterial blood gases, and body temperature were monitored at regular intervals. The left MCA was exposed using a subtemporal approach (Tamura et al., 1981), occluded by electrocoagulation midway between the inferior cerebral vein and olfactory tract, then severed to prevent recanalization. At 1, 2, or 4 h post-MCAO, rats were killed by transcardial perfusion with heparinized saline followed by 4% paraformaldehyde.
Adjacent vibratome sections were cut for H&E staining (25 n.m) and MAP2 immunohistochemistry (50 μm). Immunohistochemistry was performed on free-floating sections using the avidin-biotin peroxidase complex technique and 3,3′-diaminobenzidine as peroxidase substrate. Preliminary blocking was carried out by incubation (2–3 h) in normal horse serum. The primary and secondary antibodies were monoclonal mouse anti-MAP2 (1:4,000 dilution, 15-h incubation; Amersham) and biotinylated anti-mouse IgG (1:200 dilution in 5% rat plasma to minimize cross-reactivity, 1-h incubation; Vector). For controls, the primary antibody was omitted or replaced with mouse IgG. Stained sections were mounted onto glass slides and examined by light microscopy.
Volumetric analysis was subsequently performed on MAP2-stained sections using the method originally described by Osborne et al. (1987). Lesion borders from sections corresponding to eight preselected stereotaxic levels were transcribed onto scale drawings. Lesion areas were then measured from these drawings using an image analyzer and converted to total volume of ischemic damage using the known distance between each of the eight chosen levels.
RESULTS
Macroscopic examination of sections stained for MAP2 revealed that a clear reduction in MAP2 immunostaining was already apparent 1 h post-MCAO. The staining was characteristically lost in narrow bands that appeared to define the lower and upper borders of the lesion (Fig. 1A). Within the region enclosed by these borders, the majority of staining was preserved at the 1-h time point. Under light microscopy, these borders were found to show good agreement with the borders of the lesion in adjacent H&E-stained sections, although the lesion was more readily discernible in the MAP2 sections (Fig. 1B and C). While