
Abstract
Global dissemination of mobilized colistin resistance (mcr)-1-carrying plasmids has been reported. This study aimed to investigate the global dissemination of these plasmids using whole genome sequencing to provide better understanding on genetic characteristics. Sixty-seven complete plasmid genomes harboring mcr-1 were obtained. Phylogeny was built against full plasmid genomes. Different replicon types of plasmid were compared in terms of antimicrobial resistance genes (ARGs), insertion sequence, and other functional genes. Five different replicon types of plasmid (IncX4, IncI2, IncP1, IncHIA, and IncFIB) were found to harbor mcr-1. IncX4 and IncI2 types of plasmid were well clustered in accordance with the country where they were isolated (and not as IncHIA and IncFIB). Three insertion sequences (ISApl1, ISKpn26, and IS1294) were identified in up- and/or downstream of mcr-1. Plasmids IncX4 and IncI2 were observed across the sample origin. Plasmids IncX4 showed high uniformity regardless of the origin of isolates and harbored H–NS coding genes, a facilitator for successful plasmid transfer. All three insertion sequences were observed in IncI2 plasmids. IncHI2 plasmids harbored various ARGs in addition to mcr-1. Our results elucidate the characteristics and phylogenetic relationships of complete mcr-1-harboring plasmids, indicating that global dissemination of mcr-1 is primarily owing to plasmid transfer rather than clonal spread.
Introduction
Plasmid-borne colistin resistance is the most complex public health concern. 1 Since the first report of the mobilized colistin resistance (mcr)-1 gene from China in 2015, several studies have reported nine new variants (mcr-2, -3, -4, -5, -6, -7, -8, -9, and -10) from the Enterobacteriaceae family, such as Escherichia coli, Salmonella, and Klebsiella pneumoniae.1–10 Among them, the original variant, mcr-1, was identified in the most diverse species, and several studies have reported the global spread and genetic characteristics of mcr-1-harboring plasmid from various sources, including humans, animals, foods, and sewages.11–14
Horizontal gene transfer by plasmid is considered to be the main responsibility for the worldwide spread of antimicrobial resistance genes (ARGs), including mcr-1, and many studies are making efforts to identify the relationship between mcr-1 and a specific plasmid type.15,16
The most common plasmid types carrying ARGs in Enterobacteriaceae are IncF, IncI, IncA/C, IncL, IncN, and IncH. 15 Among them, IncF type has the highest detection frequency and is associated with various ARGs unlike others. In contrast, the mcr-1 gene was mainly located in the IncI2 and IncX4 type plasmids, and was also identified in the IncHI2, p0111, and IncP1 types.17,18 Recently, researchers have focused on generating and analyzing whole genome sequencing (WGS) data using various platforms that produce short or long reads to understand the dynamics of worldwide spread of mcr-1-harboring-plasmids with diverse genetic backgrounds.19,20
However, conventional multilocus sequence typing 13 or short-read WGS 21 does not have an adequate resolution or cannot completely resolve the molecular structure of mcr-1-harboring plasmid genomes.
To address this concern, in this study, we first attempted to build phylogeny against the complete plasmid genome harboring mcr-1 using PopPUNK, k-mer-based phylogenetic tree maker, and evaluate the potential transmission of mcr-1-harboring plasmids in a global context. Moreover, we compared different plasmid backbones containing mcr-1 to provide better understanding on the characteristics of mcr-1-harboring plasmids.
Materials and Methods
Ethics
The two newly sequenced isolates (Escherichia coli strains EC8 and EC9) in this study were collected through the Korea national surveillance system (Kor-GLASS) for antimicrobial resistant bacteria, and available for study without approval from the research ethics committee of the Korea Disease Control and Prevention Agency.
Data collection
We obtained 64 mcr-1 carrier plasmids in plasmid Atlas (pATLAS) 22 (v2018_09_20) and plasmid database (PLSDB) 23 (v. 2019_03_05) by browsing resistance gene “mcr-1.” The ZIP archive containing metadata of all plasmids was downloaded from PLSDB. The corresponding metadata not properly registered in both database was obtained from Biosample and Bioproject. Three isolates collected in this study were newly sequenced.
Whole genome sequencing
WGS of an isolate (strain KV4) from effluent was carried out with the Miseq platform (Macrogen, Seoul, Korea). WGS data for two isolates (strains EC8 and EC9) from Kor-GLASS were obtained with the PacBio platform (Pacific Biosciences, Menlo Park, CA).
Low-quality sequences (below Q30) were trimmed (ngsShorRT), and PlasmidSeeker 24 was used to determine the closest reference plasmids. Trimmed raw sequences were assembled using SPAdes 25 version 3.13.0, and contigs <500 bp were discarded. Scaffolds were mapped to reference plasmids, and consensus sequences were constructed using Geneious 26 version 11.1.5.
