
Abstract
Rapid adaptation to changing environments is key in determining the outcome of infections caused by the opportunistic human pathogen Streptococcus agalactiae. We previously demonstrated that the RofA-like protein (RALP) regulators RogB and Rga activate their downstream divergently transcribed genes, that is, the pilus operon PI-2a and the serine-rich repeat encoding gene srr1, respectively. Characterization of the Rga regulon by microarray revealed that the PI-2a pilus was strongly controlled by Rga, a result confirmed at the protein level. Complementation experiments showed that the expression of Rga, but not RogB, in the double ΔrogB/Δrga mutant, or in the clinical strain 2603V/R displaying frameshift mutations in rogB and rga genes, is sufficient to restore wild-type expression levels of PI-2a pilus and Srr1. Biofilm formation was impaired in the Δrga and Δrga/rogB mutants and restored on complementation with rga. Paradoxically, adherence to intestinal epithelial cells was unchanged in the Δrga mutant. Finally, the existence of several clinical isolates mutated in rga highlights the concept of strain-specific regulatory networks.
Introduction
During the last decade, the key virulence factors of GBS involved in the colonization, survival, spread, and persistence of the bacterium within the human host have been identified (for a recent review, see Ref. 32 ). In addition, the availability of eight genome sequences accelerated our understanding of the pathogen's strategy. 40 The rapid adaptation to the changing environments encountered during infection is essential for the GBS survival that is needed to colonize various tissues, obtain nutrients, or evade the host immune response. The genome sequences of GBS strains have revealed the presence of 17–20 two-component systems (TCS) that can respond to changes in the external environment. One of the best-characterized TCS is the CovS/CovR system that plays a major role in virulence.15,21 The regulatory activity of CovR is modulated not only by its cognate sensor CovS but also by a serine/threonine kinase Stk1 that inactivates CovR by phosphorylation on threonine residue 65. 22
GBS NEM316 encodes 6 “stand-alone” transcriptional regulators, 11 among which RogB, Rga, and Gbs1426 belong to the RofA-like protein (RALP) family (for a review, see Ref. 19 ). RogB and Rga are the closest relatives of this family exhibiting 47% identity, whereas Gbs1426 is more distantly related, displaying 23% and 24% identity to RogB and Rga, respectively. RofA, the founding member of this family, was first identified in Group A Streptococci (GAS) as a positive transcriptional activator of the adjacent prtF gene encoding the fibronectin binding protein SfbI. 30 Nra, the second RALP regulator identified in GAS, was found to be a negative regulator for an adjacent gene that encodes the collagen-binding protein Cpa, as well as an unlinked gene encoding the fibronectin-binding protein PrtF2. 30 RALP proteins exhibit strong similarities with another global regulator of M protein synthesis in GAS called Mga (Hondorp and McIver). Mga and RALP proteins are about 500 amino acids long and contain 3 conserved Pfam domains: (i) HTH_Mga (PF08280); (ii) Mga (PF05043); and (iii) PRD (PF08270). Both Helix-Turn-Helix found in PF08280 and PF05043 were involved in DNA binding 27 (see Supplementary Fig. S1; Supplementary Data are available online at www.liebertonline.com/mdr).
In the present work, we demonstrate the central role of Rga in controlling the synthesis of two important GBS NEM316 surface adhesins, namely PI-2a pilus and Srr1. Surprisingly, the adherence of an rga knock-out strain to epithelial cells was similar to that of the wild-type (WT) strain, suggesting that the display of surface proteins is a highly dynamic process which is dependent on the expression of multiple adhesins.
Materials and Methods
Bacterial strains, media, and growth conditions
The bacterial strains and plasmids used in this study are listed in Supplementary Table S1. S. agalactiae NEM316 is a ST-23 serotype III strain whose genome has been sequenced. 11 The NEM316Δrga and NEM316ΔrogB mutants were previously constructed.6,28 The Escherichia coli DH5α (Invitrogen) used for the cloning experiments was grown in Luria-Bertani (LB) medium. Unless otherwise specified, S. agalactiae was cultured in Todd-Hewitt (TH) broth (standing filled flasks) or agar (Difco Laboratories). Erythromycin was used at 150 μg/ml for E. coli and at 10 μg/ml for S. agalactiae. Lactococcus lactis strain NZ9000 20 was grown in an M17 medium supplemented with 1% glucose (GM17). Heterologous expression of PI-2a pilus in the L. lactis strain was realized as follows: The five genes gbs1478-1474 were amplified from NEM316 genomic DNA and cloned in the lactococcal vector pOri23, a high copy number erythromycin resistance plasmid expressing the cloned gene from the strong constitutive promoter P23. 31 The primers used for the amplification of gbs1478 (pilA), gbs1477 (pilB), gbs1474 (pilC), and the entire operon PI-2a (gbs1478-1474) are shown in Supplementary Table S1. The various PCR products were then cut by the appropriate enzymes (New England Biolabs), purified, and cloned into pOri23. The resulting plasmids were verified by DNA sequence and introduced into electrocompetent L. lactis NZ9000.
General DNA techniques
Standard recombinant techniques were used for nucleic acid cloning and restriction analysis. 36 Plasmid DNA from E. coli was prepared by rapid alkaline lysis using the QIAprep Spin Miniprep Kit (Qiagen). Genomic DNA from S. agalactiae was prepared using the DNeasy Blood and Tissue Kit (Qiagen). PCR was carried out with Phusion Taq polymerase as described by the manufacturer (Finnzymes).
Construction of mutants and complemented strains
The double-mutant ΔrogB/Δrga was constructed as described for NEM316ΔrogB but in the NEM316Δrga genetic background.
The primer pairs RgaF-RgaR and RogBF-RogBR were used to amplify the sequence encoding the rga and rogB genes, respectively (Supplementary Table S1). The resulting PCR fragments were digested with BamH1-Pst1 and cloned into the shuttle vector pTCVErm-Ptet to generate pRga and pRogB, respectively. The resulting plasmids were introduced by electroporation into the various strains.
Expression and purification of recombinant Sag1407
The Sag1407 DNA coding region was amplified by PCR using genomic DNA of GBS 2603V/R as a template and primers Sag1407Nco and Sag1407Bam (Supplementary Table S1). The PCR product was digested with NcoI and BamHI enzymes and cloned into pET2817 (Tazi et al.). The resulting plasmid was introduced into E. coli BL21λDE3/pDIA17 for protein expression. Recombinant 6xHis-SAG1407 protein was purified under native conditions by affinity chromatography on an Ni-NTA column according to the manufacturer's recommendation (Qiagen) followed by exclusion chromatography on a Superdex 200 column (GE Healthcare). Protein purity was checked on sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and accurate protein concentration was determined using the Bradford protein assay. Rabbit polyclonal antibodies were produced and purchased from Covalab.
Microarray analysis
We used oligo arrays carrying three oligonucleotides per gene, and this was demonstrated to be a much more sensitive method than the previously used DNA macro arrays. 21 RNA was reverse transcribed with Superscript indirect cDNA kit (Invitrogen) and labeled with Cy5 or Cy3 (Amersham Biosciences) according to the supplier's instructions. The microarray containing 6835 60-mer oligonucleotides specific for 2134 predicted genes of the genome of the NEM316 strain and for all intergenic regions longer than 100 nucleotides has been designed using the program OligoArray (http://berry.engin.umich.edu/oligoarray/). Based on these sequences a custom oligonucleotide array was manufactured (Agilent Technologies) with a final density of 15K. For hybridization, Cy3 and Cy5 target quantities were normalized at 150 pmol.
The arrays were scanned in an Axon 4000B dual laser scanner connected to the GenePix Pro 6.0 image acquisition and analysis software package. Slides were scanned at 635 and 532 nm wavelength (respectively for Cy5 and Cy3). An initial prescan of the slide was performed to determine the success of hybridization and to allow a definition of the boundaries of the array features. The photomultiplier (PMT) gain settings were adjusted during this step to ensure that the overall red-to-green ratio is similar (PMT settings are 635 nm=460, 532 nm=425). Spots were excluded from the analysis in the case of high local background fluorescence slide abnormalities or weak intensity. No background was subtracted. For the purpose of a statistical analysis, RNA was prepared from three independent cultures, and each RNA sample was hybridized twice to the microarrays (dye swap), leading to six values. Data normalization and differential analysis were conducted using the R software (www.r-project.org). A global intensity-dependent normalization using the LOESS procedure 41 was performed on a slide-by-slide basis (BioConductor package marray; www.bioconductor.org/packages/bioc/html/marray.html) to correct the dye bias. To determine the differentially expressed genes, we performed a paired t test using the VM method (VarMixt package) 4 together with the Benjamini and Yekutieli p-value correction method. 33 For each gene, 3 probes were present in the microarray. The cut-off for the expression ratio was set to either superior/equal to 2 or inferior/equal to 0.5, and the general ratio of expression of each gene was calculated as the average expression ratio from the different probes. The data for which at least two of the three probes gave a significant and noncontradictory result were taken into account.