the most marked reduction in MAP2 immunostaining at the 1-h time point occurred at the lesion borders, changes in H&E-stained sections could be observed throughout the area enclosed by these borders. Therefore, the area of this entire region enclosed by the bands of reduced MAP2 immunostaining was measured for the quantification of lesion volume at the 1-h time point.
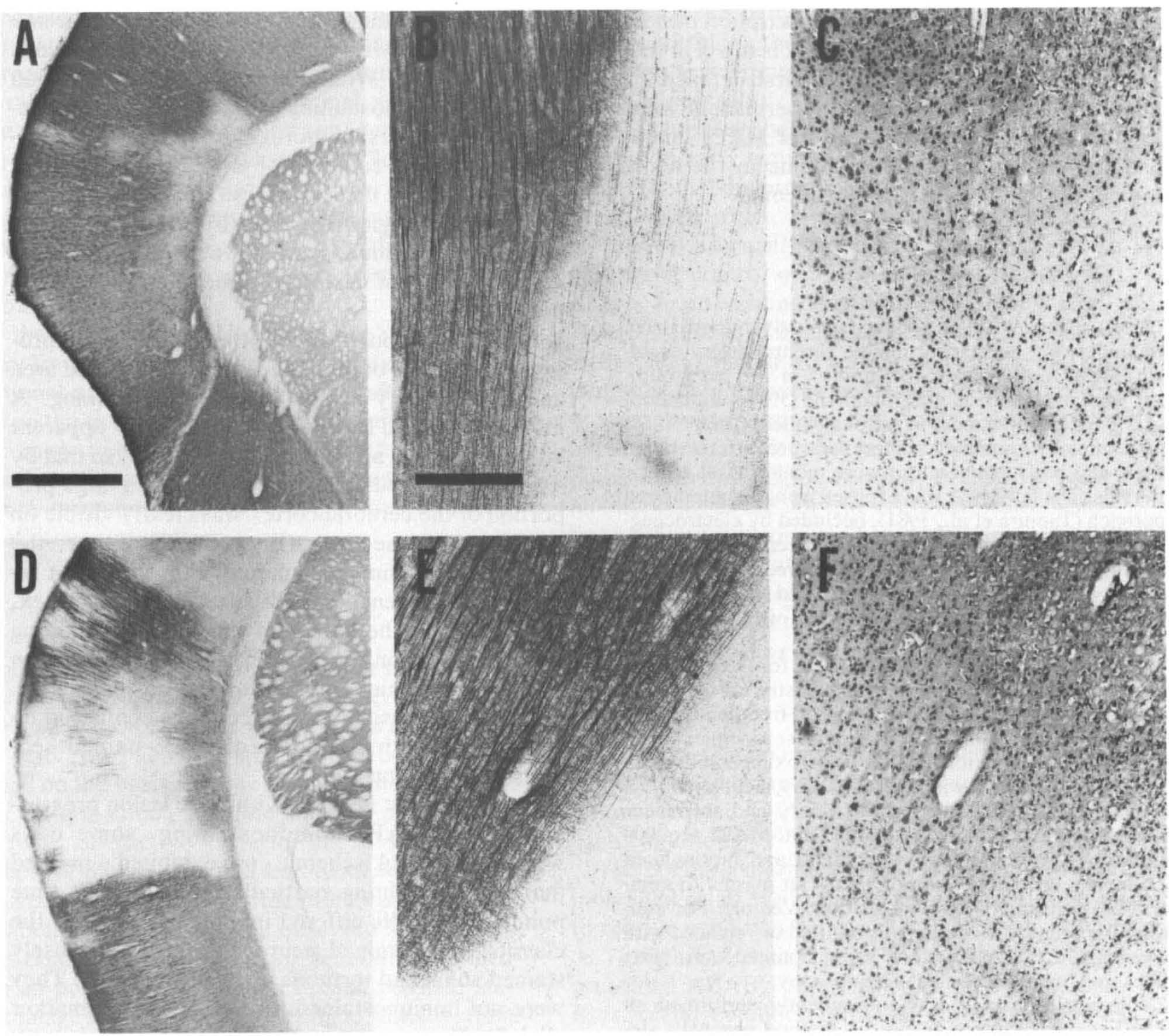
Alterations in microtubule-assisted protein 2 (MAP2) immunostaining following permanent middle cerebral artery occlusion (MCAO) in rats. Reduction in MAP2 immunostaining was apparent both 1
From 2 to 4h post-MCAO, the loss of MAP2 progressed in terms of both the size of the affected area and the intensity of the reduction in staining. A more widespread loss of staining was now apparent within the lesion as well as at the borders, so that by 4 h a well defined lesion encompassing a large proportion of the cerebral cortex was clearly visible on MAP2-stained sections (Fig. 1D). However, the loss of MAP2 immunostaining within the lesion remained heterogeneous with some areas of cortex, particularly in the center of the lesion, exhibiting smaller reductions in MAP2 immunostaining than neighboring tissue. Once again, under higher magnification, the lesion borders could be confirmed by comparison with the adjacent H&E-stained sections (Fig. 1E and F).
Although most neurons within the lesion progressively lost MAP2 immunostaining, some cells within the central ischemic core exhibited a marked increase in staining particularly at the 4-h time point. These cells differed in morphology from the classic MAP2-stained neurons with more densely stained soma and tortuous cellular processes. They were not immunostained for the astrocytic marker glial fibrillary acidic protein (results not shown), suggesting that despite the different morphological characteristics, they were, in fact, neurons rather than astrocytes.
The temporal evolution of lesion volume defined by the observed loss of MAP2 immunostaining is illustrated in Fig. 2. Statistical analysis (one-way analysis of variance followed by t tests with Bonferroni correction) revealed a significant increase in lesion volume from 1 to 4h post-MCAO.
