
Abstract
Mycobacteria species include a large number of pathogenic organisms such as Mycobacterium tuberculosis, Mycobacterium leprae, and various non-tuberculous mycobacteria. Mycobacterial membrane protein large 3 (MmpL3) is an essential mycolic acid and lipid transporter required for growth and cell viability. In the last decade, numerous studies have characterized MmpL3 with respect to protein function, localization, regulation, and substrate/inhibitor interactions. This review summarizes new findings in the field and seeks to assess future areas of research in our rapidly expanding understanding of MmpL3 as a drug target. An atlas of known MmpL3 mutations that provide resistance to inhibitors is presented, which maps amino acid substitutions to specific structural domains of MmpL3. In addition, chemical features of distinct classes of Mmpl3 inhibitors are compared to provide insights into shared and unique features of varied MmpL3 inhibitors.
Introduction
Mycobacterium tuberculosis (Mtb) is the bacterium that causes tuberculosis (TB) in humans. In 2020, the WHO estimated that 10 million people became sick with TB and 1.5 million people died from the disease. 1 Currently, no vaccine protects against pulmonary TB. 1 In the absence of an effective vaccine, antibiotic therapy requires patients to take a daily combination of four drugs, including rifampin (RIF), isoniazid (INH), ethambutol (EMB), and pyrazinamide, for 6 months. However, the long course of treatment and incomplete therapy have led to the selection and evolution of multidrug-resistant (MDR) and extensively drug-resistant (XDR) Mtb strains, which are currently spreading person to person. 2 Therefore, additional therapeutic targets and strategies need to be identified. In addition, other pathogenic non-tuberculous mycobacteria (NTM), such as Mycobacterium avium complex (MAC) and Mycobacterium abscessus (MAB), are emerging as common causes of infections, particularly in the immunocompromised, the elderly, and those with predisposing conditions, such as cystic fibrosis. These NTMs are resistant to most Mtb drugs3,4 and therefore new drugs are also needed to control NTM-mediated diseases. 5
Over the last few decades, high-throughput screens (HTS) were conducted to identify the next Mtb drug. Subsequent studies into the mechanism of action of hits identified from these screens have identified QcrB,6–14 DprE1,15–20 and mycobacterial membrane protein Large 3 (MmpL3)20–37 as recurring targets. Of these three targets, the essential mycolic acid (MA) flippase MmpL320–37 is the most commonly identified with 18 studies reporting over 30 chemical scaffolds targeting MmpL3.20–38 While several of the MmpL3 inhibitors have overlapping chemical groups (discussed further below), the specific structural differences between them make predicting MmpL3 inhibitors within a chemical library difficult. However, the essential nature of MmpL3 makes it a highly sought-after therapeutic target for Mtb. 39 Owing to its high therapeutic potential, several studies over the last decade have increased our understanding of MmpL3 in terms of function, regulation, and protein-substrate/inhibitor interactions. This review will discuss the function of MmpL3 in mycobacteria as well as the molecular insights gained from studying MmpL3 and the inhibitors proposed to target this essential flippase.
Synthesis and Transport of TMM and TDM
MmpL3 is the flippase and the sole transporter for the essential branched long chain (C60–90) glycolipid TMM (illustrated in Fig. 2a) synthesized in the cytoplasm or cytoplasmic membrane (CM). The complete synthetic pathway of TMM is still not fully understood and some differences exist between species of mycobacteria. An overview of the TMM synthetic pathway is outlined in Fig. 1 and a description is as follows: acetyl-CoA (C2) and malonyl-CoA (C3) generated from catabolic pathways serve as primers for the synthesis of short chain (C24–26 or C16–18) fatty acids (FAs) by the eukaryotic like fatty acid synthase (FAS)-I enzyme Fas (Rv2524c).40,41 From here, MA synthesis diverges along two paths to form the α-branch and the meromycolate chain.

The biosynthetic pathway of TMM and TDM. The illustration of the biosynthetic route of TMM and TDM in mycobacteria. Gene names of each step include corresponding gene numbers from Mycobacterium tuberculosis H37Rv. Enzymes highlighted in red are genes that are downregulated following MmpL3 disruption. Δψ, membrane potential; AG, arabinogalactan; CM, cytoplasmic membrane; MmpL3i, unspecified MmpL3 inhibitor; PG, peptidoglycan; yellow sphere attached to TMM is an acetyl group.?—an unknown transport system that shuttles TMM to the mycomembrane. MmpL3, mycobacterial membrane protein large 3; TDM, trehalose dimycolate; TMM, trehalose monomycolate.
TMM and MmpL3 Structures.
The α-branch consists of carboxyacyl-CoA (C24–26) (Fig. 2a, red) and is formed by the acyl-CoA carboxylase complex consisting of AccA3 (Rv3285) and AccD4 (Rv3799c). 42 Along the other branch, C16–18 short chain FAs are elongated by the β-ketoacyl acyl carrier protein (ACP) synthase mtFabH (Rv0533c), which condenses a malonyl substrate, carried by the ACP AcpM (Rv2244), to form β-keto-thioester.43–45 Malonyl-AcpM is formed by the transfer of malonyl from malonyl-CoA to AcpM by the enzyme mtFabD (Rv2543).46–48 The FAS-II system is composed of several enzymes, including the β-ketoacyl-reductase MabA (Rv1483),49–52 the β-hydroxyacyl-ACP dehydratases HadAB/HadBC (Rv0635-0637),53,54 the trans-2-enoyl-ACP reductase InhA (Rv1484),55–57 and β-ketoacyl-ACP synthases KasA/KasB (Rv2245/Rv2246).58,59
It is hypothesized that to form the fully mature long chain FAs, up to three unique FAS-II complexes exist and are composed of specific combinations of the FAS-II enzymes.60,61 From here, long chain FAs undergo several reactions independent of the FAS-II complex, including modifications, such as desaturation and cyclopropanation,62–69 to form the meromycolate chain (C48–62). FadD32 (Rv3801c) then activates the meromycolate chain42,70–72 to undergo a multistep reaction carried out by the polyketide synthase Pks13 (Rv3800c). Pks13 induces a condensation reaction between the meromycolate chain and the α-branch to form mycolic β-ketoesters.70,73 Pks13 then carries out an additional reaction to add the disaccharide trehalose to the β-ketoester to form the α-alkyl β-ketoacyl trehalose glycolipid. 74 In the final synthesis step, CmrA (Rv2509) reduces α-alkyl β-ketoacyl trehalose to form the mature MA TMM (Fig. 2a). 75
Several steps in the synthesis of TMM are still not understood, including double bond formation in MAs. In this review, we illustrate MA modifications such as cyclopropanation occurring after exiting the FAS-II complex; however, when exactly these steps occur is unknown and may occur after TMM is transported into the CM. In addition, not all TMM synthetic pathways are the same and can result in different chain lengths and structural modifications.76,77 For example, TMM of Mtb includes cyclopropyl, keto-, methoxy, and hydroxyl groups, while TMM of Mycobacterium smegmatis contains cis-, trans-, or epoxy groups. 77 Furthermore, both meromycolate and α-chain length can vary between species. 78 Chain length regulation is also not fully understood, but is likely regulated by higher order protein-protein and protein-cofactor interactions.40,79,80 TMM is essential for mycobacteria cell viability, and therefore many proteins involved in the synthesis of TMM are also essential.81–85 Consistent with a model of essentiality, enzymes involved in MA synthesis are often identified as targets of anti-TB inhibitors. Notable targets include InhA and HadAB, which are inhibited by the TB drugs INH 56 /ethionamide86,87 and isoxyl 88 /thiacetazone, 88 respectively.
The above section describes the synthesis of TMM, which has been fairly well characterized over the last half century. However, only recent efforts have managed to characterize the flipping of TMM across the CM, but some gaps do still exist. One essential step required before transport by MmpL3 is that TMM must first be acetylated (acTMM) by TmaT (Rv0228).89,90 TmaT is an essential integral membrane protein82,83,85,89 and it is hypothesized that TmaT plays a role in intercalation of (ac)TMM into the inner leaflet of the CM. 90 However, how this occurs is unclear and additional enzymes may be involved in this step. For example, two recent studies identified a putative methyltransferase MtrP (NCgl2764, Rv0224c) and the membrane protein MmpA (NCgl2761, Rv0226c) as being required for efficient transport of the Corynebacterium TMM equivalent trehalose monohydroxycorynomycolate from the CM to outermembrane in Corynebacterium glutamicum.91,92 Both of these genes are conserved in mycobacteria, and were identified as essential based on transposon mutagenesis studies82,83,85,89; however, their functions have yet to be studied in mycobacteria. Also, TtfA (Rv0383c), a protein of unknown function, was demonstrated to interact with MmpL3 and is required for TMM transport, but its role in TMM transport is still unknown. 93 Taken together, the identification of three essential proteins for (ac)TMM transport suggests that MmpL3 alone may not be the sole protein involved in MmpL3 flipping.