All transcriptome data are MIAME compliant. The RAW data have been submitted to the ArrayExpress database maintained at www.ebi.ac.uk/microarray-as/ae/under the Accession number: E-MEXP-3236 for the rga experiment, E-MEXP-3237 for the rogB experiment, and E-MEXP-3304 for the covSR experiment. It is worth mentioning that the Rga microarray analysis revealed a number of genes that were strongly upregulated in the Δrga mutant, such as gbs0189-0190 (118- and 31-fold respectively), an observation that was not confirmed by quantitative RT-PCR. We noticed that they belong to a set of about 140 genes for which a strong discrepancy was repeatedly observed for the WT strain between independant microarrays experiments and/or qRT-PCR analysis. Our hypothesis is that the expression of these genes is unexpectedly variable from one experiment to another, even though the growth conditions were cautiously monitored.
Real-time quantitative PCR
Total RNAs extracted from NEM316, NEM316Δrga, and NEM316ΔrogB were harvested at an OD600nm=0.5 as previously described. 28 cDNA synthesis was performed on DNase I treated RNA (5 μg) with random hexamers (Roche) using Superscript II RT (Invitrogen) as recommended by the manufacturer. Real-time quantitative PCR was performed as previously described 28 in a 25 μl reaction volume containing cDNA, 12.5 μl iQTM SYBR Supermix (BioRad), and 1 μl gene-specific primers (10 μM each) (Supplementary Table S2). Briefly, amplification, detection, and analysis were performed with the MyiQ Single-Color Real-Time iCycler PCR Detection System and the MyiQ Optical System Software (Bio-Rad). The specificity of the amplified products and the absence of primer dimers formation were verified by generating melting curves. The absence of contaminating genomic DNA was verified by testing each sample in control reactions without a previous reverse-transcription step. The critical threshold cycle was defined for each sample. The expression levels of the tested genes were normalized using the NEM316 gyrA gene, whose transcription level did not vary under our experimental conditions. Each assay was performed in quadruplicate and repeated with two independent RNA samples. The change (n-fold) in the transcript level was calculated as described. 23
DNase I footprinting assays
DNA fragments corresponding to the promoter regions of srr1 (409 bp amplified with primers rga/srr1-F and -R), pilA (295 bp, rogB/pilA-F and -R) were generated by PCR using Pwo polymerase (Roche) and the indicated oligonucleotide pairs (for sequence see Supplementary Table S1). The radiolabeling of DNA fragments was performed as previously described. 5 Either purified Rga-6His or crude extracts (from L. lactis/pAT18 and L. lactis/pAT18-Rga) binding to DNA (5×104 cpm per reaction) was performed at room temperature in a buffer containing 25 mM NaH2PO4 pH 8, 50 mM NaCl, 2 mM MgCl2, 1 mM DTT, and 10% glycerol. DNaseI treatment and migration of the reaction mixtures were then performed as previously described. 5
Primer extension reactions
Total RNA was used as templates for primer extension reactions using radiolabeled specific primers of rogB (gbs1479), pilA (gbs1478), rga (gbs1529), and srr1 (gbs1530), respectively (Supplementary Table S1), as previously described. 7 The corresponding Sanger DNA sequencing reactions were carried out by using the same primers and PCR-amplified fragments containing the genes upstream regions with the Sequenase PCR product sequencing kit (USB).
Immunoblotting analysis
Cell-wall protein extracts were prepared by harvesting 50 ml of bacteria in an exponential phase (0D600=0.5). The bacterial pellet was washed in ice-cold phosphate-buffered saline (PBS) and then in Tris-HCl buffer (50 mM, pH 7.3), and, finally, the pellet was resuspended in the mutanolysin digestion buffer (Tris-HCl 50 mM pH 7.3 supplemented with 20% sucrose and a complete protease inhibitor cocktail [Roche]). Mutanolysin (Sigma) dissolved in 5,000 U ml−1 in a potassium phosphate buffer (pH 6.2) was then added to the bacterial suspension to give a final concentration of 100 U ml−1 and the samples were rotated for 2 hr at 37°C. After centrifugation at 13,000 g for 15 min at 4°C, the supernatants corresponding to the cell wall extracts were analyzed on SDS-PAGE or kept frozen at −20°C. For western blotting, proteins were boiled in Laemmli sample buffer, resolved on Tris-Acetate Criterion XT gradient gels 4%–12% SDS-PAGE gels (BioRad), and transferred to nitrocellulose membrane (Hybond-C, Amersham). For dot-blot analysis on whole bacteria, exponentially growing bacteria were washed in PBS and resuspended in adjusted volumes of PBS to obtain similar OD600 values. The bacteria were loaded on a nitrocellulose membrane, dried up for 20 min at room temperature, and then blocked in PBS-milk 5% for 30 min. PilB and Srr1 were detected using specific rabbit polyclonal antibodies previously obtained6,28 at a 1:1,000 dilution and as loading control, S. agalactiae was detected using a mouse antibody raised against formaldehyde-killed BM110. The secondary horseradish peroxidase-coupled anti-rabbit secondary antibody (Zymed) was used at a 1:20,000 dilution, whereas the goat anti-mouse antibody was used at a 1:10,000 dilution. Detection was performed using the Western pico chemiluminescence kit (Thermo Scientific). Image capture and analysis were performed on a GeneGnome imaging system (Syngene).
Biofilm formation assays
Bacterial attachment and surface growth on polystyrene microtiter plates were analyzed during the growth of S. agalactiae in an LB medium with 1% glucose supplemented with erythromycin when necessary. Overnight cultures grown in TH were used to inoculate the LB glucose medium at 0.1 OD600 and after a brief vortexing, 180 μl of the cell suspension were dispensed into 96-well plates (Costar 3799; Corning, Inc.) and incubated at 37°C for 2 or 24 hr. The OD600 of each culture was measured to ensure that they had reached the stationary phase at similar cell densities, and the wells were washed twice in PBS using an ELx50 washer (Biotek) and air dried for 15 min. Biofilms were stained with 0.1% crystal violet for 30 min (100 μl per well), and the wells were washed twice with PBS and air dried. The stained biomass was resuspended for quantification in ethanol/acetone (80:20), and A595 was measured. The assay was performed in quadriplate.
Cell culture and adherence assays
The human cell lines A549 (ATCC CCL-185) from an alveolar epithelial carcinoma and the TC7 clone 3 established from the parental colon adenocarcinoma Caco-2 were cultured in Quantum 286 Medium (PAA). The cells were incubated in 10% CO2 at 37°C and were seeded at a density of 2 to 5×105 cells per well in 24-well tissue culture plates. Monolayers were used after 24–72 hr of incubation. Bacterial cultures from overnight cultures OD600 of 2 (∼6×108 CFU/ml) were washed once in PBS and resuspended in DMEM. The cells were infected at a multiplicity of infection of 10 bacteria per cell for 1 hr at 37°C in 10% CO2. The monolayers were then washed four times with PBS, and the cells were disrupted by the addition of 1 ml sterile deionized ice-cold water and repeated pipetting. Serial dilutions of the lysate were plated onto TH agar for the count of viable bacteria. The percent of adherence was calculated as follows: (CFU on plate count/CFU in original inoculum)×100. Assays were performed in triplicate and were repeated at least thrice.
