
Abstract
For a long time, colicin M was known for killing susceptible Escherichia coli cells by interfering with cell wall peptidoglycan biosynthesis, but its precise mode of action was only recently elucidated: this bacterial toxin was demonstrated to be an enzyme that catalyzes the specific degradation of peptidoglycan lipid intermediate II, thereby provoking the arrest of peptidoglycan synthesis and cell lysis. The discovery of this activity renewed the interest in this colicin and opened the way for biochemical and structural analyses of this new class of enzyme (phosphoesterase). The identification of a few orthologs produced by pathogenic strains of Pseudomonas further enlarged the field of investigation. The present article aims at reviewing recently acquired knowledge on the biology of this small family of bacteriocins.
Introduction
Colicin M (ColM) is the smallest colicin identified to date (271 residues, 29.5 kDa), the molecular masses of the others ranging from 39 to 90 kDa. 35 It is also the only one known to interfere with peptidoglycan (murein) biosynthesis and to provoke bacterial cell lysis. 40 The uptake of this toxin by cells first needs specific binding to the FhuA outer membrane receptor 11 and, as a class B colicin, its subsequent energy-dependent translocation into the periplasm by both the TonB machinery (TonB, ExbB and ExbD) and the proton motive force of the inner membrane.10,13 The latter protein partners are required for internalization of the toxin but are dispensable for expression of its activity. Indeed, osmotic shock treatments applied to import-deficient strains (the so-called “by-pass” experiments) enable ColM to pass through the outer membrane and fully exert its toxic effect. Interestingly, ColM can be readily overproduced in a fhuA or tonB mutant in the absence of the immunity protein, without any toxic effect, indicating that ColM should enter the cell from the outside to be bactericidal.14,21
Mode of Action of ColM
Early studies performed in the 1980s already demonstrated that ColM kills susceptible E. coli bacteria by inhibiting peptidoglycan biosynthesis, but its exact mode of action was not elucidated.20,40 It was shown to be devoid of peptidoglycan-degrading activity and to act synergistically with β-lactam antibiotics that inhibit the last cross-linking steps of peptidoglycan synthesis. Accumulation of soluble UDP-MurNAc-pentapeptide and depletion of the pools of lipid intermediates mapped the block induced by ColM in membrane steps of the peptidoglycan assembly pathway20,40 (Fig. 1). Since ColM also blocked the synthesis of the O-antigen moiety of lipopolysaccharides (LPS), Braun and coworkers proposed that this toxin should interfere with the regeneration of undecaprenyl-phosphate (C55-P), the essential carrier lipid that is shared between the LPS and peptidoglycan biosynthesis pathways. 22 An inhibition of the dephosphorylation step allowing formation of C55-P from its precursor C55-PP was particularly suspected. The recent identification of multiple C55-PP phosphatase activities that participate in C55-P synthesis and recycling in E. coli prompted us to revisit the mode of action of ColM.15,16 A 6×histidine-tagged form of ColM was purified to homogeneity and shown to be bacteriolytic at ng·ml−1 concentrations (∼50 molecules per cell) 14 , as previously observed with the wild-type toxin. 39 Inconsistent with previous assumptions, recombinant ColM did not inhibit enzymes involved in the synthesis (UppS) or the dephosphorylation (UppP) of C55-PP. 14 The effect of ColM was, therefore, tested on the activities of MraY and MurG, which catalyze the formation of peptidoglycan lipid intermediates I and II from C55-P, UDP-MurNAc-pentapeptide, and UDP-GlcNAc (Fig. 1). 9 Although no inhibition of the latter enzymes was detectable, these assays revealed ColM-mediated degradation of their reaction products. The site of cleavage of lipid intermediates I and II was identified as the phosphoester bond connecting the C55 and pyrophospho-MurNAc moieties of the peptidoglycan precursors 14 (Fig. 2). In vivo whole cell assays, based on specific incorporation of [ 3 H]diaminopimelic acid, indicated that ColM-induced arrest in peptidoglycan polymerization was due to hydrolysis of lipid II. The products of the reaction, undecaprenol (C55-OH) and 1-pyrophospho-MurNAc(-pentapeptide)-GlcNAc, that usually do not exist in E. coli cells, were shown to accumulate in ColM-treated cells.6,14 Two additional degradation products were also detected under these conditions, which were identified as 1-pyrophospho-MurNAc-pentapeptide and 1-pyrophospho-MurNAc-tripeptide. 14 Since lipid intermediate I is not thought to be translocated through the cytoplasmic membrane, it was proposed that these compounds result from processing of the lipid II degradation product [1-pyrophospho-MurNAc(-pentapeptide)-GlcNAc] by glucosaminidase to form 1-pyrophospho-MurNAc-pentapeptide, and sequential cleavage of the C-terminal D-Ala residues of the stem pentapeptide by carboxypeptidases to form 1-pyrophospho-MurNAc-tripeptide. These activities are known to be involved in peptidoglycan maturation and recycling in E. coli. 32 The fact that ColM also concomitantly inhibited the biosynthesis of the O-antigen moiety of LPS, therefore, appeared to be an indirect consequence of the depletion of the pool of C55-P carrier lipid that is shared between the two pathways.