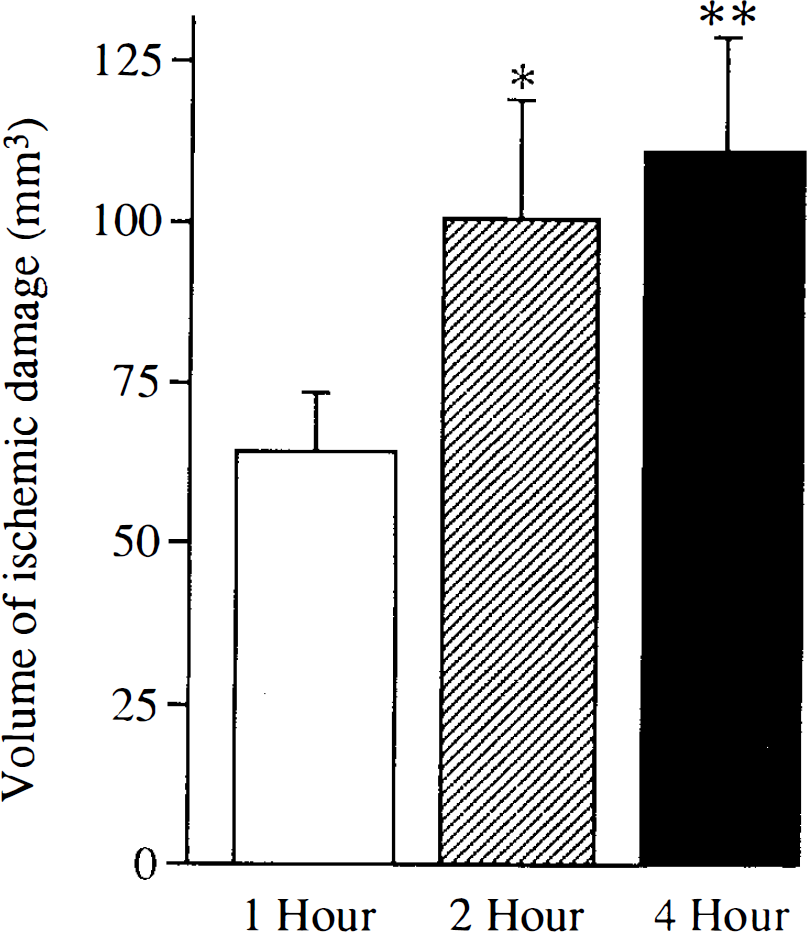
Temporal evolution of acute focal ischemic damage assessed by loss of microtubule-associated protein 2 immunostaining. The lesion volume (mean ± SD) increased significantly from 1 to 4 h postpermanent middle cerebral artery occlusion. *p < 0.05, **p < 0.01 comparison with 1-h group, unpaired t tests with Bonferroni correction, n = 5 per group.
DISCUSSION
In this study we have demonstrated reduced MAP2 immunostaining as early as 1 h following the onset of permanent focal ischemia. Furthermore, the observed reduction in MAP2 immunostaining could be used to delineate the borders of the acute ischemic lesion since (a) the borders between areas of preserved and reduced MAP2 immunostaining were sharp and well defined and (b) the lesion borders on the MAP2 sections were shown to be in reasonable agreement with the ischemic lesion borders on adjacent H&E-stained sections for each time point investigated. The considerable practical advantage of this novel MAP2 immunostaining technique over conventional H&E staining lies in the relative ease with which lesion borders could be identified on MAP2-immunostained sections (without the necessity of rigorous microscopic examination), particularly with regard to the acute 1-h time point.