Results
Rga microarray analysis
To define the respective roles of the paralogous regulators Rga and RogB, we carried out a genome-wide transcriptional analysis of the WT NEM316 strain and its isogenic Δrga and ΔrogB mutant strains. In the Δrga strain, 60 genes were found to be down-regulated, and 36 were found to be up-regulated compared with the parental NEM316 using a twofold cut-off threshold. These regulated genes are often grouped into operons and encode proteins associated to a large variety of unrelated functions, suggesting that Rga is a pleiotropic regulator. Among the down-regulated genes, we identified its cognate target gene srr1, but, most importantly, the regulator-encoding gene rogB and its target operon gbs1478-1474 (named PI-2a in Ref. 35 ) (Table 1). In the ΔrogB strain, 96 genes were found to be down–regulated, and 82 genes were found to be up-regulated. The down-regulated genes included the PI-2a operon and the two component regulatory systems ciaRH and relPR, whereas srr1 was among the most up-regulated genes. RogB, similar to Rga, may have an impact on the expression of other regulatory genes, making it difficult to identify the genes that are directly regulated by either regulators from those which are indirectly regulated.
RNAs were extracted from the bacteria collected at the mid-exponential phase of growth in Todd–Hewitt broth (OD600=0.3).
Asterisk (*) indicates the few chosen genes whose differential expression has been validated by quantitative reverse transcription polymerase chain reaction, thereby confirming the microarray results.
The functional classification is that of the SagaList server: http://genolist.pasteur.fr/SagaList/help/function-codes.html.
Rga-dependent CovRS regulation.
RogB-dependent Rga regulation.
RALP, RofA-like protein; GBS, group B streptococcus; WT, wild-type.
A previous macroarray analysis performed in our laboratory showed that rga (gbs1530), but not rogB, was twofold induced in the NEM316ΔcovSR mutant. 21 This observation was confirmed in this study by using a microarray analysis (E-MEXP-3304). We, thus, compared the Rga regulon, which included 96 differentially regulated genes (twofold cut-off threshold), with the CovSR and RogB regulons using Cluster 3 and TreeView programs. The subset of 61 genes differentially regulated in at least two microarrays is shown in Fig. 1. The most striking group of genes likely regulated through a CovSR-dependent Rga regulation includes srr1 and the PI-2a pilus operon (gbs1478-1474), the glycosyltransferase encoding genes gbs1605-1606, and that encoding the surface adhesin BibA (gbs2018) (Table 1).
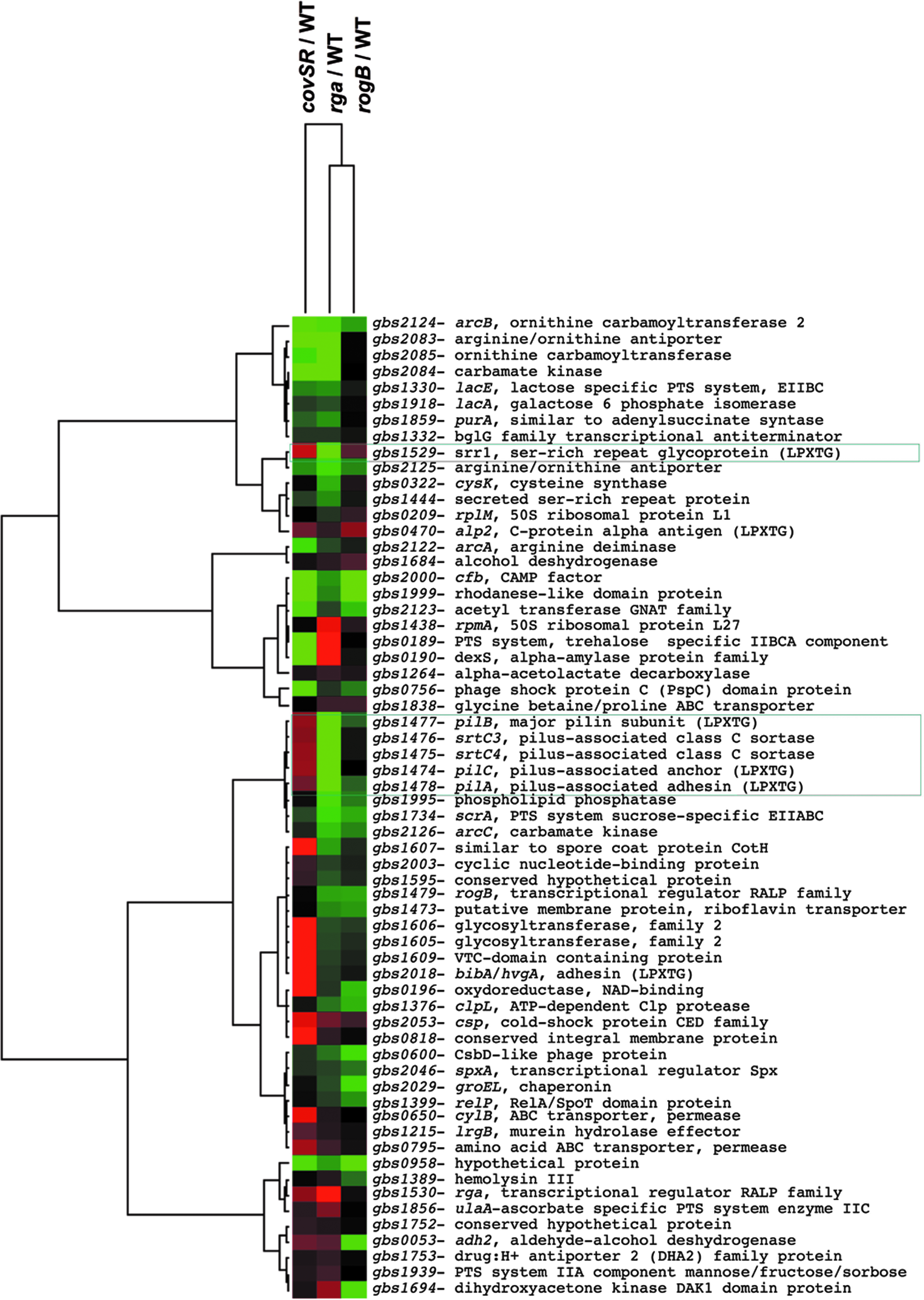
Comparison of the Rga regulon to the CovSR and RogB regulons using Cluster 3.0 and Java Tree View representation. Among the 97 differentially regulated genes in the Rga regulon, a subset of 61 genes whose expression varies in the CovSR or RogB microarrays are shown. Upregulated genes are indicated in red, downregulated genes are indicated in green, and nonvariable genes are shown in black.
A quantitative analysis of the results shows that the expression of the PI-2a operon is positively regulated by both Rga and RogB, but that the impact of the rga deletion is 10 times that of the rogB mutation.
Rga is the master regulator of the pilus operon
In order to determine the hierarchy between Rga and RogB in the transcriptional activation of the PI-2a pilus operon (gbs1478-1474) and srr1 (gbs1529), quantitative reverse transcription polymerase chain reaction (RT-PCR) experiments on srr1 and pilB were performed using the WT strain NEM316 and its isogenic Δrga and ΔrogB mutant strains. The results shown in Fig. 2 demonstrated that Rga is the major transcriptional activator of PI-2a pilus and srr1 transcription (>100-fold on pilB, the second gene of the operon, encoding the major pilin). As previously described, RogB only slightly activates pilB transcription two- to threefold.6,13 We also observed a slight repression in srr1 transcription by RogB, confirming the transcriptomic data.