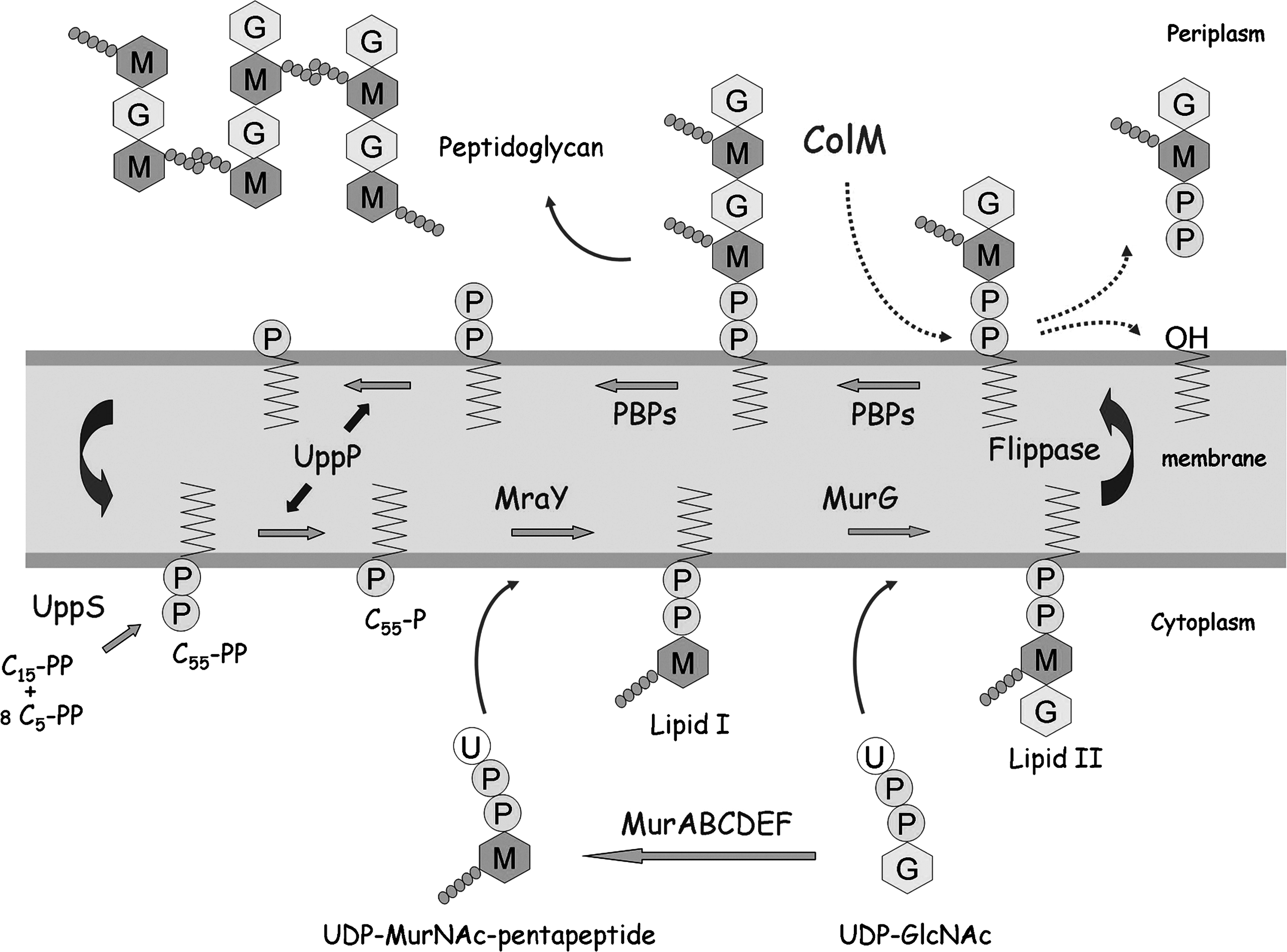
Synthesis of peptidoglycan lipid-linked intermediates and mode of action of ColM. The C55-PP precursor synthesized by the UppS synthetase is dephosphorylated by the UppP enzymes to yield the C55-P carrier lipid. MraY and MurG enzymes catalyze successive transfers of MurNAc-pentapeptide (M-•••••) and GlcNAc (G) motifs from the soluble nucleotide precursors to C55-P, generating the lipid I and II intermediates, respectively. Lipid II is then translocated (flippase) to the outer side of the membrane where polymerization reactions catalyzed by penicillin-binding proteins (PBPs) occur. At the end of this process, the carrier lipid, which is released in C55-PP form, should be dephosphorylated before being reused for de novo peptidoglycan synthesis. 9 ColM molecules internalized into the periplasmic space block this cycle of reactions by specifically degrading lipid II. The site of cleavage was identified as the phosphoester bond connecting the C55 and pyrophosphoryl-disaccharide-peptide moieties of this precursor. 14 ColM, Colicin M.