Antibiotic resistance genes and plasmid-replicon types were annotated using the ResFinder version 2.1 and PlasmidFinder version 1.6 databases, respectively.27,28 Sequence homology was investigated using the National Center for Biotechnology Information data bank. Integrons, transposons, and prophages were annotated with GenBank, ISFinder, and PHASTER.29,30 Mutations were investigated using a BLAST search (https://blast.ncbi.nlm.nih.gov/Blast.cgi). Polished contigs from the PacBio platform were annotated using these same tools.
Phylogeny building
Phylogeny against 67 mcr-1 carrying plasmids (64 downloaded; 3 newly sequenced; Supplementary Table S1) was built using PopPUNK, based on k-mer differences. The k-mer mash distance among plasmid genomes was calculated with the following options (—min-k 13, —plot-fit 5, —max-a-dist 1.0, —ignore-length) and maximum sketch size (106), followed by model fitting and refining of default settings, yielding a final network score of 0.9448.
Bacterial conjugation
Conjugation assays were performed using azide-resistant Escherichia coli J53 as a recipient. 31 Overnight cultures of donor and recipient bacterium were mixed (10:1, recipient:donor) in tryptic soy broth (Oxoid) and incubated at 37°C for 18 hours with rotating (150 rpm). One hundred microliters of serially diluted enrichments were inoculated onto tryptic soy agar (TSA) containing sodium azide (Oxoid; 100 μg/mL) and colistin (Sigma, St. Louis, MO; 1 μg/mL for EC8, and EC9). Maximum five colonies per plate were subcultured onto TSA (Oxoid), and minimum inhibitory concentration assays and PCR were conducted to confirm the transfer of plasmid from donor to recipient.
Results and Discussions
Clustering whole plasmids is quite challenging because of frequent recombination and insertion/deletion of genes and size discrepancies among plasmids. To date, such a limited number of genes (replicons, relaxosome, etc.) has been selected for plasmid phylogeny construction, of which resolution power might be insufficient to fully understand the epidemiology of plasmid transfer. Some researchers adopted core genomes of plasmid to increase resolution. Nevertheless, they are not largely shared among different replicon types of plasmid, indicating that this strategy also has some limitations. 2 Recently, a previous study reported the MinHash algorithm-based phylogeny builder for whole plasmid genomes, namely pATLAS. 22
It is a web-based tool and provides information on resistance genes and plasmid replicons with high-quality visualization. The problem with this tool is that users must set up local server to analyze their own data because it allows the analysis on their own database. It might be an obstacle for many researchers who are not familiar with Docker environment. Instead, we applied PopPUNK to investigate genetic relationships of whole plasmid genome possessing mcr-1. 32 PopPUNK, a user-friendly phylogenetic tree maker, also includes the MinHash algorithm with an available option for building phylogeny among highly variable plasmid genomes.
The limitations of this study are that the number of whole genomes was not large enough to support our conclusion, owing to a dearth of studies. Furthermore, clonal background of mcr-1 carrier bacteria was not investigated owing to nonavailability of corresponding genomes. Nevertheless, to our knowledge, this is the first study to compare all available plasmid genomes carrying mcr-1 using a long-read sequence platform.
Figure 1 shows the phylogeny against 67 mcr-1-carrying plasmids, including samples from 15 countries, 7 origins, and 5 replicons (Supplementary Table S1). Plasmids were located in accordance with their replicons. IncP1 was clustered alone. IncX4 plasmids showed high genetic uniformity regardless of country, origin, and host bacterial species except nodes of phylogroup 3, suggesting that it was almost identical to IncX4 (>99% nucleotide identity) and is widely available from among humans, animals, and sewage.