Following full maturation and acetylation, TMM is then transported across the CM by the dedicated flippase MmpL3 (Rv0206c). This flipping action occurs in a two-step process powered by membrane energetics (discussed further in the next section). Following flipping, (ac)TMM is associated with the porter domains of MmpL3, where it is hypothesized that (ac)TMM is handed off to a yet identified chaperone system to be transported across the peptidoglycan (PG) and arabinogalactan (AG) layers to the mycomembrane (MM) (Fig. 1). Once in the MM, TMM can either accumulate or undergo two different acyltransferase reactions carried out by the Ag85 complex (FbpA, FbpB, and FbpC2).94–98 The first acyltransferase reaction removes the trehalose moiety and covalently links the MA to the terminus of the AG layer forming mycolyl-arabinogalactan-peptidoglycan (mAGP), a single molecule covalently linking the PG, AG, and MM. 96 This acyltransferase reaction is essential for cell viability,96,97 and is the reason why MmpL3, as the sole transporter of TMM, is essential. The second possible acyltransferase reaction moves the MA moiety of one TMM molecule to the 6′C of a second TMM molecule to form trehalose dimycolate (TDM) (Fig. 2a). 96 TDM is also known as “Cord Factor” due its role in the formation of mycobacterial cords, 99 a multicellular aggregate characteristic of mycobacteria. 100 TDM plays roles as a protective barrier,69,97,101 biofilm formation, 102 granuloma formation,103–108 and macrophage stimulation.106–109 While mAGP faces the periplasm space, the localization of TDM in the inner or outer leaflet of the MM following synthesis is not clear. Future studies may seek to understand the (a)symmetry of TDM across the MM.
Preceding the acyltransferase reactions in the MM, several questions revolving around the fate of acTMM following transport across the CM still remain. For one, it is still unclear when TMM undergoes de-acetylation (acTMM to TMM) and whether or not this is an essential step. Second, one of the main pressing questions concerning the fate of (ac)TMM is how it is transported across the PG and AG layers. Surface plasmon resonance (SPR) data from Belardinelli et al demonstrated that MmpL3 does not directly interact with proteins from the Ag85 complex. 110 This is consistent with distance measurements that the 35 Å porter domains protruding from the CM would not reach across the 30–40 nm gap between the CM and the MM.110–112 Two studies have attempted to find periplasmic interacting partners of MmpL3 using the bacterial adenylate cyclase-based two-hybrid (BACTH) system and protein co-precipitation methods, but neither identified any periplasmic or cell wall localized candidates.93,110 Further studies into (ac)TMM transport may identify additional drug targets, as well as further our understanding of the physiology and cell wall biogenesis of mycobacteria.
MmpL3 Protein Structure and Function
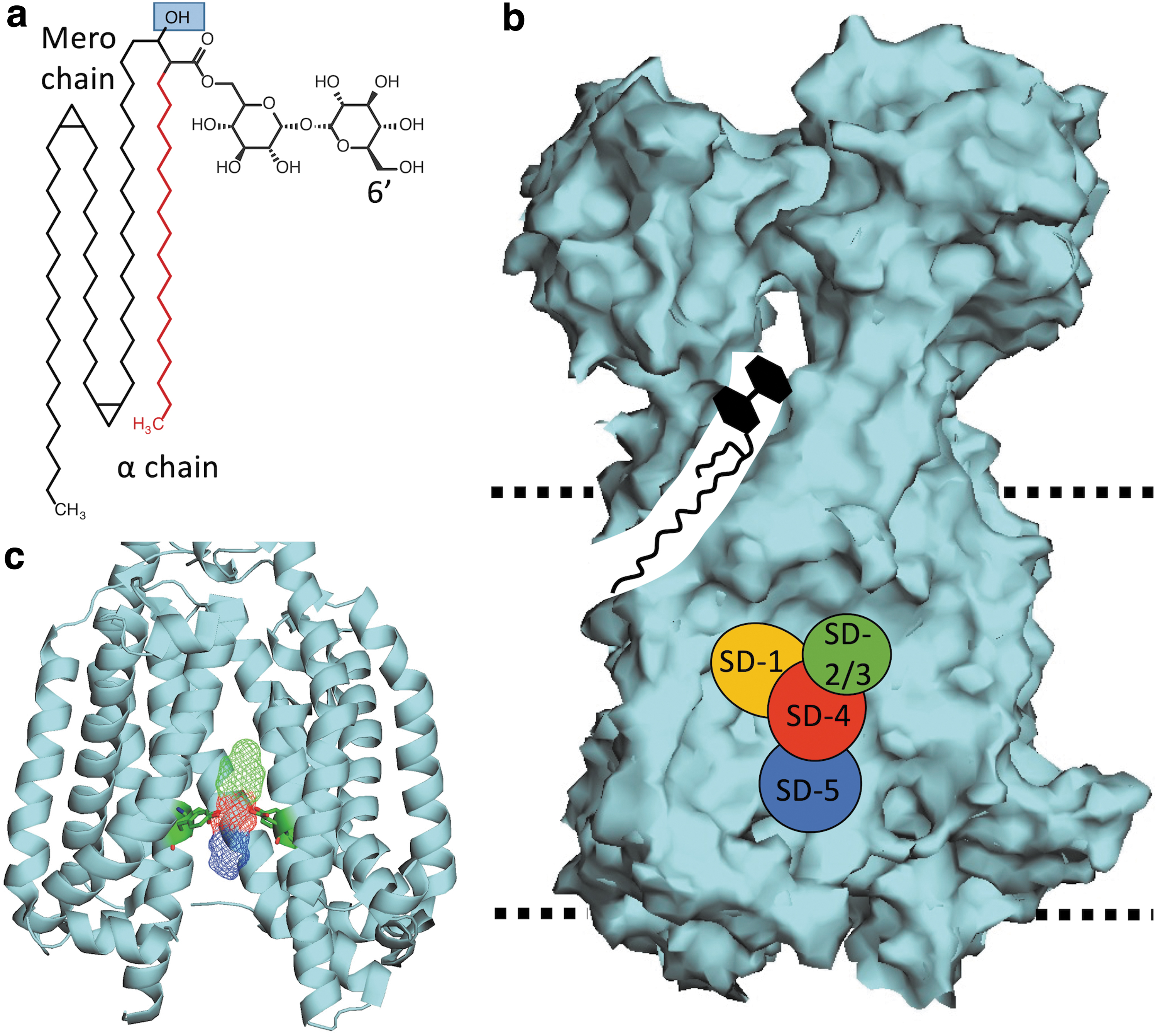
MmpL3 (illustrated in Fig. 2b) is one of several MmpL proteins found in mycobacteria 113 and is a member of the resistance nodulation and division (RND) superfamily of proteins.89,114–121 MmpL3 has three main structural regions, a transmembrane region that spans the CM, a porter domain consisting of two large loops in the periplasmic space, and a cytoplasmic C-terminal domain. The transmembrane region is made up of 12 transmembrane α-helix domains (TMD 1–12).89,116,117,120–122
Like other RND proteins, MmpL3 is powered by proton translocation through a central vestibule made up of TMDs 4, 5, 6, 10, 11, and 12.89,123 Proton translocation is guided by conserved Asp/Tyr pairs on TMD 4 and 10 (Fig. 2c).89,115,117,119–122 Furthermore, the transmembrane region also serves to interact with lipid substrates. Recently, TMD 7–10 were shown to play a role in lipid binding based on cryo-EM structures of TMM bound to MmpL3Mtb. 124 The 12 TMDs are conserved among the 13 MmpL proteins of Mtb, with the exception of MmpL13, which seems to have undergone a genetic cleavage event in which the protein was split into 2 genes (mmpL13a/Rv1145 and mmpL13b/Rv1146). 125
While the domain architecture of MmpL3 is similar to the other 12 MmpL proteins in Mtb, 116 one key difference is the presence of a large cytoplasmic C-terminal domain, found only in MmpLs 3, 11, and 13114,116 and rarely found in canonical RND proteins.114,116 The C-terminal domain is involved in protein localization, 110 protein-protein interactions,93,110 and post-translational phosphoregulation (all discussed further below).126,127 This C-terminal domain is nonessential 89 and is not found in other canonical RND proteins.114,116 The tertiary structure of MmpL3 from M. smegmatis (MmpL3Msm) has been resolved and crystal structures have suggested that MmpL3 is a monomer.120–122 However, these structures were from truncated forms of MmpL3 lacking the C-terminal domain.120–122 Recently, a BACTH system study suggested that full-length MmpL3 forms an oligomer. 110 A complexed MmpL3 is consistent with previous modeling predictions 89 and observations for canonical RND proteins such as AcrB of Escherichia coli. 123
Interestingly, full-length MmpL3 from either M. smegmatis or Mtb has been difficult to purify120,121,124,128 and the model confidence in the predicted AlphaFold structure of MmpL3 drops dramatically for the cytoplasmic domain.129,130 A brief analysis using the Intrinsically Unstructured Protein Prediction server (IUPred2A)131,132 suggests that the C-terminal domain is highly disordered (Supplementary Fig. S1), which would explain the inability to purify this domain of the protein. Alternatively, post-translation modification, such as phosphorylation (discussed in a separate section) may be required to stabilize the structure before purification.