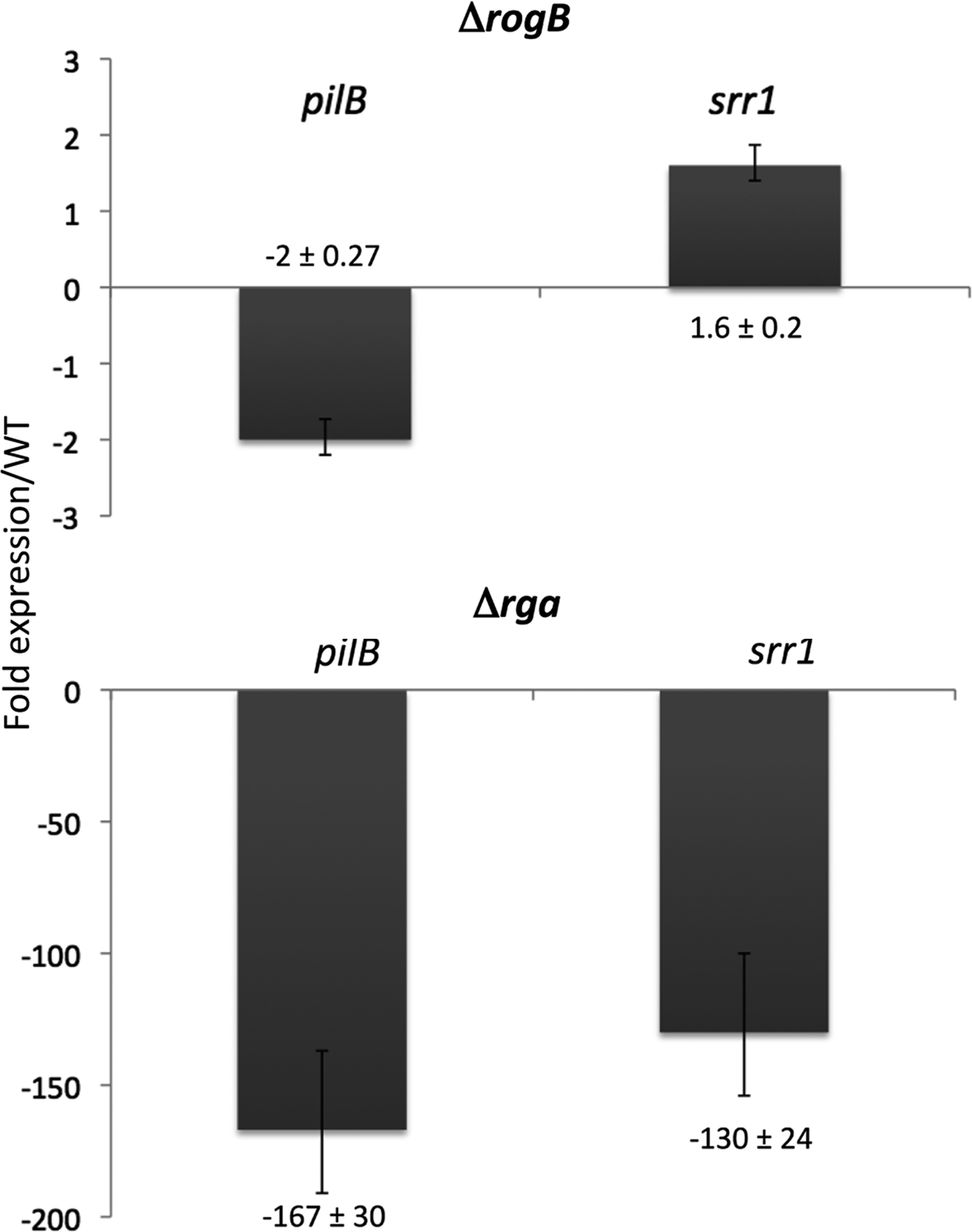
Expression of the genes pilB and srr1 in Streptococcus agalactiae ΔrogB (top panel) and Δrga (bottom panel). The transcriptional analysis was performed by quantitative reverse transcription polymerase chain reaction and normalized relative to the gyrA gene. The figure indicates the calculated ratio of the expression in the mutant strains relative to that in the NEM316 WT strain. WT, wild-type.
By dot-immunoblotting with specific polyclonal antibodies against Srr1 and PilB on exponentially growing bacteria, we determined Srr1 and PilB protein levels in the WT strain NEM316 and the two isogenic mutants Δrga and ΔrogB. As a control for bacterial loading, we used an antibody against formaldehyde-killed GBS bacteria. No signal could be detected in the Δrga mutant, indicating that Rga is the major activator of both Srr1 and PilB synthesis (Fig. 3). Trans-complementation with a plasmid-borne rga restored both Srr1 and PilB synthesis (Fig. 3).
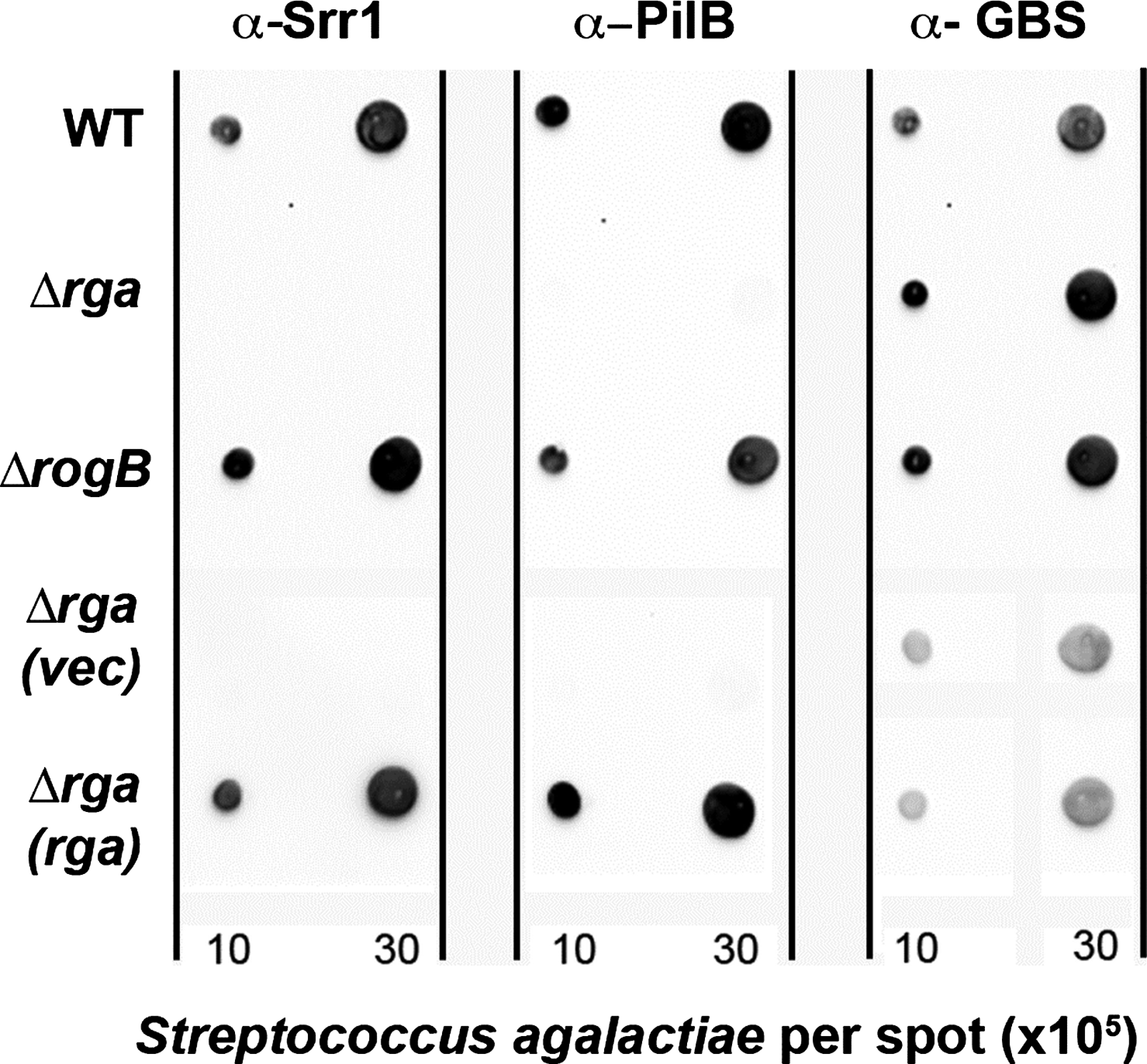
Analysis of surface exposure of Srr1 and PilB proteins in WT NEM316, Δrga, and ΔrogB strains by immunodot-blot. 2 and 6 μl of whole bacterial cells suspension in phosphate buffer saline (OD600nm=1) harvested in the exponential phase (OD600nm=0.5) were spotted on nitrocellulose. Membranes were hybridized with specific anti-Srr1 and anti-PilB rabbit polyclonal antibodies. Anti-GBS antiserum raised in mice was used as a loading control. The membranes were next incubated with horseradish peroxidase-conjugated secondary antibodies and revealed using the Western pico chemiluminescence kit. GBS, group B streptococcus.