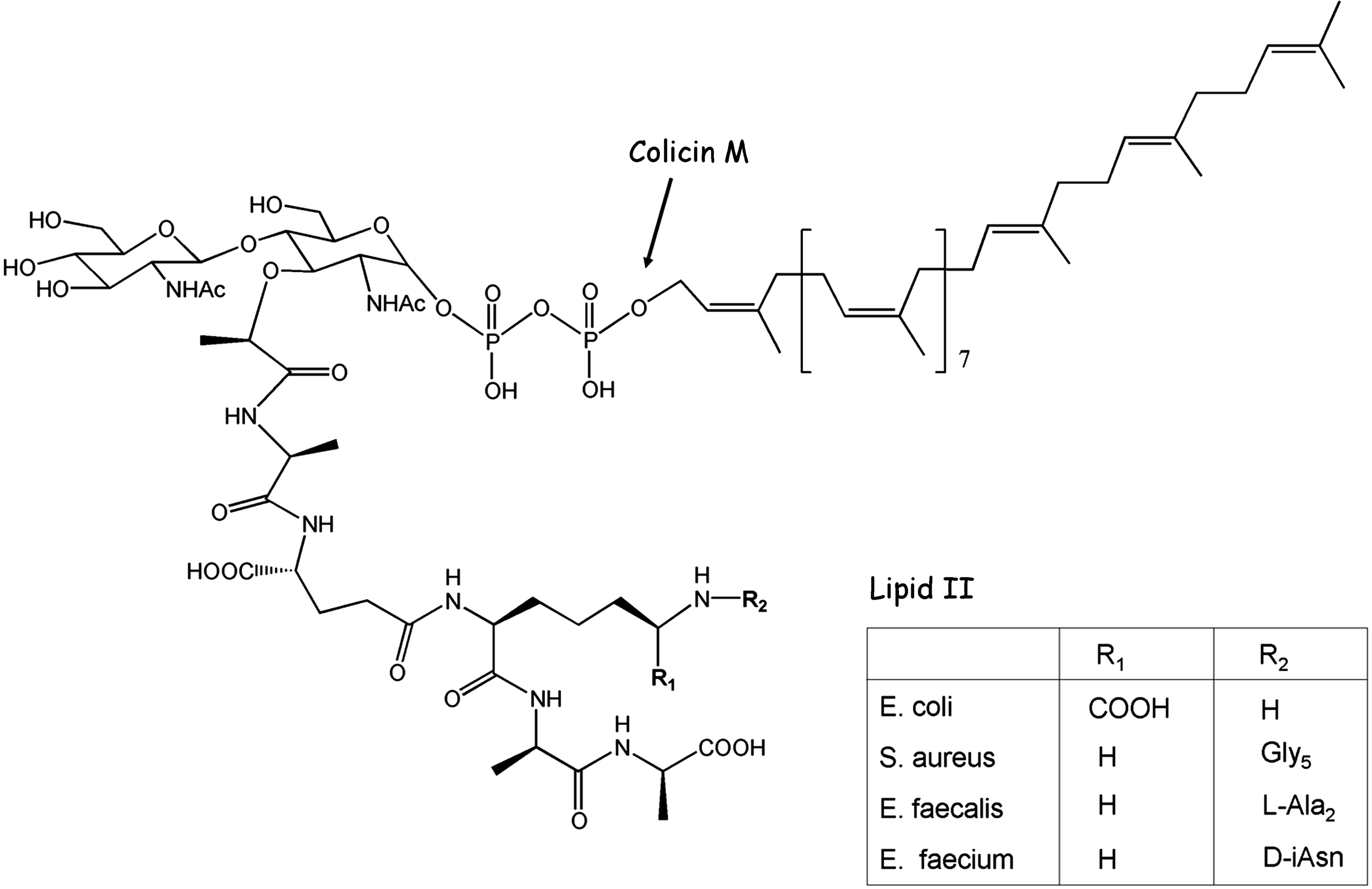
Chemical structure of lipid II. Main variations of lipid II peptide moiety encountered in the bacterial world are indicated (for more details, see reference 44).
Identification and Characterization of ColM Homologs
Genes encoding proteins that exhibit similarity to the C-terminal domain of E. coli ColM were identified in the genomes of Pseudomonas aeruginosa, Pseudomonas syringae and Pseudomonas fluorescens, Burkholderia spp., and Pectobacterium carotovorum species. 4 In all cases, these genes were not a part of the core genome, as they were not detected in all members of the species and could be mapped in the pathogenicity islands in some strains. For instance, the P. aeruginosa colM homolog was located within the 80-kb exoU-containing genomic island A, a large horizontally acquired genetic element and virulence determinant. 28 The P. syringae gene was specifically detected in the genome of the bacterial pathogen P. syringae pv. tomato strain DC3000, which causes bacterial speck disease to tomato and can also infect Arabidopsis and Brassica species. 12 In P. fluorescens, the colM homolog was identified by exploring genome diversity using a substraction method, among several genes contributing to the exceptional rhizosphere competence of D-genotype strains that colonize roots and suppress soil-borne diseases much more efficiently than others. 29 An alignment of the amino-acid sequences of ColM homologs revealed a significant identity principally in the C-terminal region, that is, in the activity domain in ColM. 34 From 35% to 45% sequence identity was observed in the latter region, which roughly comprises half of the proteins.4,5 In contrast, no homology was detected in the N-terminal regions, which presumably participate in receptor recognition and translocation.