The structure of MmpL3 is further characterized by two large periplasmic porter domains located between TMD 1 + 2 and 5 + 6.89,114,120–122 These porter domains were demonstrated to interact with the lipids phosphatidylethanolamine 120 and TMM. 124 Purified porter domains of MmpL3 were also shown to interact with heme, 133 but it is unclear what role MmpL3 may play in iron metabolism. It is hypothesized that these porter domains play a role in handing off exported TMM to the aforementioned unidentified chaperone system to transport TMM to the MM; however, other proteins could also be involved in this process.
As mentioned above, following acetylation, acTMM is flipped across the CM in a two-step mechanism. The first step relies on the proton motive force (PMF), which flips acTMM from the inner to the outer leaflet of the CM (Fig. 1). The evidence for this first step primarily comes from a spheroplast assay developed by Xu et al. This assay measures the abundance of (ac)TMM that is fluorescently labeled once deposited into the outer leaflet of M. smegmatis spheroplasts. 117 Xu et al observed that labeled (ac)TMM accumulates in the outer leaflet in untreated cells; however, following treatment with MmpL3 inhibitors, such as AU1235 or BM212, or PMF uncouplers, such as CCCP or nigericin, fluorescence is diminished. 117 These data clearly demonstrated that MmpL3 is involved in (ac)TMM flipping across the CM and that (ac)TMM transport may be a two-step mechanism. The second step in (ac)TMM flipping and transport involves MmpL3 shuttling (ac)TMM from the outer leaflet of the CM to be transported to the MM. This step is carried out by TMDs 7–10, which seem to relay TMM to the porter domains. Evidence for this primarily comes from a recently published cryo-EM study by Su et al, which captured TMM in both the transmembrane domain and the porter domain (Fig. 2b). 124 This study was remarkably informative in how MmpL3 transports (ac)TMM from the CM to the porter domains, which are then hypothesized to hand (ac)TMM off to an unidentified chaperone system, as described above.
Taken together, the results of the spheroplast assay and cryo-EM structures generally agree with a two-step model that was predicted, but never demonstrated. While these studies have elucidated much of the mechanism that drives TMM flipping, two important questions remain unanswered. The first is how MmpL3 recognizes acTMM before flipping and what protein domains drive flipping from the inner to the outer leaflet of the CM. A recent photoactivatable probe was created by Kavunja et al that structurally resembles TMM. 134 Upon photoactivation, this probe is covalently linked to interacting proteins and allows for enrichment of proteins that associate with the probe. This probe enriched several proteins in M. smegmatis cells previously demonstrated to interact with TMM, including MmpL3. The authors identified the enriched proteins through peptide-based MS; however, which residues the probe specifically linked to were either not identified or not reported. Additional experiments using these probes with pre-enriched MmpL3-expressing cells or inverted membrane vesicles may identify which MmpL3 residues TMM interacts with in the inner leaflet of the CM.
The second unanswered question is following incorporation of acTMM to the porter domain, how is acTMM handed off to the next protein and which protein(s) is/are responsible for chaperoning acTMM to the MM. Several studies have attempted to identify periplasmic MmpL3 interactors; however, neither reported any major hit.
Furthermore, while this review and many studies have focused on the role of MmpL3 in TMM transportation, a recent study reported that MmpL3 interacts with additional lipids, including cardiolipin, diacylglycerol, phosphatidylglycerol, and phosphatidylinositol. 120 More importantly, this study also reported co-crystal structure of the lipid phosphatidylethanolamine bound to the porter region of MmpL3 in a way that was highly similar to what was reported for TMM. This suggests that MmpL3 is not solely dedicated to TMM transport; however, the biological consequences of these findings have yet to be investigated.
The number of mmpL genes varies between mycobacteria. For example, Mycobacterium leprae encodes only 5 mmpL genes, while Mtb encodes 13 mmpL genes, and Mycobacterium immunogenum, of the MAB complex, encodes 29 mmpL genes. 116 However, only mmpL3 and mmpL11 are conserved in all species of mycobacteria 116 and only mmpL3 is essential for growth and viability both in vitro and in vivo in most mycobacteria.29,81–85,89,135–140
Comparative protein structure prediction studies show that Mtb MmpL proteins fall into two distinct clusters. 114 MmpL3 is a member of the Cluster II MmpL proteins, along with MmpL11 and MmpL13, with the remaining 10 Mtb MmpL proteins falling into Cluster I. Cluster II MmpL proteins are distinguished from Cluster I MmpL proteins by the inclusion of the cytosolic C-terminal domain and the lack of a docking domain in the second porter loop. 114 It should be noted that some MmpL proteins have associated mycobacterial membrane protein small (MmpS) proteins, which are typically encoded adjacent to their cognate mmpL genes. 113 MmpS proteins have been hypothesized to help in MmpL function, although how they do this is not clear. A study looking into the role of the semiredundant MmpS4 and MmpS5 proteins indicates that they play a role in assisting the cognate MmpL4 and MmpL5 proteins in iron scavenging. 141 In addition, not all MmpL proteins have associated MmpS proteins, including MmpL3. 142 While the exact function of all MmpL proteins in mycobacteria is not yet clearly defined, it is largely believed that they serve as substrate exporters, including xenobiotic efflux. 143
MmpL3 Localization and Interactome
Incorporation of TMM into the cell wall of mycobacteria during cell division is an essential process. Mycobacteria undergo asymmetric cell division and extend from the old pole. 144 Therefore, it stands to reason that proteins involved in cell wall synthesis, including MmpL3, would localize to the same area. Indeed, fluorescently labeled MmpL3 was demonstrated to localize at the dividing pole during cell division.33,93,110,118,145,146 Localization of MmpL3 to the dividing pole is guided, in part, by the C-terminal domain, as mycobacteria expressing truncated MmpL3 lacking the C-terminal domain show decreased MmpL3 polar localization during cell division. 110 In addition to the C-terminal domain, the DivIVA homolog Wag31 (Rv2145c) may also play a role in MmpL3 localization. 147 While it is not clear if Wag31 directly interacts with MmpL3, 147 Wag31 does coordinate cell division machinery to the pole during cell division, including enzymes involved in PG synthesis148,149 and MA biosynthesis, such as AccA3. 147
Recent reports have identified additional proteins that directly interact with MmpL3, including additional proteins involved in MA transport, as well as proteins involved in PG and AG substrate transport.93,110 MmpL3-interacting proteins involved in TMM transport included the TMM acetylation protein TmaT, 110 as well as TtfA (Rv0383c) and Msmeg_5308 (Rv1057). 93 The function of TtfA is not fully defined, but is essential in mycobacteria and is required for the transport of TMM, 93 while MSMEG_5308 is not required for TMM transport and dispensable for cell viability, 150 but may stabilize TtfA-MmpL3 interactions during cell stress. 93 MmpL3 was also demonstrated to interact with MmpL11 (Rv0202c), 110 which is involved in the transport of MA-containing lipids, monomeromycolic diacylglycerol, and mycolate wax ester. 151
Additional proteins of unknown or ill-defined function were also identified as possible MmpL3 interactors based on a BACTH screen, including Rv0204c, Rv0207c, Rv0625c, Rv1275 (LprC), Rv1337, Rv1457c, Rv1799 (LppT), Rv2169c, Rv3064c, Rv3271c, Rv3483c, Rv3909, and MT2653, 110 which may allude to the existence of a large cell wall transport complex; however, these hits have yet to be verified using alternative methods. Notably MmpL3 does not interact with the Ag85 complex or enzymes involved in MA synthesis enzymes such as FAS-II enzymes or Pks13.93,110 While a lack of interaction between the Ag85 complex and MmpL3 could be predicted based on distance measurements as discussed above, 110 the lack of interaction between MA synthesis enzymes and MmpL3 was surprising, given they are co-coordinated by Wag31147 to localize to the dividing pole 146 and are co-regulated by PknAB/PstP.126,127,148
Current studies of MmpL3 localization and co-localization have focused on dividing cells to gain better insights into cell wall biogenesis and the mycobacterial divisome.33,93,110,118,145,146 However, questions remain for where MmpL3, and other divisome proteins, localizes during states of nonreplication when MmpL3 is dispensable for cell viability,89,137 and some MmpL3 inhibitors do not kill nonreplicating mycobacteria.22,152 Using scanning electron microscopy, Lun et al observed that Mtb treated with an MmpL3 inhibitor developed dimples at the dividing pole where MmpL3 localizes. 153 They hypothesized that these dimples were actually holes forming at the site of MmpL3 localization leading to cell death. 153 If MmpL3 does not localize to the dividing pole during states of nonreplication, these holes may not develop, which could explain why MmpL3 inhibitors do not kill nonreplicating cells. Future studies may seek to address where proteins involved in cell wall synthesis and division, including MmpL3, localize during states of nonreplication.