The complex interaction between RogB and Rga prompted us to construct a double Δrga/rogB mutant derivative of the NEM316 strain. We then introduced plasmid-borne rga or rogB under the control of a constitutive promoter in both the WT and double-mutant strains. Western-blot and dot-blot analyses showed that (i) the overexpression of rogB or rga in the WT strain increased the formation of PilB polymers; and (ii) the expression of rga only, but not rogB, in the Δrga/rogB strain was sufficient to restore WT levels of pilus synthesis (Fig. 4). Similarly, overexpression of rga in the WT strain increased the expression level of Srr1 at the cell surface and was sufficient to restore the WT levels of Srr1 in the Δrga/rogB strain (data not shown).
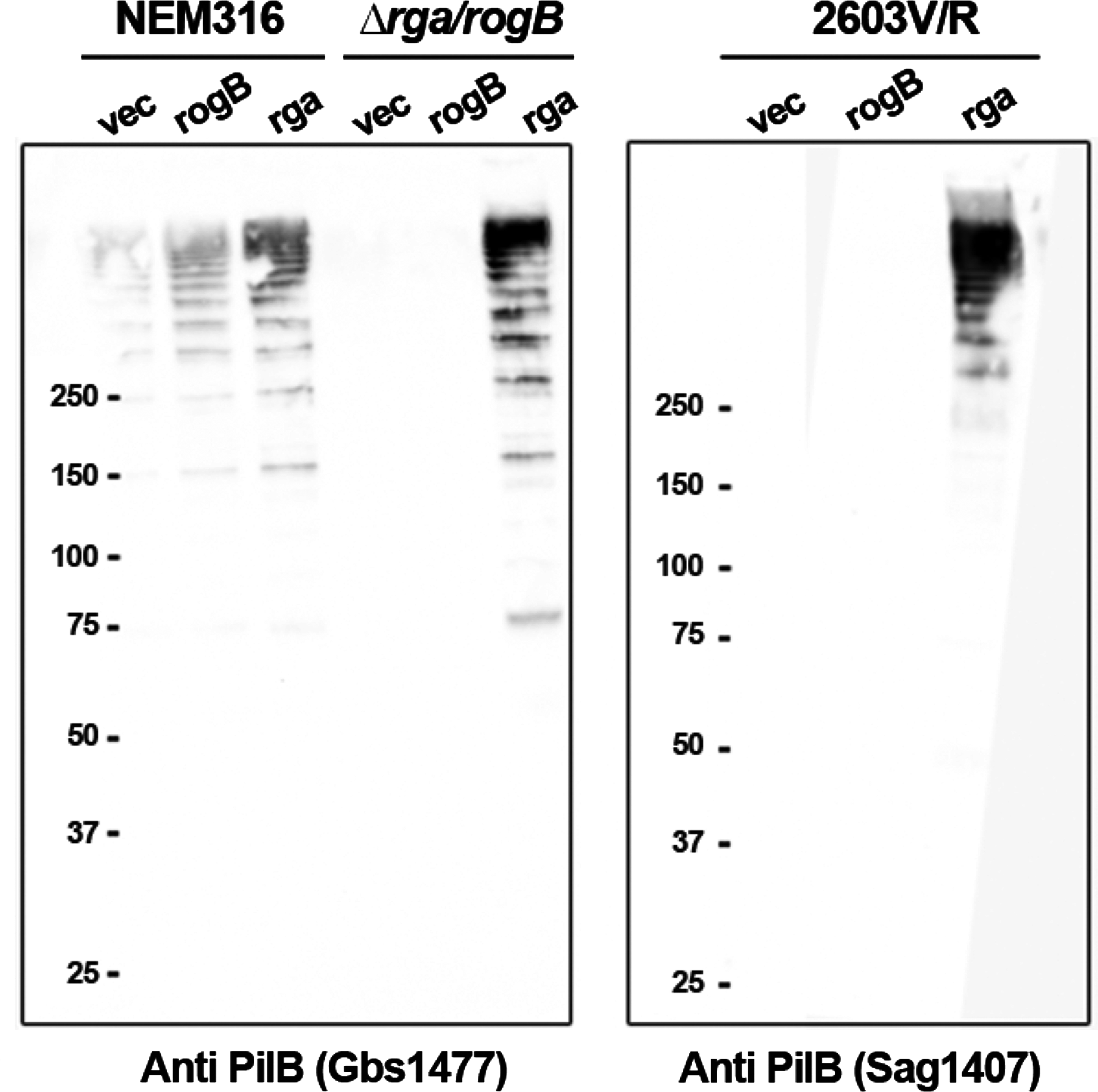
Western blot analysis of cell-wall anchored pili in three S. agalactiae strains overexpressing rogB or rga. NEM316 and its isogenic Δrga/rogB mutant strains
The analysis of the rga sequence in the three complete GBS genomes and four partially sequenced genomes revealed that strains A909, 2603V/R, and CJB111 likely produced truncated Rga, resulting from frameshift mutation and/or mutation creating an internal stop codon (Supplementary Fig. S1). Interestingly, the GBS strain 2603V/R responsible for adult invasive disease also exhibited a frameshift mutation in the rogB gene, thus constituting a natural rga/rogB double mutant. We first showed by immunoblotting analyses that 2603V/R did not synthesize any detectable levels of Srr1 and PilB proteins (not shown). By introducing in trans a functional copy of rga (pRga) in the 2603V/R strain, we restored the expression of both PilB polymers and Srr1, whereas the trans-expression of rogB did not do so (Fig. 4 for PilB, not shown for Srr1). Altogether, these results indicate that the expression of rga is necessary and sufficient for both PI-2a pilus and Srr1 expression.
Characterization of PI-2a and Srr1 promoters
To gain further insights in the molecular mechanisms underlying the Rga-dependant regulation, transcription start sites were localized in the rogB-pilA and rga-srr1 intergenic regions (Supplementary Fig. S2). Single nonoverlapping transcriptional start sites (TSS) were found upstream pilA (the first gene of the PI-2a operon), rogB, srr1, and rga genes, suggesting no transcriptional interference between the divergently transcribed genes (Supplementary Fig. S2).
In vitro binding assays were performed to test whether the Rga-dependant activation results from the direct interaction of Rga with its target promoters. Our many attempts to express and purify the recombinant Rga protein in E. coli using different systems (Rga-6His or MBP-Rga or RgA-intein) have failed. In all cases, the protein was either insoluble or degraded (data not shown). We, therefore, cloned the entire rga with a C-terminal histidyl tag in the multicopy vector pAT18 giving rise to pRga-6His. This plasmid, able to complement the Δrga mutant (not shown), was expressed in the L. lactis strain NZ9000 under the control of a constitutive promoter. The recombinant protein Rga-6His was purified from L. lactis, and its identity was verified by mass spectrometry analysis (data not shown). DNAse I footprinting assays were carried out using either the purified Rga-6His protein (20 μg) or total protein extracts (25–100 μg) from L. lactis (pRga-6His) or L. lactis (pAT18). As shown in Fig. 5, two close DNA sequences in the srr1 promoter region from −100 to −69 and from −64 to −45 relative to the TSS were protected with the total extracts of L. lactis (pRga-6His). Protection was detected with neither the control L. lactis (pAT18) extracts, nor the purified protein Rga-6His. The same experiment was done with the PI-2a promoter region, and no protection against DNase I digestion was observed (data not shown). These results show that (i) srr1 is directly regulated by Rga, while the PI-2a operon is probably indirectly regulated; and (ii) that purification of Rga is detrimental to its DNA-binding activity, either because of protein instability or due to the loss of a cytoplasmic cofactor.