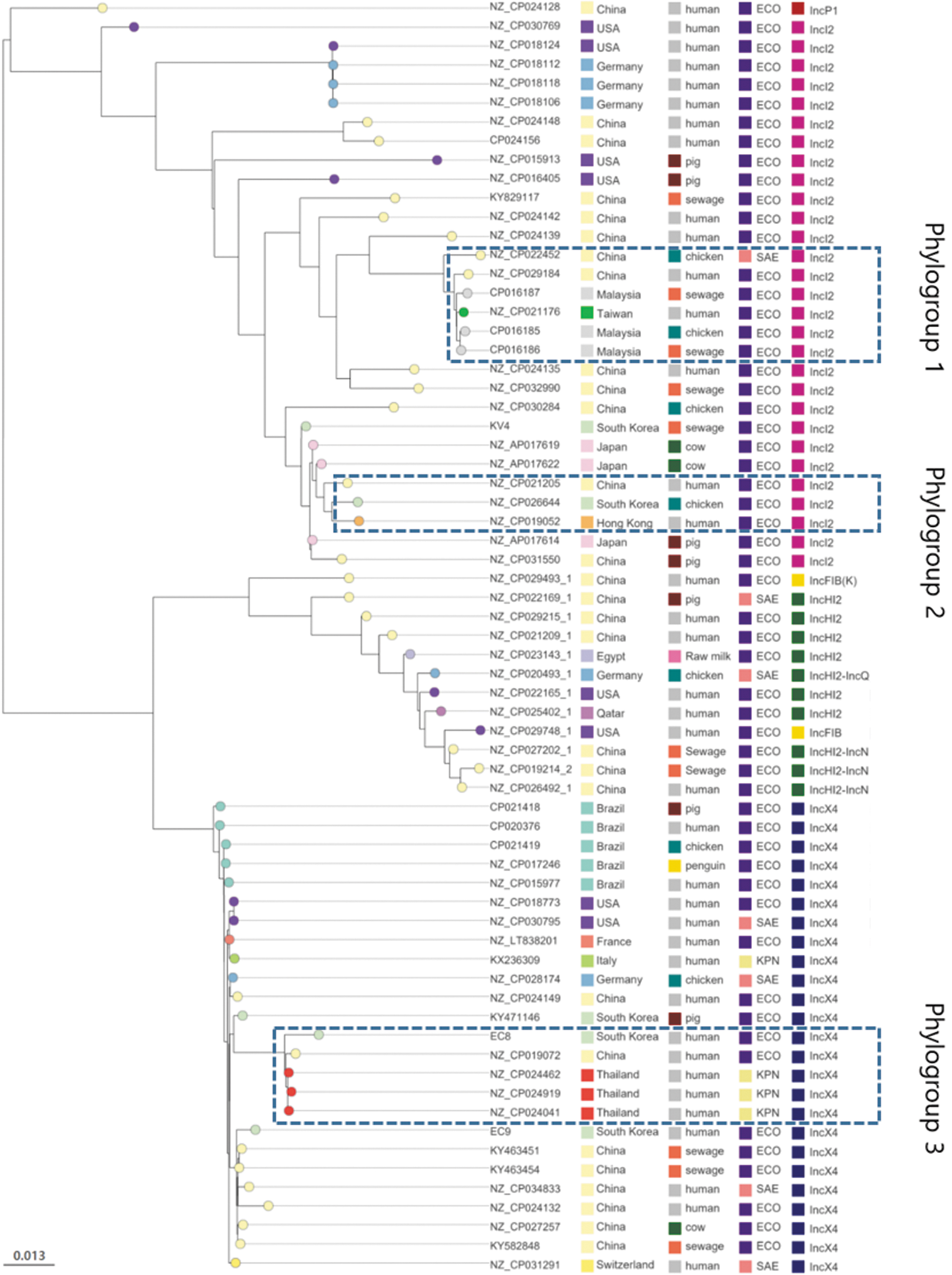
A phylogenetic tree of 67 mcr-1-harboring plasmids of Enterobacteriaceae isolated from humans and animals. ECO, Escherichia coli; KPN, Klebsiella pneumoniae; SAE, Salmonella enterica.
This plasmid is not likely to acquire mobile genetic elements, owing to its low genetic diversity compared with that of other plasmids. IncI2 plasmids displayed the highest diversity and most of the nodes were placed on the basis of their country of origin. Among them, plasmids of phylogroups 1 and 2 displayed 1 scale bar of genetic distance (i.e., >98.7% identity) although they were isolated from different sources and countries, indicating potential circulation among them. Plasmids of IncF and IncHI2 also exhibited high genetic diversity, probably owing to the presence of mobile genetic elements and diverse ARGs.
Plasmid genomes were aligned against reference genomes consisting of ARGs and insertion sequences (Fig. 2). All IncX4 plasmids harbored an hns-coding gene, which is known to facilitate horizontal plasmid transfer.33,34 This might promote the wide propagation of this plasmid. Although not shown as a result, we performed conjugation with two isolates (EC8 and EC9) with mcr-1-harboring-IncX4 plasmid as donor and J53 as recipient, and it was observed that the mcr-1 gene was successfully transferred from donor to recipient.