Regulation of MmpL3
mmpL3 is encoded in a monocistronic operon in mycobacteria. 136 To date, the transcriptional regulation of mmpL3 is not fully understood. However, Chromatin Imunnoprecipitation sequencing and electrophoresis mobility shift assays identified Rv1816 and Rv3249c as repressors of mmpL3, as well as all other mmpS/mmpL genes, with the exception of mmpL6.154,155 The observation that so many mmpS/mmpL genes share transcriptional regulators with mmpL3 suggests that a coordinated regulatory pathway is needed for Mtb to adapt to new environments. In addition, Rv1816 also regulates kasA of the FAS-II pathway. 154 This finding suggests that mmpL3 regulation is co-coordinated with MA synthesis, despite no direct protein-protein interaction. 110 This finding is consistent with the observation that FAS-I/FAS-II genes are downregulated following mmpL3 knockdown or MmpL3 inhibition (Fig. 1).22,138,156
Repression by Rv1816 and Rv3249c is relieved upon binding to either palmitic acid or isopropyl laurate. 154 The identification of palmitic acid as an inducer was serendipitous, 154 but is consistent with the model that links MA synthesis with transport regulation as Mtb Fas is biased to stearic acid (C18).40,41 One model that may link Fas with MmpL3 is that as Fas begins to generate short-chain FAs, mmpL3, as well as other genes regulated by Rv1816/Rv3249c, is induced to ready the cell for replication. However, additional experiments would be needed to test this model.
In addition to transcriptional regulation, MmpL3 activity is post-translationally phosphoregulated by PknA, PknB, and PstP at the C-terminal domain.126,127 In two separate studies, Zeng J. and Adams O. demonstrated that overexpression of PknB led to increased phosphorylation of MmpL3Msm at T920 and T984, 126 while depletion of pknA resulted in decreased phosphorylation of the MmpL3Mtb residues T893 and T910. 127 Consistent with a repressive model, overexpression of PknB is lethal and mirrored MmpL3 perturbation lipid profiles resulting in increased TMM as MmpL3 transport was lost and decreased TDM due to lower TMM substrate. 126 In accordance with this model, both genetic depletion of pknA and pknB and small molecule inhibition of PknA and PknB resulted in increased TMM, but no alteration in TDM levels.127,157 The lipid profiles of the PknA/B disruption studies are consistent with a model of unregulated MmpL3 activity in which TMM is synthesized through FAS-II upregulation and TMM export at increased rates, but without TDM conversion due to lower Ag85B expression following PknA/B disruption.127,157 These studies were conducted in two mycobacterial species, Msm and Mtb,126,127,157 suggesting MmpL3 phosphoregulation is conserved in other mycobacteria.
The phosphorylation-based inhibition of MmpL3 activity is relieved by the serine/threonine phosphatase PstP. 126 Repression of pstP resulted in the loss of viability and a decrease in TMM abundance consistent with the loss of MmpL3 activity through phosphorylation. 126 That phosphorylation of the MmpL3 C-terminal inhibits protein activity is consistent with observations that the C-terminal domain is not essential for MmpL3 activity or cell viability. 89 Regulation by PknAB/PstP is not limited to MmpL3, and includes other lipid/MA synthesis enzymes, including Fas, FabG, HadA, AccD5, FadD32, AccA3, and Pks13, 126 as well as Wag31,148,149 further linking MmpL3 activity with cell wall synthesis and division. PknAB/PstP also regulates PG synthesis, 158 linking MA transport regulation with PG biosynthesis.
Interestingly, while depletion of pknA led to decreased phosphorylation of MmpL3, depletion of pknB did not. 127 This may suggest differential regulatory roles of PknA and PknB, despite pknB being encoded immediately downstream of pknA in the same operon. 127 Although PknB localizes to the dividing pole during cell division, PknB was not identified as a not identified as a part of the MmpL3 interactome,93,110 suggesting a transient association with MmpL3. Fascinatingly, depletion of both pknA and pknB in a double knockdown strain led to increased phosphorylation of MmpL3Mtb at residues S823, T840, S868, T872, T893, and T910. 127 As only Ser/Thr residues were phosphorylated in the pknA/B double knockdown strain, this suggests that other Ser/Thr kinases are involved in MmpL3 phosphoregulation. Mtb has 11 Ser/Thr kinases, 113 including PknA and PknB, suggesting that the other 9 Ser/Thr kinases may play a role in phosphoregulating MmpL3. A study by Prisic et al identified Thr residues as the preferred phosphorylation sites for PknA, PknB, PknD, PknE, PknF, and PknH. 160 This would suggest that the Ser residues identified to be phosphorylated by Zeng et al in the pknA/B double knockdown strain may be phosphorylated by PknG, PknI, PknJ, PknK, or PknL.
Additional studies are required to understand the transcriptional and post-translational regulation of MmpL3. While MmpL3 is post-translationally regulated through phosphorylation of the C-terminal domain,126,127 the residues identified are not conserved in all mycobacteria. These differences may result in differential post-translation regulation of MmpL3 between species. In addition, how phosphorylation of the C-terminal domain results in decreased MmpL3 activity is not clear, but may result in (i) dissociation of predicted MmpL3 homotrimers,89,110 (ii) dissociation of MmpL3 from other interacting proteins such as TmaT 110 and TtfA, 93 or (iii) delocalization from the dividing pole.
The Therapeutic Potential of MmpL3
MmpL3 is conserved in mycobacteria as well as the closely related Corynebacterium spp. (CmpL1).89,136 MmpL3 was initially demonstrated to be essential in two studies by Lamichhane et al in a transposon screen, 161 and by Domenech et al, who could not generate an mmpL3 knockout. 143 Since then, several additional lines of genetic evidence have validated this finding, including the observation that mycobacteria rapidly lose viability upon mmpL3 knockdown or MmpL3 protein depletion.137,138,140 In addition, saturating transposon mutagenesis studies have failed to identify null mutants in Mtb, Mycobacterium bovis Bacillus Calmette-Guérin (BCG), or Mycobacterium paratuberculosis.81–85,135 However, recently, Xiong et al reported an mmpL3 knockout in M. neoaurum (ATCC 25795). 139 The method used to create this knockout strain failed to generate knockouts in Mtb 143 and M. smegmatis. 29 Therefore, why Xiong et al were able to knockout mmpL3 in M. neoaurum is unclear. Some possibilities include (i) the presence of an additional (ac)TMM transporter or (ii) a lowered dependency on TMM for MM anchoring in M. neoaurum. Precedent for either scenario exists for closely related Corynebacterium species, which are viable without the TMM equivalent trehalose corynemycolate (TMCM).90,136 To date, the effects of MmpL3 inhibitors have not been tested in M. neoaurum, which may give some insights into the essentiality of MmpL3 and TMM transport in this mycobacterial species. However, in most species of mycobacteria, including clinically relevant Mtb, MAC, and MAB, mmpL3 remains classified as an essential gene in replicating cells. This was recently highlighted in a whole genome CRISPRi knockdown screen by Bosch et al, which identified mmpL3 as one of the most vulnerable genes in Mtb to genetic perturbation. 162
The essentiality of MmpL3 in vitro translates to both ex vivo and in vivo infection models. Genetic knockdown and protein depletion models have demonstrated that Mtb rapidly loses cell viability in both infected macrophages138,163 and mice (C57Bl/6) 137 following MmpL3 depletion. These observations make MmpL3 an attractive target for TB chemotherapy and efforts to identify MmpL3 inhibitors have been remarkably successful through both untargeted20–35,37 and targeted25,36 screening approaches. To date, at least 30 parental chemical scaffolds have been proposed as MmpL3 inhibitors.20–38
Treatment of Mtb and other mycobacteria with these inhibitors results in bactericidal effects in vitro,20–38 as well as growth inhibitory effects in ex vivo models.21,22,25,26,35,164–166 Follow-up structure–activity relationship (SAR) studies have resulted in the development of hundreds of active analogs,22,31,35,37,165–168 with some demonstrating activity against Mtb and MAB in vivo.27,28,35,153,164,167–174 However, it should be noted to what extent these parental compounds and their active analogs have been investigated to directly target MmpL3 varies. In addition, in many cases, SAR studies are not followed up with target validation to ensure that MmpL3 remains a target. Furthermore, several MmpL3 inhibitors have demonstrated additional mechanism of action (MOA), and few studies have been conducted to test for such off-target effects in most proposed MmpL3 inhibitors.
Adding to the success of MmpL3 inhibitors in preclinical models, SQ109 has had success in clinical trials175,176 and recently completed a Phase IIb clinical trial. 177 In humans, SQ109 is well tolerated175–177 and leads to decreases in viable Mtb in sputum samples when taken in combination with RIF over 14 days. 176 While SQ109 is active against MDR-TB in patients, 177 SQ109 has a short half-life due to host drug metabolism,178,179 which is exacerbated when taken in combination with RIF. 175 However, the success of SQ109 thus far validates MmpL3 as a clinical target for Mtb therapy. Moving forward, clinical trials involving other MmpL3 inhibitors with altered pharmacokinetic profiles may find additional success.
The success of SQ109 as an MmpL3 inhibitor in human TB patients shows promise for other MmpL3 inhibitors being studied and developed elsewhere. However, MmpL3 as a target is not without its limitations. Despite being a commonly identified target for inhibitors active against Mtb, these same inhibitors are not active in all mycobacterial species. Using a wide selection of previously described MmpL3 inhibitors, Li et al demonstrated that some MmpL3 inhibitors, including SQ109, have low activity against MAB and MAC species. 180 The low activity in NTM species was scaffold specific, and some MmpL3 inhibitors such as the indolecarboxamides NITD-304 and NITD-349 demonstrate high activity against NTMs. 180
Another limitation of MmpL3 as a target is the nonessential nature of MmpL3 in nonreplicating mycobacteria.89,137 In the granuloma, the growth rate of Mtb exists along a spectrum of actively replicating to nonreplicating due to factors including nutrient starvation, host derived stresses, and hypoxia. 181 Using an inducible MmpL3 depletion strain, Li et al demonstrated that during states of nonreplication, depletion of MmpL3 did not significantly reduce the viability of Mtb. 137 This observation is consistent with observations that the treatment of nonreplicating Mtb with indocarboxamides, AU1235, and HC2091 did not lead to significant losses in viability22,152; however, others do kill nonreplicating bacteria (discussed further below). Taken together, these observations indicate that strategies must be developed to overcome therapeutic limitations of targeting MmpL3.