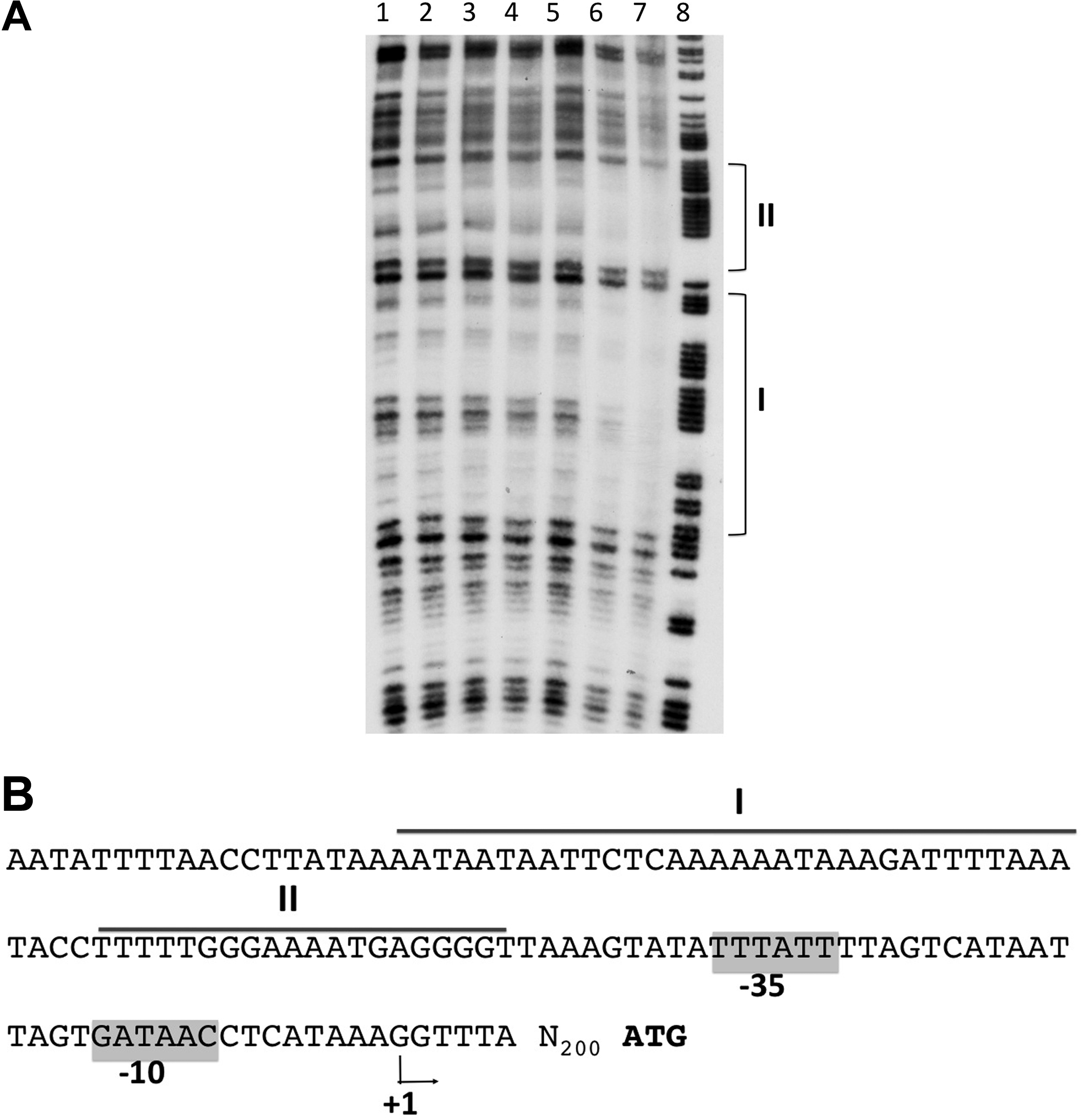
DNase I footprinting analysis of Rga binding to the srr1 promoter region.
Biofilm formation is abolished in the Δrga mutant and restored on complementation
A major role of GBS PI-2a pilus is its implication in biofilm formation.18,34 Indeed, expression of the NEM316 PI-2a pilus operon in the nonpathogenic L. lactis strain NZ9000 conferred to the recombinant bacteria the ability to form biofilm (Supplementary Fig. S3 and Ref. 34 ). We, therefore, tested the isogenic Δrga and ΔrogB mutants for their ability to form biofilm on polystyrene plates using LB glucose medium. As shown in Fig. 6A, biofilm formation is slightly reduced in the ΔrogB mutant but strongly impaired in the Δrga mutant as compared with the parental NEM316 strain. The complementation of ΔrogB and Δrga by plasmid overexpression of rogB and rga, respectively, restored biofilm formation (Fig. 6B). Finally, we showed that the overexpression of rga in the Δrga/rogB was sufficient to confer to the strain the ability to form biofilm, whereas the overexpressing vector alone or rogB did not (Fig. 6C). Similarly, the overexpression of rga in GBS strain 2603V/R conferred to this GBS clinical isolate the capacity to form biofilm in our experimental conditions. Finally, the overexpression of rga in the WT strain increased the levels of biofilm as compared with the WT strain carrying the vector alone or the plasmid carrying rogB (Fig. 6C).
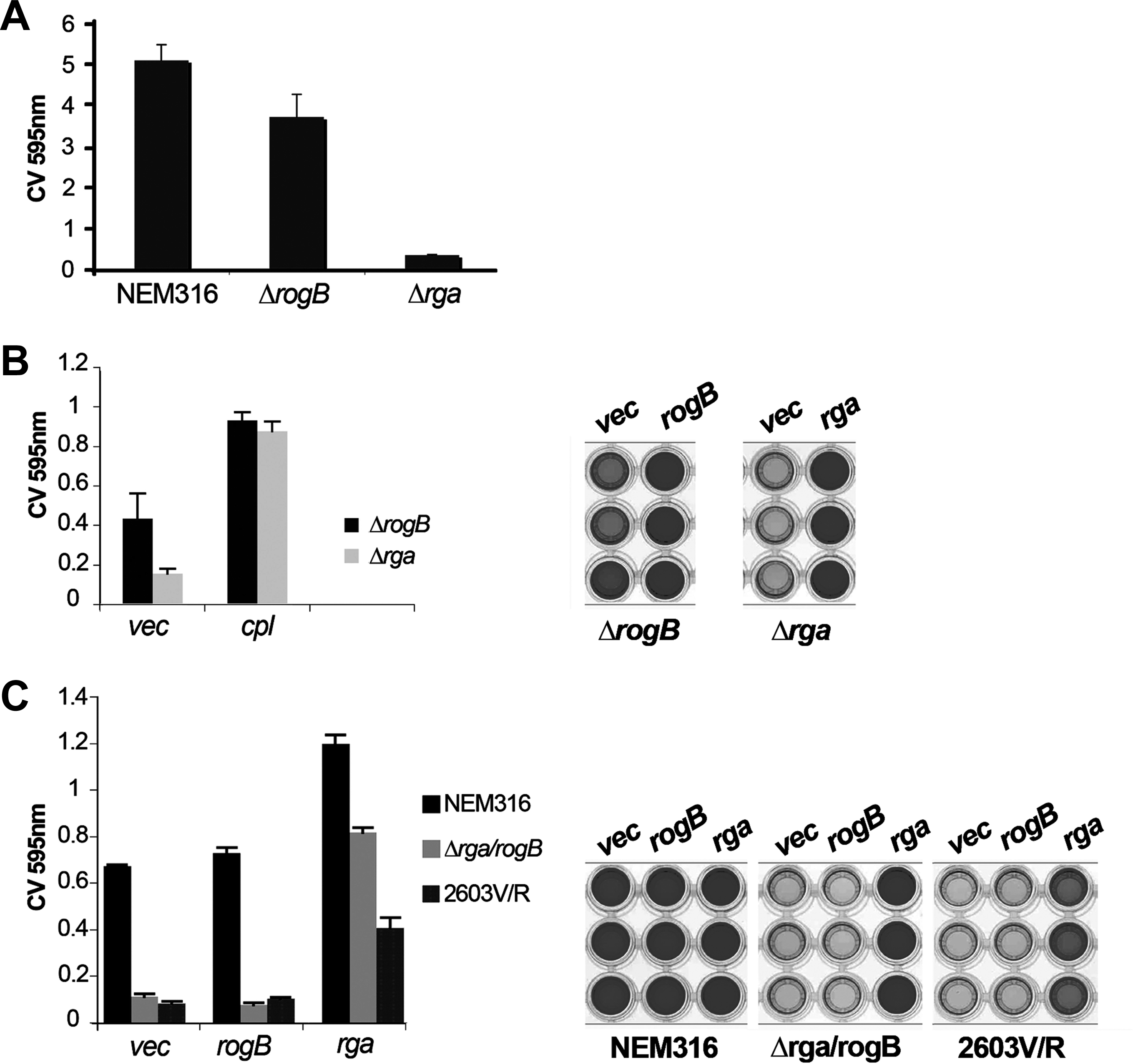
Role of transcriptional regulators Rga and RogB in biofilm formation. S. agalactiae NEM316 and its isogenic Δrga and ΔrogB mutants
The second important role assigned to GBS pilus PI-2A is the promotion of adhesion to alveolar epithelial cells A549,6,18 whereas Srr1 was shown to be a surface adhesin. 28 We, thus, tested the adhesive capacity of the Δrga mutant to A549 alveolar cells and TC7 intestinal cells. Surprisingly, we did not found any significant difference for adherence to these cell lines between the Δrga mutant and the WT strain (Fig. 7 and data not shown). This led us to hypothesize that expression and/or display of other surface proteins in the Δrga mutant compensate for the absence of GBS adhesins Srr1 and PI-2a pilus.