The three Pseudomonas genes were cloned, and the encoded proteins (namely PaeM, PsyM, and PflM) were purified to homogeneity. In vitro assays confirmed that all of them cleaved lipid II at the position previously identified for ColM. The P. aeruginosa enzyme PaeM was the most efficient homolog, with a kcat/Km ratio for lipid II being about 600-fold higher than that of ColM. 4 These enzymes required Mg2+ for activity. As this is the case for colicins, ColM homologs have a narrow antibacterial spectrum. For instance, in spite of its high catalytic activity, PaeM did not show any cytotoxic effect toward E. coli cells, suggesting a failure for this toxin to parasitize the ColM receptor and import systems. However, purified PaeM was shown to affect the growth of 2 out of 14 tested P. aeruginosa strains, which included reference strains and clinical isolates. 4 The effective production of this bacteriocin by P. aeruginosa strains carrying the exoU genomic island gene was confirmed by Western-blot experiments using antibodies raised against PaeM. Its expression was greatly increased when the strains were treated with the fluoroquinolone antibiotic ciprofloxacin, demonstrating that PaeM production was under SOS control in P. aeruginosa (unpublished data), as observed for colicins in E. coli. 13
Mapping of ColM Functional Domains and Characterization of the Activity Domain
In spite of its small size, ColM was assumed to adopt the three-domain structural organization shared by all other colicins. A typical “TonB box” sequence (ETLTV) that is characteristic of class B colicins was found at the protein N-terminal extremity. Amino-acid substitutions in this motif abolished ColM import without altering binding to the FhuA receptor.23,34 Braun and coworkers also earlier reported the isolation of several spontaneous point mutations that affected ColM cytotoxicity but not its uptake.11,27,34
The X-ray structure of ColM, recently determined by Zeth and co-workers, 46 revealed a very compact fold lacking the well-individualized domains observed in all other colicins (Fig. 3). Nonetheless, ColM was tentatively divided into three functional regions: an N-terminal, 35-residue translocation region corresponding to an unstructured and flexible random coil region wrapping around the central domain, a receptor-binding domain delineated by residues 36 to 140, and a C-terminal activity domain (141 to 271). 46 Importantly, the predicted activity domain of ColM did not exhibit a significant similarity in sequence or folding with already known phosphatases.
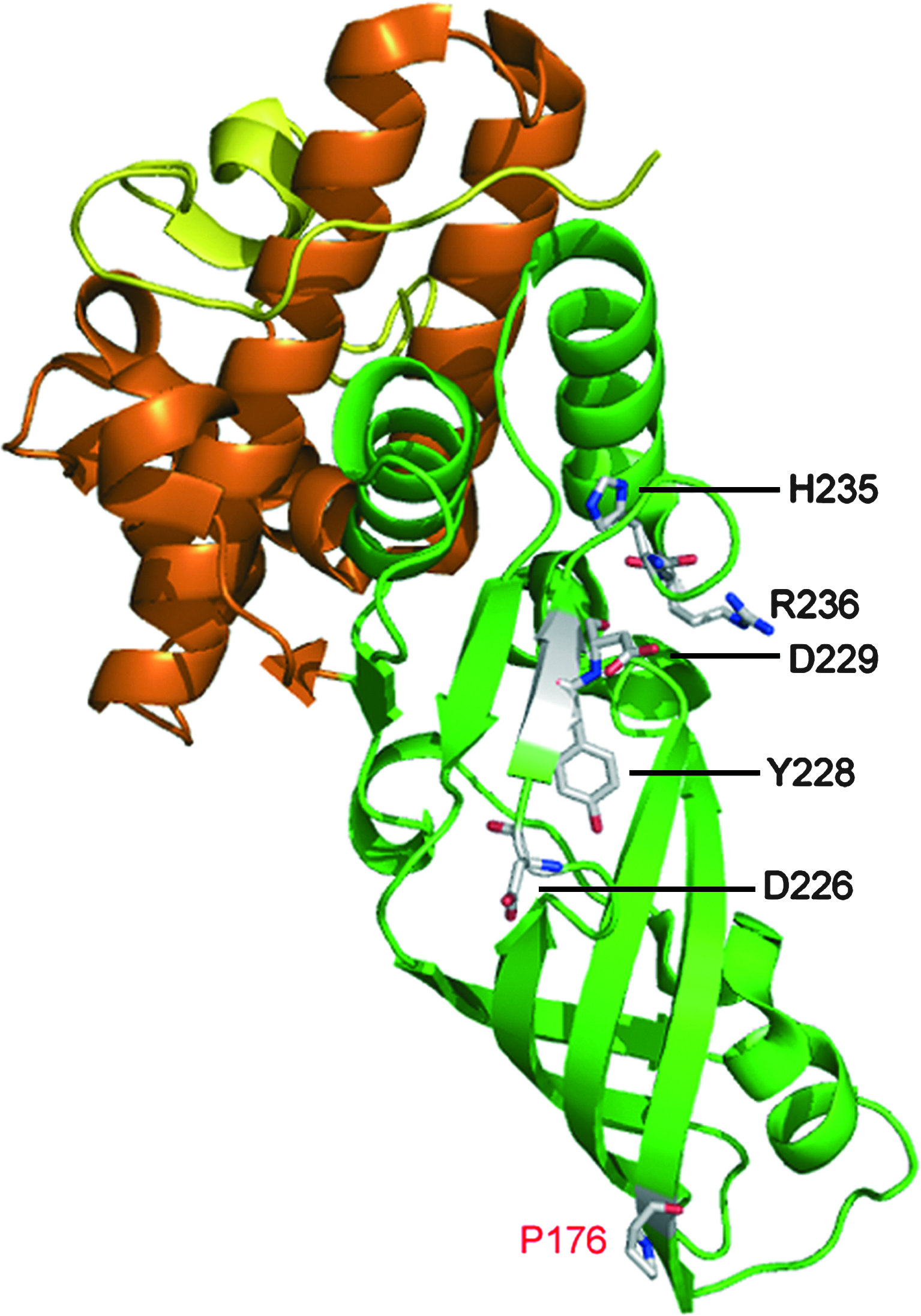
Three-dimensional structure of ColM. Based on structural data from Zeth et al. 46 and biochemical data from Barreteau et al., 5 the three functional domains of ColM are illustrated as follows: the N-terminal translocation domain (residues 1–35), the central receptor-binding domain (residues 36–123), and the C-terminal toxicity domain (residues 124–271) are shown in yellow, red, and green, respectively. Residues that likely belong to the enzyme active center (D226, Y228, D229, H235, and R236),5,23 and the P176 residue expected to be the main target of the FkpA chaperone/PPiase, 24 are indicated. The figure was prepared with the atomic coordinates deposited by Zeth et al. 46 (PDB accession code: 3DA4) by using the Pymol program (http://www.pymol.org).