While MmpL3, as a target, has limitations for nonreplicating mycobacteria, similar “lack of activity” observations for nonreplicating bacteria have been made for the first-line drug INH,22,152,182–185 which has been used clinically for nearly 70 years. 186 In addition some MmpL3 inhibitors, including SQ109, BM212, C215, TBL-140, E11, HC2032, HC2134, HC2138, HC2149, HC2178, and HC2184, have additional effects, including PMF uncoupling,25,31,152,165 and some have been demonstrated to be active against nonreplicating persistent Mtb.22,31,152,165 While these off-target effects do not add to the therapeutic potential of MmpL3, they do suggest that limitations in MmpL3 inhibition can be overcome through secondary MOA. In addition, mycobacteria are treated using drug combinations, and inclusion of drugs active against nonreplicating mycobacteria such as Bedaquiline, 187 RIF, and others 188 could overcome therapeutic limitations of MmpL3. MmpL3 inhibitors have also demonstrated high activity against clinical monodrug-resistant, MDR, and XDR-Mtb strains.26,27,29,32,35,165,171
Furthermore, there have been no report of resistant mutants isolated from patients who received SQ109 during clinical trials. However, analysis of >45,000 whole genome sequences from clinical samples biased toward drug-resistant strains identified nonsynonymous mutations, although at low frequencies. 128 These mutations included ones known to cause resistance to preclinical MmpL3 inhibitors, including a V210A mutant that has been demonstrated to be resistant to SQ109 in vitro. 34 While follow-up studies to this finding have not been conducted to determine the level of resistance such strains have against SQ109 and other MmpL3 inhibitors being developed, these findings do emphasize the need for active surveillance of clinical strains resistant to MmpL3.
One possible silver lining, although, is the observation that mmpL3 mutants resistant to MmpL3 inhibitors are hypersusceptible to RIF,25,189 suggesting a co-therapy of RIF and an MmpL3 inhibitor could reduce the frequency of resistance (FoR) to MmpL3 inhibitors in the clinic. Taken together, while MmpL3 does have limitations as a target, strategies are available to overcome them and MmpL3 remains a viable therapeutic target.
How are Putative MmpL3 Inhibitors Identified and Validated?
MmpL3 remains a common target, owing to its continued identification as the target of a broad number of compounds with varying chemical scaffolds (Fig. 5a–f). However, most MmpL3 inhibitors have been identified through two methods; (i) the isolation and whole genome sequencing of mmpL3 mutants resistant to an inhibitor20–37,164–169,171,172,189,190 and (ii) the observation that treatment of mycobacteria with proposed MmpL3 inhibitors leads to the accumulation of TMM and a decrease in TDM in whole cell lipid extracts.21,22,25,27,29,31–33,35,164,166
While these two methods have served as early indicators for the MOA of these compounds, they are confounded by the additional observations that (i) MmpL3 inhibitors can have multiple MOA and can kill Mtb in nonreplicating states31,152,165 and (ii) disruption of the PMF can lead to similar lipid abundance profiles as cells treated with proposed MmpL3 inhibitors. 152 These two observations previously brought into question the true target of proposed MmpL3 inhibitors.
The isolation and whole genome sequencing of mutants resistant to novel inhibitors can act as an early indicator of the cellular target. Mutants resistant to MmpL3 inhibitors have been isolated in multiple species, including Mtb, M. smegmatis, M. bovis (BCG), and M. abscessus (Fig. 3).20–37,164–169,171,172,189,190 The FoR to MmpL3 inhibitors generally ranges from 10−7 to 10−9 (Fig. 4),20–37,164–169,171,172,189,190 but can vary between species. 30 Further, mutations to MmpL3 inhibitors primarily occur in regions encoding TMD surrounding the central vestibule (Fig. 3) 121 where inhibitors bind to MmpL3.121,122

Amino acid substitution localization for MmpL3 inhibitor-resistant mutants. A matrix that demonstrates the amino acid position and substitution for nonsynonymous mutations found in MmpL3 inhibitor-resistant mutants. The matrix includes inhibitors for which resistant mutants have been identified in four species, including Mycobacterium tuberculosis (gray), Mycobacterium smegmatis (orange), Mycobacterium bovis BCG (purple), and Mycobacterium abscessus (green). MmpL3 protein sequences were aligned and indicate orthologous positions between species. *Substitutions in position 581 of the aligned protein for PIPD-1 were discovered in either the M. tuberculosis (P) or M. abscessus (Δ), A† indicates substitution in M. tuberculosis background at the 299 position of the aligned sequence (M. tuberculosis—V285). aIndicates secondary substitutions made in the M. tuberculosis F255L background isolated from IDR-0033216, b,cIndicate tertiary substitutions sequenced from AU1235 resistant mutants in M. tuberculosis F255L/L567P and F255L/V646M backgrounds, respectively. BCG, Bacillus Calmette-Guérin; L, loop; TM, transmembrane.

While the isolation of mmpL3 mutants against proposed MmpL3 inhibitors has a good track record as an early indicator that a compound targets MmpL3, it is neither proof that MmpL3 is either the target nor the only target of an inhibitor. For example, resistant mutants isolated against THPP-based MmpL3 inhibitors by both Ioerger as well as Remuinan and their respective colleagues indicated that THPP inhibits MmpL3.24,35 However, a protein pull-down study conducted by Cox et al using chiral enantiomers of THPP, GSK729 (active), and GSK730 (inactive) identified EchA6, but not MmpL3, as a strong binder of THPP (GSK729). 191
Targeted mutagenesis of echA6 conferred resistance to THPP both in vitro and in vivo (murine), indicating that EchA6 was, at least, an additional target of THPP. 191 These observations led Cox et al to hypothesize that MmpL3 acted as a drug importer, and that mutations in mmpL3 blocked this function. 191 However, mmpL3 mutants resistant to AU1235 and BM212 demonstrated no difference in cellular accumulation of these inhibitors, indicating MmpL3 is not an importer for these inhibitors.29,30 Later studies would also validate MmpL3 as a target of THPP based on protein–inhibitor interaction studies, including biolayer interferometry (BLI) and SPR. 118 In addition to THPP, SQ109 also has multiple proposed targets, including MmpL3, 34 PMF uncoupling, 152 and menaquinone biosynthesis by targeting MenA. 192 As an additional limitation to this primary method, no Mtb resistant mutant has been identified against SQ109 either in vitro or reported from clinical trials. FoR studies in Helicobacter pylori have indicated that the FoR for SQ109 is 10−9 to 10−11 (Fig. 4), which is likely due to the multitarget nature of SQ109. 193 Due to this limitation, resistance studies for SQ109 have utilized mmpL3 mutants isolated against other inhibitors that are cross-resistant to SQ109. 34 However, taken together, observations for THPP and SQ109 demonstrate how isolation of mmpL3 mutants is not enough to validate MmpL3 as a target, nor does it rule out the possibility of additional MOAs.
The most common method used to validate MmpL3 as the target following mmpL3 mutant sequencing is to perform lipid profiling of mycobacteria treated with an MmpL3 inhibitor. This method relies on the comparison of relative lipid abundance of TMM and TDM in MmpL3 inhibitor treated versus untreated control cells. Following MmpL3 disruption, mycobacteria accumulate TMM as it can no longer be transported across the CM.29,117 This leads to a decrease in TDM due to a lack of TMM substrate in the MM. These lipid profiles are consistent between mycobacteria treated with MmpL3 inhibitors 152 and inducible MmpL3 depletion strains.89,118,137
However, MmpL3 is powered by proton translocation and PMF uncoupling can lead to MmpL3 perturbation. Studies by Li et al generated similar lipid profiles from M. smegmatis cells treated with MmpL3 inhibitors or PMF uncouplers such as CCCP and nigericin. 152 Further, studies conducted by multiple groups have indicated that MmpL3 inhibitors such as SQ109, BM212, C215, TBL-140, E11, HC2032, HC2134, HC2138, HC2149, HC2178, HC2184, and SIMBL-2 can disrupt one or more components of the PMF.25,31,33,117,152,165 These observations brought into question whether proposed inhibitors inhibit MmpL3 through direct protein binding or indirectly through PMF uncoupling.