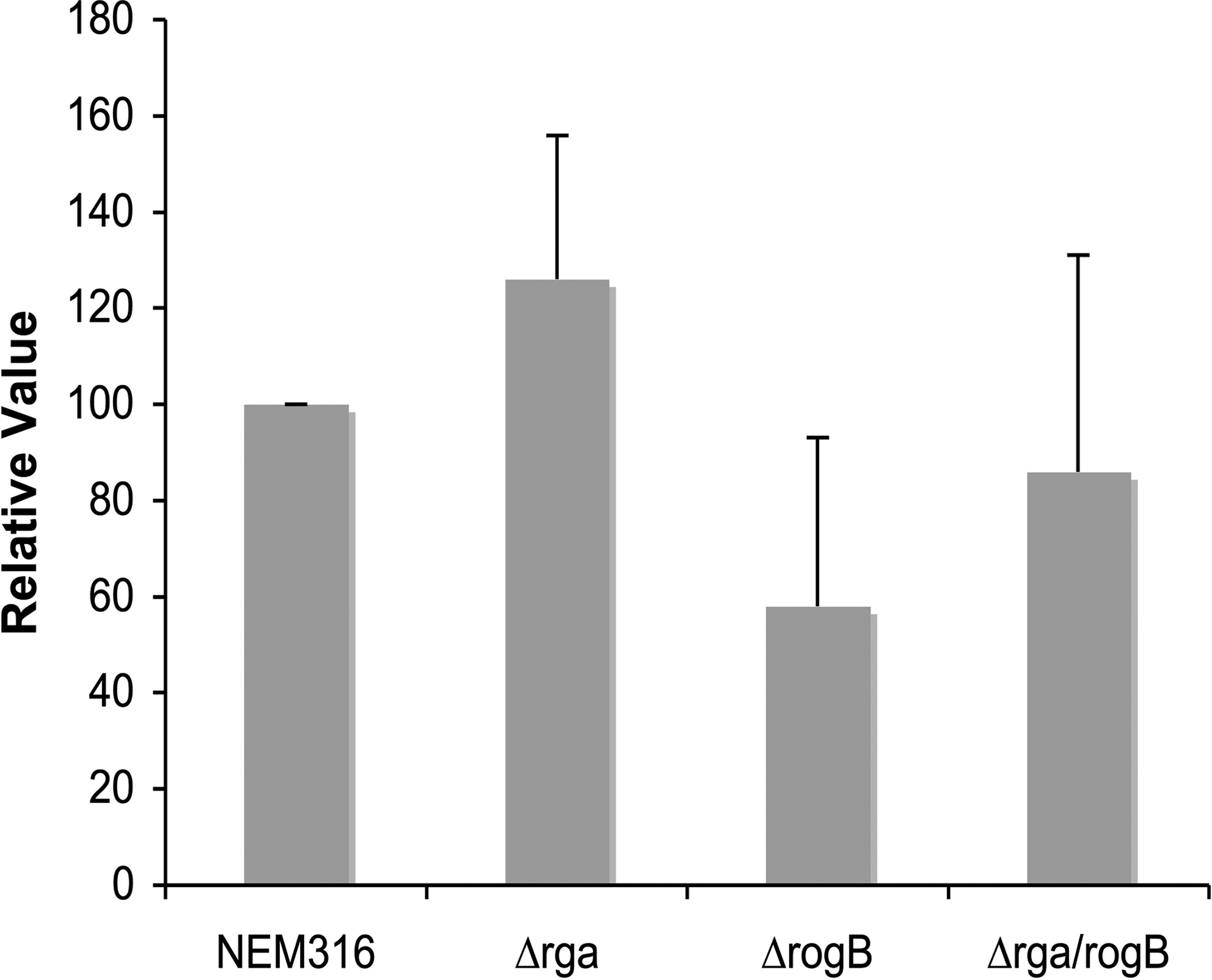
Adhesion of S. agalactiae NEM316 and its isogenic ΔrogB, Δrga, and Δrga/rogB mutants to the alveolar epithelial A549 cell line. Monolayers were infected at a multiplicity of infection of 10 bacteria per cell for 1 hr at 37°C. The adherence frequencies were calculated from the number of bacteria that remained attached to the cell after the incubation period and three washes with regard to the number of inoculated bacteria. The level of adherence of the WT strain is arbitrarily reported as 100, and the level of adherence of the various mutants are relative values. The results are presented as mean value (±SD) from three independent experiments, and each strain is tested in triplicate.
Discussion
We show here that the PI-2a pilus and Srr1, two major surface components previously characterized in our laboratory, are under the positive control of a common transcriptional regulator Rga belonging to the RALP family. We demonstrated the pre-eminence of Rga over RogB, the second closest RALP paralog exhibiting 47% identity with Rga and previously shown to activate the transcription of the pilus operon PI-2a.6,13 It is worth noting that the synthesis of these adhesins is partially dependent on the TCS CovSR, which acts by negatively controlling the transcription of rga. A working model of regulation is depicted in Fig. 8.
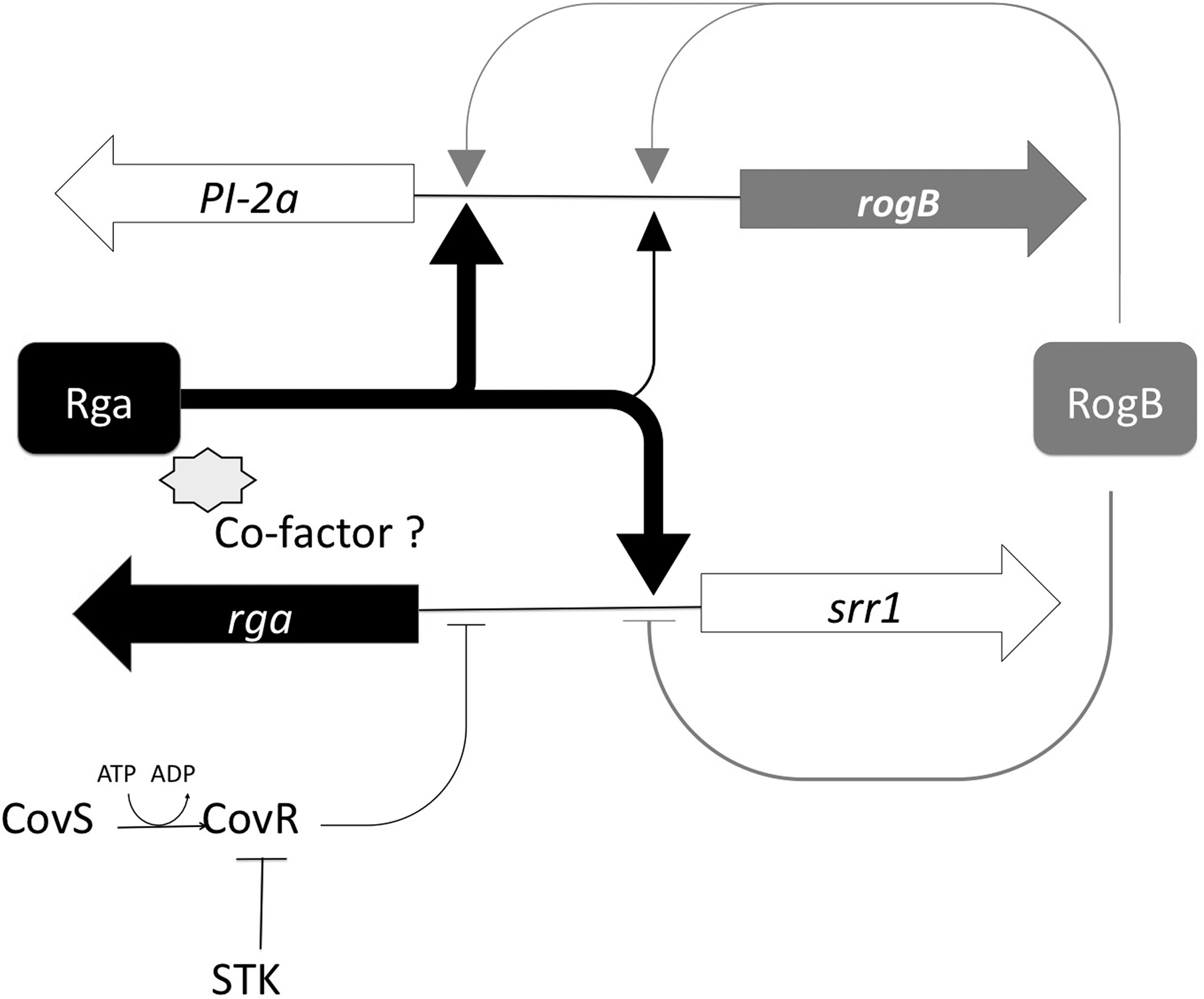
Working model for Rga regulation and its integration in other regulatory networks. Strong transcriptional activation of srr1 and PI-2a operon by Rga (black thick lines) and low activation of the PI-2a operon by RogB (gray thin lines). RogB also activates its own transcription and slightly represses srr1. At an upper level, the CovS/R two-component system, central for virulence in GBS, negatively regulates rga transcription. STK, serine threonine kinase.