The alignment of sequences of the ColM orthologs revealed several invariant charged residues in their C-terminal regions, whose role in the catalytic process was investigated by site-directed mutagenesis. 5 As judged by a strong effect of alanine replacement with regard to the ColM activity, five of these residues, D226, Y228, D229, H235, and R236, likely belonged to the active site. Indeed, both the cytotoxic effect on susceptible E. coli cells and the in vitro enzyme activity on lipid II of these mutant proteins appeared dramatically reduced or totally abolished. 5 A by-pass of the translocation process by osmotic shock further confirmed that the substitutions affected ColM catalytic efficiency. Significantly, these five residues formed a cluster at the surface of the ColM structure, 46 with their side chains protruding toward the exterior and positioned relatively far from each other, therefore, constituting an atypical, particularly extended, active site (Fig. 3), which might be relevant when considering the large size of the lipid II substrate (Fig. 2). Alanine replacement at five additional conserved positions (T200, R222, N231, E241, and T244) also affected ColM activity, but to a much lower extent. These residues that surrounded the central patch formed by the first group of critical residues might, thus, also be a part of the active site. Although the precise reaction mechanism of ColM still remains to be elucidated, the role of specific residues was inferred from the results of site-directed mutagenesis and from the catalytic mechanism of other phosphatases. The surface-exposed aspartate residues D226 and D229 could be involved in the coordination of the Mg2+ ion, which together with the R236 residue could then bind the negatively charged pyrophosphate group of lipid II. A subsequent nucleophilic attack of the phosphoester bond likely takes place, involving H235 or the essential D226/D229 residues.5,23
In order to precisely localize the activity domain of ColM, protease (chymotrypsin) accessibility assays and protein dissection experiments were performed. A series of truncated variants were purified and tested both in vitro (lipid II-degrading activity) and in vivo (cytotoxicity). To by-pass requirements for the FhuA receptor and the TonB/ExbB/ExbD machinery, these variants were artificially addressed to the periplasm, either imported from the external medium by using the osmotic shock method, or exported from the cytoplasm by using the Sec system.3,5 These experiments allowed the delineation of an independent toxicity domain which comprised exactly the C-terminal region that is highly conserved in the ColM homologs (residues 124–271). The minimal fragment required for full ColM catalytic activity (ColM-Δ7) 5 was 16.2 kDa in size (Fig. 3). It started at M122, that is, was by 18 residues larger than the catalytic domain predicted from the 3D-structure. 46 Interestingly, the in vitro activity of the truncated variant was by 45-fold higher than that of full-length ColM, indicating that the presence of the N-terminal translocation and reception domains hindered in some way, at least in vitro, enzyme activity. This observation also suggested that protein maturation may modulate activity in vivo.