More recently, several protein binding assays have indicated that MmpL3 inhibitors bind to MmpL3 directly,118,121,122 and that PMF uncoupling is likely a secondary effect either independent of MmpL3 inhibition or as a result of conformational changes following binding (discussed further below). 122 Taken together, while the isolation of mmpL3 mutants coupled with lipid profiling can act as an early indicator that a compound is an MmpL3 inhibitor, additional studies are required to validate MmpL3 as the primary target. Some methods that have been used to test for target specificity (discussed further below) include transcriptional profiling,22,138,156 sensitivity testing in mmpL3 knockdown strains, 36 fluorophore displacement, 118 and metabolic profiling. 23
Due to the limitations of both genomic sequencing and lipid profiling, efforts were made to create assays that more directly measured protein–inhibitor interactions. The aforementioned spheroplast assay was one such assay, 117 which shows that MmpL3 inhibitors prevent (ac)TMM flipping across the CM. However, this assay is not without its limitations. The generation of spheroplasts can be difficult as mycobacteria lacking their cell wall are vulnerable to spontaneous lysis, as mentioned by the developers of the assay. 117 In addition, the confounding finding that SQ109 did not inhibit TMM flipping is still unclear, as more direct biochemical assays have demonstrated that SQ109 does interact with MmpL3 in a manner similar to the other inhibitors (discussed shortly).118,121
A separate assay developed by Li et al uses a competitive binding of MmpL3 with fluorescently labeled analogs of known MmpL3 inhibitors. 118 Briefly, fluorescently tagged MmpL3, either in cells or as purified protein, is incubated in the presence of fluorescently labeled MmpL3 inhibitors called North probes; following incubation, MmpL3 is challenged with unlabeled MmpL3 inhibitors, which competitively bind and displace the North probes. Measuring the loss of probe fluorescence following inhibitor exposure allows for the determination of MmpL3-inhibitor interaction. This method has several advantages to the spheroplast assay. First, it is amenable to both biochemical and live cell assays. 118 Second, the competitive binding assay is insensitive to PMF uncoupling, whereas the spheroplast assay is not.117,118 However, this binding assay is not without limits and could be susceptible to false positives through compounds that dislodge the North probes through nonspecific interactions with MmpL3 rather than competitive binding.
Finally, progress has been made in purifying MmpL3 form both Mtb and M. smegmatis. This has allowed for more direct protein inhibitor interactions to be generated by SPR and BLI, which allow kinetic measurements to be made. 118 However, this assay has been limited by solubility issues for some proposed MmpL3 inhibitors such as BM212, which could not used in either SPR or BLI. 118 Nonkinetic structural assays have also been conducted through X-ray crystallography. These studies have shown that several proposed MmpL3 inhibitors, including SQ109, AU1235, Rimonabant, ICA38, Spiro, and NITD-349, directly bind to MmpL3 in a similar manner, despite their broad differences in structure.121,122
Taken together, researchers now have the tools to test directly whether or not a proposed MmpL3 inhibitor directly binds to and inhibits MmpL3 both biochemically and in live bacteria. However, these assays do not preclude the possibility of secondary and off-target effects. We have compiled the summary of proposed MmpL3 inhibitors identified to date, not including the expansive analogs generated by SAR studies, and what assays have provided evidence that each of these compounds targets MmpL3 (Supplementary Table S1).
Impacts of MmpL3 Disruption
Because MmpL3 is involved in MM synthesis, it would be expected that perturbation of MmpL3 would put mycobacteria into a state of cell wall stress. Mycobacterial reporter strains for cell wall stress were generated through -gfp and -lacZ fusions to iniB, which, along with downstream genes iniAC, are highly upregulated following INH and EMB treatment.194,195 These reporter systems are largely insensitive to non-cell wall inhibitors194,195 and were used in screens that identified the MmpL3 inhibitors DA-5, DA-8, and E11.31,34 These observations are consistent with transcriptional profiles of Mtb treated with SQ109, HC2091, and following mmpL3 knockdown, which resulted in increased iniBAC expression.22,138,156 These findings support a cell wall stress model for MmpL3 inhibitor treatment even in the case of E11, which has the added effect of PMF disruption. 31
As described earlier, the inhibition of MmpL3 leads to a decrease in TMM transport resulting in TMM accumulation in the inner leaflet of the CM. This was most clearly demonstrated by Xu et al using M. smegmatis spheroplasts and dual TMM fluorescent metabolic probes and TMM degradation assays. 117 They demonstrated that treatment of M. smegmatis spheroplasts with MmpL3 inhibitors BM212 and AU1235 resulted in decreased TMM flipping to the outer leaflet of the CM.
Consistent with alterations in the makeup of the cell wall, it was reported that MmpL3 inhibition leads to increased cell hydrophobicity in M. smegmatis treated with AU1235189 and cell permeability following mmpL3 silencing in Mtb. 36 Drug combination studies have demonstrated that MmpL3 disruption leads to RIF hypersusceptibility25,36,103,196; additionally, MmpL3 perturbation leads to increased susceptibility to PG synthesis inhibitors.36,196 Scanning electron and transmission electron micrographs have also shown alterations to cell morphology following MmpL3 inhibition34,153 which is consistent with cell wall alterations. RIF susceptibility is tied to cell permeability 197 and the observation that MmpL3 inhibition leads to PG dysfunction suggests association between MA biosynthesis and PG synthesis as discussed above.
Treatment of bacteria can lead to whole cell changes and alter transcriptional and metabolic profiles. Early studies into the MOA of SQ109 included a large transcriptional profiling study by Boshoff et al, who conducted over 400 microarray experiments of Mtb cultured and treated under different conditions. 156 The resulting profiles placed SQ109 (then diamine 109) with the AG biosynthesis inhibitor EMB. However, the gene expression profiles of SQ109 included the downregulation of genes involved in MA biosynthesis, including fas, fadA2, pks16, and the fabD-acpM-kasA-kasB-accD6 operon (Fig. 1, red highlight). While these genes were downregulated in the SQ109 profiles, they were upregulated in the EMB and INH profiles, suggesting SQ109 had a MOA unique from other cell wall inhibtiros. 156 Later, similar patterns for downregulated genes were observed in our laboratory from RNAseq profiles of Mtb treated with SQ109 or HC2091. 22
Similarly, the RNAseq profile from an Mtb mmpL3 knockdown strain also identified the downregulation of MA biosynthesis genes. 138 The profiles generated in these three studies were highly similar and generated in the presence of SQ109, which decreases the Δψ, 152 HC2091, which does not affect the Δψ, 22 and following mmpL3 depletion, which, presumably, leads to an increased Δψ, as observed by Li et al. 118 This suggests that the transcriptional responses identified were highly specific to MmpL3 disruption and independent of secondary membrane energetic effects.
The consistent differential profiles generated by MmpL3 disruption not shared by other cell wall inhibitors suggest that mycobacteria can sense when MmpL3 specifically is inhibited. As a consequence of these expression changes, the associated metabolites should also decrease in abundance. Consistent with this model, metabolomic profiles generated by Zampieri et al of M. smegmatis treated with GSK2623870A identified a decrease in trehalose 6-phosphate, 23 the activated form of trehalose that serves as a substrate to make TMM. 74 In addition, Zampieri et al identified that FAS proteins, such as Fas, were primarily affected in a proteomic analysis. 23
Of the three profiles generated to date, the mmpL3 knockdown profile was the most robust, with several regulatory pathways identified as differentially expressed, in addition to MA biosynthesis. 138 The regulatory pathways may be involved in the sensing of changes to the cell envelope following MmpL3 perturbation. However, it is also possible that this profile includes genes responding to the increase in the Δψ following mmpL3 knockdown. 118 Based on the transcriptional profiles, we know that Mtb represses genes involved in both the FAS-I and FAS-II pathways. Mtb also upregulates expression of the cell wall stress operon iniBAC in response to MmpL3 disruption.22,138,156
How Mtb specifically senses MmpL3 disruption, and differentially regulates its genes from other cell wall inhibitors such as EMB and INH, is not clear. One model may suggest that Mtb senses changes in CM fluidity following TMM accumulation or TMM accumulation in the inner leafelt. 117 Such changes would not occur in INH-treated cells, as FAS-II disruption from INH would not directly affect CM fluidity as CM lipids are generated through FAS-I. 198 If so, then this may be through either the alternative sigma factor, SigE (Rv1221), or the two-component system regulator MprAB (Rv0981 and Rv0982), which responds to cell wall stress199,200 and whose regulons were upregulated in the mmpL3 knockdown profile. 138
Of note, the transcriptional effects following MmpL3 disruption are primarily repressive, and few genes are observed to be upregulated in response to MmpL3 inhibition.22,138,156 One notable change was the induction of osmotic stress genes, oprA (Rv0516c) in the mmpL3 knockdown profile, 138 suggesting that the bacteria are experiencing osmotic stress. Other genes upregulated in these profiles include SigE and MprAB regulated genes in the mmpL3 knockdown profile, and the iniBAC cell wall stress signature genes in all three profiles.22,138,156 However, these transcriptional signatures are not specific to MmpL3 disruption and are upregulated in Mtb in other stresses. 156
Several reporter strains were built around genes that are upregulated following specific stresses.194,195 However, limited genes that are upregulated in response to MmpL3 disruption in Mtb does not lend itself for the use of building a reporter strain. While the gene repression signature following MmpL3 disruption is unique compared to FAS-II or AG synthesis inhibition, 156 disentangling this gene repression signature from cell death or transcriptional repression from RNA polymerase and DNA gyrase inhibitors is difficult.