Relatively little is known about the regulation of virulence factors in GBS besides the central role of CovSR, a TCS. We and others previously showed that the response regulator CovR is a pleiotropic regulator of virulence genes which binds to the promoter regions of the genes that it controls.15,21 A consensus CovR recognition sequence has been determined (5′-TATTTTAAT-3′) using both DNAse I footprinting and computational analyses. 21 Interestingly, the rga gene was twofold induced in the ΔcovRS strain, indicating that a part of the response of S. agalactiae to the covR mutation may be indirect. By carefully examining the rga promoter region, we found a putative binding site for CovR located 40 bp from the TSS with two mismatches in the recognition sequence.
Rga and RogB belong to the Mga surperfamily which includes the RALP family or RALPs originally described in GAS (for a recent review, see Ref. 25 ). These transcriptional regulators coined as “stand-alone” response regulators were previously shown to control global virulence regulons in response to changing environmental conditions. 14 Similar to the other members of this family of regulators, Rga is predicted to exhibit two successive helix-turn-helix motifs at its N-terminus, which is consistent with a DNA-binding function. Only two poorly defined consensus sites have been defined for Mga and RofA.12,26 Interestingly the DNA sequence protected by Rga found upstream from srr1 (boxes I and II) is reminiscent of the RofA-binding site found within the intergenic region shared by rofA and prtF at a similar position relative to the TSS. 12 Computational analyses of the various promoter regions of Rga target genes were performed but did not reveal any clear consensus sequence, suggesting a complex regulation.
During the preparation of this article, another study reporting the role of Rga in pilus formation in S. agalactiae NEM316 was published. 37 These authors claim that recombinant Rga purified from E. coli can bind to the promoter regions of pilA and fbsA. We were unable to see any convincing gel shift in the pilA and srr1 promoter regions with purified Rga under various conditions (not shown). Using the DNAse I protection assay, a protected region in the srr1 promoter region was defined with total extracts of L. lactis expressing rga compared with the L. lactis carrying the vector alone. However, no protection was observed with the recombinant purified Rga-6His alone. Collectively, our results on Rga suggest that other bacterial components are necessary to promote the binding of Rga to certain of its target promoters.
A genome-wide transcriptional analysis indicated that Rga controls not only the expression of genes encoding the surface proteins but also additional aspects of bacterial physiology, in particular, arginine catabolism (gbs2083-2085; gbs2122-2126). Both gbs2083-2085 and gbs2122-2126 operons encode genes showing homologies to those found in the arginine deiminase pathway (ADI). This multi-enzyme complex catalyzes the conversion of arginine to ornithine, ammonium, and carbon dioxide with the concomitant production of ATP. ADI-expressing bacteria were shown to be better not only in overcoming oxygen and nutrient starvation but also in surviving in acidic environments. 24 From the microarray data (Table 1), it appears that Rga strongly activates the ADI pathway. We, therefore, tested the survival of GBS NEM316 and its isogenic mutant strain Δrga in various pH conditions (6.0, 5.5, 5 and 4). We found that the growth of the Δrga mutant was strongly impaired in acidic conditions, with a 2-log reduction in viability after 2 hr at pH 4 compared with the WT strain (data not shown). It is, thus, tempting to speculate that Rga may play a major role during vaginal colonization of the GBS strain NEM316 and that the pilus PI-2A and Srr1 are the major adhesins involved in this process.
We and others previously showed a major role of GBS PI-2a pilus in biofilm formation.18,34 Here, we further strengthen this result by demonstrating a strict correlation between the PI-2a expression and the ability of the different mutants tested to form biofilm (Fig. 6 and Supplementary Fig. S3). We previously showed that both PI-2a pilus and the surface glycoprotein Srr1 are bacterial adhesins for host epithelial cells.6,18,28 To our surprise, the Δrga mutant, which is strongly impaired for both PI-2a and Srr1 synthesis, was found to be as adherent as the parental strains on human epithelial cells A549 and TC7. We suggest a profound remodeling of the bacterial surface in the Δrga mutant with the exposition of other potential adhesins such as FbsA (Samen et al.), a major fibrinogen binding protein, that could potentially compensate for the loss of PI-2a and Srr1. Global surface proteome changes might also explain the behaviour of two other NEM316 derivatives, Δgbs1478-74 and ΔsrtC3C4 mutant in the PI-2a locus, which were found to be as adherent as the WT strain to epithelial cells (S. Dramsi, unpublished data). Overall these results suggest that the adherence of GBS to epithelial cells is probably mediated by multiple adhesins and that the removal of one or several adhesins from the GBS cell surface is often compensated by the display of other surface proteins involved in the same function, proving the robustness of the system.
In contrast, the biofilm formation in LB glucose is strictly dependent on PI-2a pilus synthesis in GBS. Moreover, the recombinant L. lactis strains overexpressing the PI-2a pilus operon are able to form biofilm 34 (Supplementary Fig. S3). We also noticed that overexpression of rga in the Δrga strain that increased biofilm formation decreased the adherence level to epithelial cells, suggesting, in these conditions, a fine-tuned balance between bacteria-bacteria and bacteria-host interactions (data not shown). Of note, the recombinant L. lactis strains overexpressing the PI-2a pilus or the pilus-associated adhesin PilA were unable to adhere to epithelial cells as compared with the control L. lactis strain carrying the empty plasmid (Supplementary Fig. S3).
Our study was performed on the NEM316 strain, but we noticed that, among the other available GBS genomes, at least three strains (A909, CJB111, and 2603V/R) display a frameshift mutation in the rga gene at various positions, leading to a truncated protein. Similarly, the GBS strains 2603V/R and 515 display a frameshift mutation in the rogB gene (Supplementary Fig. S1). It is worth noting that in the GBS strain COH1, which belongs to the hypervirulent clonal complex ST-17, the chromosomal locus PI-2a is replaced by PI-2b and that the srr1 locus is missing; consequently, this strain expresses neither Rga nor RogB regulators. 1 These observations reveal that the GBS clinical isolates exhibit a diverse repertoire of surface components and associated transcriptional regulators.
This work leads us to consider the bacterial surface as a dynamic structure susceptible to unpredictable remodeling that may have important functional consequences. Thus, the concept of pathogenicity in streptococci appears more complex than previously thought.
Footnotes
Acknowledgments
This work was supported in part by grant ANR-06-PATHO-001-01 from the Agence Nationale de la Recherche “ERA-NET PathoGenoMics” (to P. T.-C.) and by the Institut Pasteur. The authors thank Tarek Msadek for his help and constant support during the purification of recombinant Rga protein and Mathieu Brochet for the preliminary results using macroarrays. They also thank Agnès Fouet for a critical reading of the article.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.