In our study the time course for loss of MAP2 closely paralleled focal ischemic injury defined by H&E staining. This is in broad agreement with rat models of transient global ischemia where loss of MAP2 immunostaining in the CAj region of the hippocampus is coincident with neuronal loss assessed by standard histological techniques (Tomioka et al., 1992; Miyazawa et al., 1993). Thus, in the postischemic rat brain, the spatial and temporal agreement between reduction in MAP2 immunostaining and histological evidence of ischemic injury suggests that loss of MAP2 immunostaining is a reliable marker of neurons that are already undergoing irreversible processes leading ultimately to cell death. This is in contrast to gerbil models of transient global ischemia, where loss of MAP2 immunostaining may precede the development of overt neuronal loss and where it has been suggested that MAP2 immunostaining may be used as an indicator of still viable neurons that will only at a later time point initiate irreversible injury (Kitagamia et al., 1989; Yanagihara et al., 1990).
The observed loss of MAP2 immunostaining following ischemia may reflect cytoskeletal breakdown in response to rapid ischemia-induced elevations in intracellular Ca2+ concentration and subsequent activation of Ca2+-dependent phosphatases (e.g., calcineurin) and proteases (e.g., calpains) leading to dephosphorylation and proteolytic degradation of MAP2 (Siman and Noszek, 1988; Halpain and Greengard, 1990). In contrast to our findings of rapid MAP2 loss, a previous report found a significant reduction in MAP2 only after 72 h of permanent MCAO (Inuzuka et al., 1990). However, this study analyzed MAP2 levels in the entire cerebral hemisphere, and thus a comparatively small, localized reduction in cortical MAP2 may have been masked at time points earlier than 3 days following MCAO.
One difference between the MAP2 and H&E techniques concerns the heterogeneity of the MAP2 staining loss within the lesion boundaries. Neurons in areas with relatively preserved MAP2 immunostaining may have also maintained cytoskeletal integrity, and thus, by reference to the MAP2-stained sections alone, we cannot unequivocally state that these cells were irreversibly damaged. However, because the areas of preserved MAP2 immunostaining were completely enclosed by regions of marked MAP2 loss, and because the corresponding areas on adjacent H&E sections showed evidence of ischemic cell change, these areas were assumed to be injured and were included in the quantification of lesion volume. Acknowledging this assumption, the volume of ischemic injury was found to increase significantly from 1 to 4 h post-MCAO. While there was a numerical increase in lesion volume between 2 and 4 h, this difference was not statistically significant, suggesting that in this model of untreated ischemia (induced by electrocoagulation of the MCA), the volume of affected tissue is already maximal within 4 h of the onset of occlusion. This is in agreement with other studies utilizing conventional histological techniques that have demonstrated maximal lesion volume with 2- to 4-h duration of MCAO for both spontaneously hypertensive and normotensive rat strains (Kaplan et al., 1991; Buchan et al., 1992; Memezawa et al., 1992; Dawson et al., 1993).
In our study, some cellular elements in the ventral core of the lesion exhibited a marked increase in staining for MAP2, a phenomenon that has also been reported following transient global ischemia (Miyazawa et al., 1993; Geddes et al., 1994) and attributed to either an increase in neuronal staining (Geddes et al., 1994) or a novel expression of MAP2 in reactive astrocytes (Miyazawa et al., 1993). Our study suggests that (at least in the acute phase) following focal ischemia, the increase in MAP2 immunostaining occurs in neurons rather than astrocytes since cells that stained for MAP2 were not labeled for glial fibrillary acidic protein. The reasons for increased MAP2 immunostaining are unclear but are unlikely to reflect merely a change in antigenicity of MAP2 in response to changes in the phosphorylation state of the protein (Halpain and Greengard, 1990; Tomimoto and Yanagihara, 1992) and may instead reflect up-regulation of MAP2, perhaps in response to nitric oxide (Johnston and Morris, 1994).
In conclusion, we have demonstrated that permanent focal ischemia results in a rapid reduction in MAP2 immunostaining. Furthermore, the region borders defined by this loss of MAP2 can be measured to give a volumetric assessment of acute focal ischemic brain injury. Thus, loss of MAP2 immunostaining may prove to be a useful, quantifiable marker of brain damage following acute focal ischemia particularly where methodological constraints (such as in experiments utilizing multiple label immunohistochemistry) preclude the use of more conventional histological techniques.