Whatever differences may exist in the exact nature of each inhibitor's MOA, it remains clear that MmpL3 inhibition leads to the accumulation of TMM and a decrease in TDM. As discussed above, TMM covalently anchors the MM to the rest of the cell wall forming mAGP, and TDM, the metabolic product of Ag85 and TMM, acts as a major penetration barrier in mycobacteria. 97 Mycobacteria treated with MmpL3 inhibitors have increased cell permeability similar to strains with lower MmpL3 function leading to increased antibiotic efficacy.36,197
MmpL3 Protein–Inhibitor Interactions
Despite the limitations of the two primary methods used to identify MmpL3 inhibitors listed in the previous section, a competitive binding assay developed by Li et al has demonstrated direct interaction for many MmpL3 inhibitors with MmpL3 (discussed in a previous section). 118 Leveraging the displacement property, these North probes can be used to determine direct MmpL3 interaction in live cell mycobacteria through competitive binding using flow cytometry by measuring the relative fluorescent intensity. This system has already been used in three separate studies to demonstrate direct interaction of MmpL3 with inhibitors SQ109, NITD-304, NITD-349, BM212, AU1235, THPP1, HC2032, HC2060, HC2091, HC2099, HC2134, HC2138, HC2149, HC2169, HC2178, HC2184, C215, SIMBL-1, and SIMBL-2.25,33,118 Further, the results of SQ109, NITD-304, NITD-349, AU1235, and THPP1 have were backed by protein binding data, including BLI and SPR. 118
However, while this competitive binding assay does allow for the simple measurement of direct interaction between MmpL3 and an inhibitor in live cells, this competitive binding assay does not rule out the possibility of additional effects. Inhibitors like SQ109, BM212, C215, TBL-140, and E11, and many of the listed HC2 compounds have the additional property of PMF disruption.25,31,152,165 Compounds like BM212 and SQ109 were both demonstrated bactericidal properties against Mtb in nonreplicating states 152 when MmpL3 is dispensable for viability. 137 In addition, evidence does support secondary protein targets for both THPP 191 and SQ109. 192
While the specific chemical structure varies between proposed MmpL3 inhibitors, the compounds can be broadly classified into seven primary categories based on shared core structures (Fig. 5a–g). The seven classes consist of diamine/acetamides, ureas/guanidines, pyrole/pyrazoles, benz-amides/indoles/imidazole/thiazoles, amides, amines, and a seventh class of scaffolds that do not share a common core structure (Fig. 5a–g).

MmpL3 inhibitors share distinguishing and overlapping features. MmpL3 inhibitors fall into seven distinct classes of inhibitors based on shared Central core chemical groups, including diamines/acetamides
For simplicity of description, we adopt nomenclature from Guardia et al 170 and described the shared core structures as the Central chemical groups (Fig. 5a–e, red). The Central cores are typically composed of nucleophilic/basic residues, and even in the seventh chemical class, which lack a shared core structure, the inhibitors share a Central core with similar chemical properties (Fig. 5g). Again, adopting nomenclature from Guardia et al, the Central cores are flanked by lipophilic/hydrophobic Northern (Fig. 5a–g Green) and Southern (Fig. 5a–g Blue) groups. The Northern group is typically in the form of a substituted aryl group, such as benzene, while the Southern group typically consists of cyclic alkyl groups.
While the shared chemical cores exist for nearly all MmpL3 inhibitors, additional groups have been identified for BM212 and Rimonabant, each of which has additional substituted benzene rings classified here as North-Western groups (Fig. 5c, Yellow). A review of the modeled and co-crystal structures indicates that the chemical domains align to specific MmpL3 binding subdomains (SD-1 to SD-5) identified by Zhang et al (Fig. 2b). 121 The Northern groups are typically found in the SD-3 toward the periplasmic side of the Asp-Tyr residues. 121 Exceptions to this are noted for BM212 and Rimonabant, where the Northern groups bind to SD-2 and the North-Western groups bind to SD-1. 121 The nucleophilic/basic Central groups interact with the essential Tyr-Asp groups in SD-4 (Fig. 2c).
The Central groups typically bind to the Tyr residues through H-bonding or noncovalent interactions. 121 This binding disrupts Asp-Tyr pairing, preventing H+ translocation that powers MmpL3 function. 121 The Southern group of MmpL3 inhibitors is typically located in SD-5 and acts as stabilizers through hydrophobic interactions. 121 Based on these binding patterns, while specific protein–inhibitor interactions exist, as exemplified through different inhibitory concentrations for structurally similar series identified in SAR studies,22,31,37,165–168,170,201,202 MmpL3-inhibitor interactions are characterized by a limited number of general protein inhibitor binding motifs.
mmpL3 Drug-Resistant Mutations May Affect MmpL3 Structure Function and Mycobacterial Growth
Forward genetic screening for mmpL3 mutants resistant to MmpL3 inhibitors has been one of the primary methods used to identify MmpL3 inhibitors. Mutations in mmpL3 are primarily located in codons encoding residues located in the central vestibule.20–37,164–169,171,172,189,190 Some researchers have noted that these mutant strains have in vitro growth defects.25,118,189 Protein gels of trypsin-digested wild-type (WT) and mutant MmpL3 demonstrated altered folding motifs in MmpL3118 and modeled substitutions in the MmpL3 protein structure suggests that substitutions lead to changes in the protein folding around the central vestibule.121,128 A recent study by McNeil et al demonstrated that mmpL3 mutants isolated against MmpL3 inhibitors have altered membrane potential (Δψ). 189 The altered Δψ had global effects on M. smegmatis cells, including lowered efflux, increased cell permeability, RIF hypersusceptibility, and altered TMM/TDM profiles similar to MmpL3 inhibition.
These altered physiologies suggested that mutations in mmpL3 lowered MmpL3 function resulting in lower growth rates. 189 Additional studies by McNeil et al demonstrated that passaging these mutants in the presence or absence of an MmpL3 inhibitor led to the selection of compensatory mutations in the form of secondary, as well as tertiary, mutations in mmpL3.189,190 These additional substitutions often arose in the central vestibule in TMD adjacent to the primary substitution. 189 The passaged mutants demonstrated a reversion to WT levels of growth, suggesting a return to normal protein function, while maintaining inhibitor resistance.189,190 While this allowed for insights into the biology and structure-function relationship of MmpL3, it also supports the potential selection of mmpL3 mutants resistant to MmpL3 inhibitors in the clinic.
Passaging the mutants in the presence of everincreasing MmpL3 inhibitor concentrations only led to additional mutations and increased resistance without a growth defect. 190 The isolated tertiary mmpL3 mutants were also cross-resistant between IDR-0033216, IDR-0334448, AU1235, and SQ109. 190 Based on this observation, it is possible that swapping between MmpL3 inhibitors in the clinic may select for additional mutations and higher resistance. The compensatory mutants also did not have increased susceptibility to RIF like primary mmpL3 mutants. 190 How these findings translate to the clinic will only be answered with time, but underscore the need for vigilant antibiotic resistance monitoring.
Screening for MmpL3 Inhibitors
MmpL3 inhibitors continue to be the most diverse class of inhibitors that share a single target for Mtb. Because of the slow growth of Mtb and M. bovis, untargeted screening approaches for MmpL3 inhibitors through the isolation and sequencing of resistant mutants are largely inefficient and time-consuming. An alternative approach of using the rapidly growing mycobacteria M. smegmatis or M. abscessus is limited by observations that not all MmpL3 inhibitors are active in these two species.25,180
Because MmpL3 is such a common target with high therapeutic potential, screening platforms to identify these inhibitors are highly sought. While reporter strains such as the iniB reporter system have identified some MmpL3 inhibitors,31,34 the low specificity for MmpL3 limits the use of this reporter system to its original intended purpose of identifying cell wall inhibitors. In addition, while reporter systems have been built around genes highly expressed following target inhibition, MmpL3 inhibition largely leads to the downregulation of signature genes 156 limiting the ability to develop a reporter around transcriptional signatures following MmpL3 disruption.
Alternatively, whole cell morphological changes were recently described to differentiate the inhibitors by their specific MOA using a system called Morphological Evaluation and Understanding of Stress (MorphEUS). 203 While this system is unique in its ability to identify both primary and secondary MOAs, it is limited by specificity, and places inhibitors in broad stressor categories like cell wall stress. Recently, however, four potential screening methods have been proposed as screening platforms for MmpL3 inhibitors. These include (i) a high-throughput metabolic screening platform, 23 (ii) a whole cell targeted mutant screen that uses mmpL3 mutants, 25 (iii) a two-way mmpL3 regulatory phenotypic reporter system, 36 and (iv) a competitive binding assay that uses MmpL3 binding fluorophores. 118
The MOA of inhibitors can lead to differential responses in bacteria, which can be measured in a number of ways. One of these measurable responses are the metabolic changes bacteria undergo following the inhibiton of specific metbaolic pathways. Using a high-throughput metabolic analysis platform, Zampieri et al identified several MmpL3 inhibitors through differential metabolic profiles. 23 They observed that 6 inhibitors from a library of 212 compounds led to repeated decreases in the metabolite α,α-trehalose-6-phosphate and neighboring lipids, precursors to TMM.