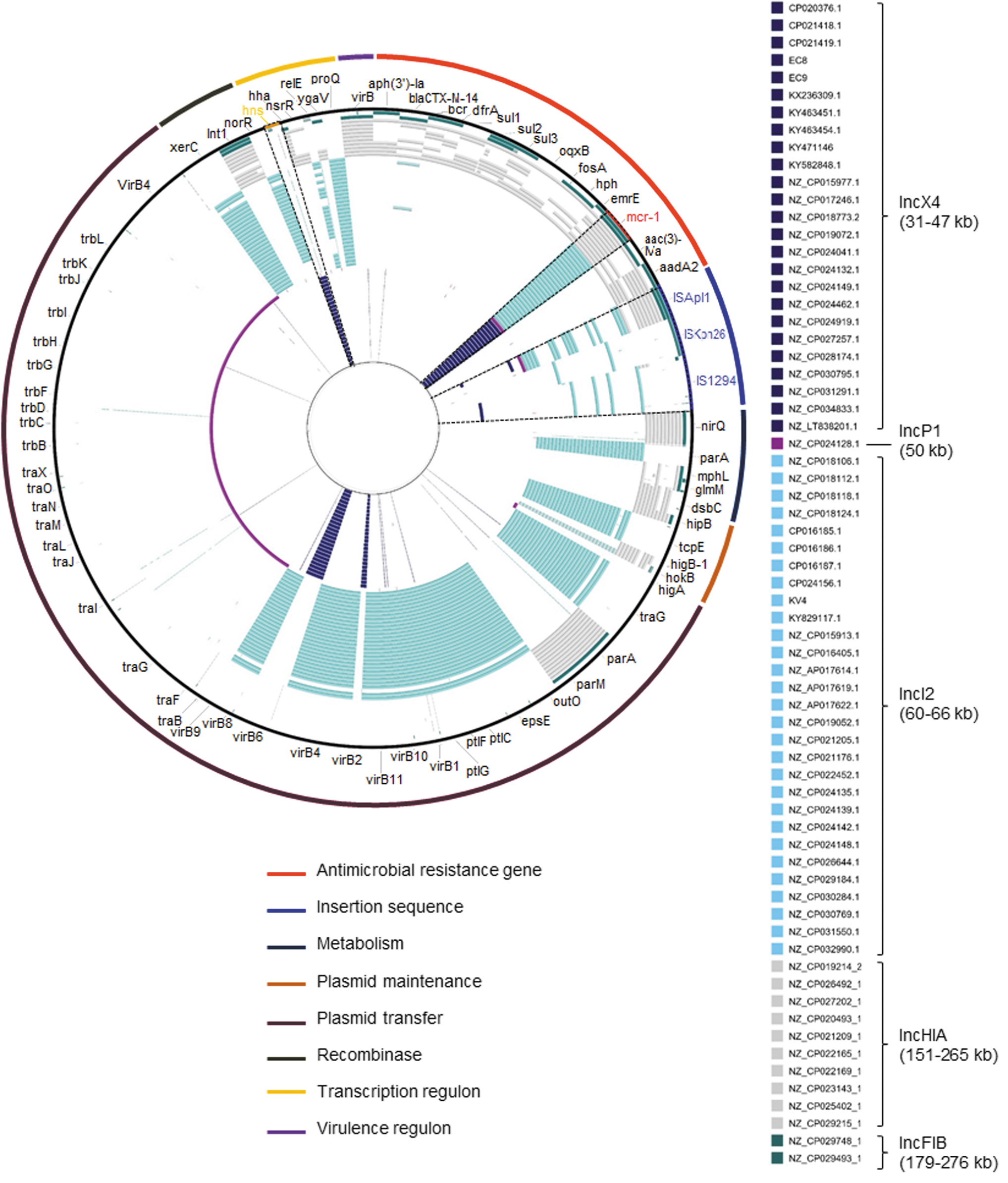
Sequence alignment of 67 mcr-1-harboring plasmids of Enterobacteriaceae strain.
Moreover, most of them (22 of 25; 88.0%) did not have up- and/or downstream insertion sequence. This indicates mcr-1 is stably located onto IncX4 without cut-and-paste transposition2/4 whereas IncI2 plasmids harbored diverse insertion sequences, including ISApl1, ISKpn26, and IS1294, thus imparting high plasticity to them. IncF and IncH plasmids contained various ARGs and mcr-1, responsible for resistance to third cephalosporins, sulfonamide, quinone, and aminoglycoside, which is a significant issue considering that its large-scale propagation would remarkably limit treatment alternatives.
In this study, we examined mcr-1-harboring plasmid genomes using a k-mer-based phylogenetic tree builder. Our results show that global dissemination of mcr-1 carrier plasmids at the whole genome level. Whole genome alignment revealed that plasmid IncX4 harbored an hns-protein coding gene, a potential plasmid transfer facilitator. They lost insertion sequence responsible for cut-and-paste transposition, imparting stability to mcr-1 in the IncX4 plasmid. On the contrary, IncI2 contained diverse insertion sequences, and IncF and IncF plasmids possessed various ARGs. Overall, the present results show that the k-mer-based alignment-free phylogeny tool is a suitable alternative to compare the genetic diversity of plasmid genomes and elucidates the genetic characteristics of mcr-1-harboring plasmids.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.