Two of the inhibitors, GSK1829729A and GSK1829728A, were reported to be chemically similar to THPP and isolation of resistant mutants to a third compound, GSK2623870A, identified mmpL3 mutants. While not specifically designed to identify MmpL3 inhibitors, this system was fast and accurate at identifying the MOA of mycobacterial growth inhibitors. This system is not specifically limited to mycobacteria and could feasibly be applied to other bacterial and pathogenic species. Although this system can be applied to larger libraries, the technological and computational requirements to perform this assay are still somewhat limiting and require specific expertise.
The second MmpL3 inhibitor screening platform was developed in our laboratory and was built on the observation that mmpL3 mutants have broad cross-resistance to MmpL3 inhibitors,22,25,28,32,34,36,118,165,168,190 but low cross-resistance to non-MmpL3 inhibitors.25,27,28,32,34,35,37,118,164,168,189 Another observation previously made was that the amount of cross-resistance depended on the specific combination of mmpL3 mutant and MmpL3 inhibitor tested. 118 To overcome potential limitations, we generated a pool of 24 unique mmpL3 mutant isolates against 5 structurally unique MmpL3 inhibitors.22,25
We hypothesized that MmpL3 inhibitors would select for the mutant(s) with the highest resistance, while non-MmpL3 inhibitors would equally affect all the mutants. The resulting Targeted Mutant Screening assay compared the effect inhibitors have on the growth (OD600) of WT versus the mmpL3 mutant pool, in which the mutants would be less effected by an MmpL3 inhibitor. Using this mutant assay, we screened a small library of 174 Mtb growth inhibitors, including previously described MmpL3 inhibitors SQ109, C215, and HC2091.20,22,25,34 The results of our assay identified controls SQ109 and C215, as well as HC2060, HC2091, HC149, HC2169, and HC2184, which were used to generate the mmpL3 mutants used in the assay. 25 In addition to these seven compounds, we also identified six other inhibitors HC2032, HC2099, HC2134, HC2138, HC2178, and HC2183. 25
Overall, the screen was highly successful and confirmed hits induced lipid profiles consistent with MmpL3 inhibition and were positive for the competitive binding assay described above.25,118 However, this system has limitations. For one, we did not observe differences in the inhibitory effects for Rimonabant between the WT and mmpL3 mutant pool. 25 Rimonabant is highly similar to BM212, which demonstrates cross-resistance to a very small number of mmpL3 mutants, 118 which were not included in our pool. Future versions of this screen may seek to include mmpL3 mutants isolated against BM212 or Rimonabant to broaden the screening potential of this system.
The third screening platform uses strains that differentially express mmpL3. An early version of this screen was originally proposed by Li et al, who observed inducible knockdown of mmpL3 to subinhibitory levels rendered Mtb sensitive to MmpL3 inhibitors. 137 While promising, this system was limited by synthetically lethal combinations with other TB drugs such as RIF. 137 Alternatively, Zhang as well as Kozikowski, and their respective colleagues, demonstrated that mmpL3 overexpression mycobacterial strains, expressing either WT or resistant mutant mmpL3 alleles, conferred high resistance to MmpL3 inhibitors.121,166 The overexpression strain created by Zhang et al did not confer cross-resistance to INH, 121 but further investigation into the screening potential of overexpression strains like these was not conducted.
However, recently, Grover et al demonstrated that a combined dual regulatory strain of Mtb that either repressed or overexpressed mmpL3 was highly accurate at identifying MmpL3 inhibitors from a library of 220 Mtb growth inhibitors. 36 The authors of this study demonstrated that mmpL3 knockdown sensitized bacteria to MmpL3 inhibitors as previously described. However, this knockdown also made Mtb hypersensitive to non-MmpL3 inhibitors such as RIF, clarithromycin, erythromycin, fidaxomicin, and fusidic acid, as well as β-lactams. 36 Conversely, overexpression of mmpL3 resulted in decreased activity of MmpL3 inhibitors, but did not lead to differential inhibitory effects for non-MmpL3 inhibitors. 36 The recognition of a bidirectional shift that only occurred for MmpL3 inhibitors indicated that this system could be used to screen MmpL3 inhibitors, which was applied to a library of 220 Mtb growth inhibitors. The results of this screen identified several previously described MmpL3 inhibitor scaffolds, including THPP-, Spiro-, Urea-, Pyrole-, PIPD-, and oxadiazole-like compounds, as well as a novel guanidine-based MmpL3 inhibitor CCI7967. 36 The identification of CCI7967 was backed by the isolation of mmpL3 mutants resistant to CCI7967. This screening platform overcomes the limitations of the Targeted Mutant Screening platform by identifying Pyrole-based MmpL3 inhibitors such as BM212. 25
However, this Two-Way Regulation screen, along with the Targeted Mutant Screen, shares a common limitation of requiring compounds to be tested against both the WT reference strain as well as the experimental mmpL3 strain(s). While these screens are not burdensome for larger pharmaceutical companies or smaller compound libraries for academic institutes, such as the ones conducted in either screen, the doubling of resources required to run these screens could become costly for larger libraries. This limitation renders both screening platforms to be used as mechanisms to identify MmpL3 inhibitors from hits from larger HTS. This limitation could be overcome in the fourth potential screening platform described next.
Finally, the fourth potential screening platform proposed uses the competitive binding assay mentioned above, using the North series of MmpL3 binding probes. 118 This competitive binding assay utilizes MmpL3 inhibitor scaffolds such as ICAs, AU125, or the SIMBL covalently linked to the commercially available fluorophore TAMRA through click chemistry. 118 These MmpL3-fluorophores co-localize with MmpL3 in dividing M. smegmatis cells and are demonstrated to interact with MmpL3 based on SPR and BLI. 118 Using flowcytometry, researchers can measure the relative fluorescence intensity of cells treated with the North probes, which decreases in the presence of a competing MmpL3 inhibitor. This competitive binding assay is insensitive to non-MmpL3 inhibitors such as RIF and INH, as well as PMF uncouplers such as CCCP. 118
This screen works in both whole cell mycobacteria, as well as isolated MmpL3 protein, adding diversity not available to the previously described Targeted Mutant and Two-Way Regulation screens described above. In addition, both the whole cell and biochemical assays can measure MmpL3 binding within hours, overcoming the slower times required for the other two screen platforms, which take place over days due to the slow growth of mycobacteria.25,36 This screen also has the potential to be used to conduct SAR campaigns directly against MmpL3 rather than whole cell bacteria. As mentioned above, SAR campaigns can result in either gain or loss in activity through the modification of parental structures. However, it has never been clear if changes in activity following structural alteration of parental compounds was due to changes in cell permeability or protein binding affinity. Utilizing in vitro and whole cell aspects of the competitive binding assay, it may be possible to delineate permeability from changes in binding affinity.
While this system has yet to be tested in a screen, the rapid nature of this assay coupled with the direct measurement of MmpL3 binding without the requirement for comparative strains suggests that this assay will likely make an efficient platform to screen MmpL3 inhibitors in a large library. However, this screen is limited and could identify noninhibitory MmpL3-interacting substrates that could also lead to probe displacement. Furthermore, biochemical screens have previously identified metabolic inhibitors devoid of whole cell activity due to factors such as cell impermeability. 204 Therefore, this screening platform will still require secondary assays to demonstrate whole cell activity.
It is clear that MmpL3 inhibitors will continue to be identified through the traditional untargeted approaches of isolating mmpL3 mutants, but with the invention of the novel screening systems listed in this study, the rate of identification will likely increase dramatically. While SQ109 has demonstrated great drug potential so far, if SQ109 were to fail in clinical trials, then it is likely that another MmpL3 inhibitor from the ones identified in the last decade could take its place.
Concluding Remarks
MmpL3 continues to be an attractive target for TB therapy. The essential nature of the protein both in vitro and in vivo and the clinical success of SQ109 so far support further development of MmpL3 inhibitors. Protein localization and interactome studies have demonstrated that MmpL3 complexes with other cell wall synthesis metabolic pathways as part of the mycobacterial divisome. While some questions still remain, including how MmpL3 complexes with TMM in the CM, live cell microscopy and biochemical evidence demonstrate that MmpL3 plays a direct role in TMM transport across the CM.
In addition, biochemical studies have clearly demonstrated that MmpL3 directly binds to many proposed inhibitors described in the literature. These direct protein–inhibitor interactions support the isolation of resistant mmpL3 mutants and lipid profiling as good early indicators of the MOA of identified inhibitors. Furthermore, the similar chemical properties and protein localization of these inhibitors indicate a general threshold of what makes an MmpL3 inhibitor and how they bind to MmpL3. While additional questions still remain for MmpL3 regarding regulation, protein-protein interaction, and function, the insights gained in the last decade have advanced our understanding of the role MmpL3 plays in cell biology of mycobacteria.
Footnotes
Acknowledgments
Authors' Contributions
J.T.W. conceptualized, wrote, and edited the article, and prepared the figures. R.B.A. wrote and edited the article.
Disclosure Statement
R.B.A. is the founder and owner of Tarn Biosciences, Inc., a company that is working to develop new antimycobacterial drugs.
Funding Information
This research was supported by a grant from the NIH-NIAID (R21 AI148909) and AgBioResearch.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.