Role of FkpA in ColM Activation
Analyses of the structure-activity relationships of ColM recently included an additional level of complexity, as the in vivo activity of the protein was found to depend on a specific periplasmic protein, FkpA, from the targeted cells. 25 The fkpA gene had been originally designated as tolM, as its inactivation led to a ColM tolerance phenotype. 41 FkpA is now characterized as a bifunctional protein carrying chaperone and cis/trans peptidyl-prolyl isomerase (PPiase) activities, each of these activities being supported by a distinct protein domain that remains functional when independently produced. 2 The use of different point mutations and truncated variants of FkpA showed that both functions of FkpA were required for ColM activation.3,25
Interestingly, the deletion of fkpA did not prevent cells from the toxic effects of the isolated catalytic domain of ColM (ColM-Δ7 construct addressed to the periplasm), contrary to what had been observed with the full-length ColM.3,5
A model for action of PPiases in protein folding has recently been postulated, in which the chaperone domain may initially bind with a low affinity to its non-native protein substrate and then transfer it to the PPiase site. 26 The data obtained are consistent with a scenario in which externally added ColM molecules, at least partially, unfold during their translocation through the outer membrane, and FkpA assists their refolding in the periplasm before they can exert their lipid II-degrading activity. A potential correlation between ColM activity and FkpA-mediated cis/trans isomerization of specific prolyl bond(s) of ColM was recently questioned by Helbig et al. 24 Mutations of four of the fifteen proline residues of ColM (P107, P129, P176, and P260) resulted in a dramatic loss of cytotoxic activity (1% to 10% residual activity as compared with the wild-type toxin). In fact, three of these mutations, P107A, P129A, and P260A, mainly affected toxin import, and only the fourth one, P176A, inactivated the phosphatase activity. Regardless of whether ColM and its four proline mutant derivatives were imported across the outer membrane or secreted across the cytoplasmic membrane, they still required FkpA to exert their activity in the periplasm. Using a novel in vitro assay consisting of measuring the cis/trans isomerization of synthetic fluorescent pentapeptides representative of ColM sequences around proline residues, these authors came to the conclusion that the main target of FkpA PPiase activity in the ColM sequence was the P176 residue, 24 which is located far from the predicted active center (Fig. 3). Interestingly, these experiments also suggested that the only cis-prolyl bond (F106-P107) detected in the X-ray ColM structure 46 was unlikely to be a site of FkpA cis/trans isomerization. Demonstration of the in vivo role of cis to trans isomerization in ColM activation is a difficult task, as the number of ColM molecules in the periplasm is very limited. The precise mode of action of FkpA, therefore, still remains to be clarified.
ColM Immunity Protein
Colicinogenic E. coli strains are protected against the toxin they produce by concomitant expression of a specific immunity protein. The ColM immunity gene cmi is found adjacent to the ColM activity gene cma on pColBM plasmids, and these two genes are transcribed in opposite directions. 30 Unlike the other enzymatic colicins, ColM is not released as a complex with its immunity protein. The latter protein, Cmi, composed of 117 amino-acid residues (13.8 kDa), remains anchored to the outer leaflet of the cytoplasmic membrane, that is, in the region where the toxin exerts its lipid II-degrading activity. It is expected to either inactivate the incoming toxin or interfere with its access to the target by a yet unknown mechanism. To get insight into the ColM inactivation mechanism, Cmi was recently purified, and its biochemical and structural properties were investigated in detail. To facilitate its purification and avoid the use of detergents, it was expressed with a C-terminal His-tag and without the N-terminal 23-amino-acid residues membrane anchor. 18 The Δ(1–23) truncated Cmi is functional, as it confers ColM immunity when addressed to the periplasm.18,19 The X-ray structure of Cmi Δ(1–23) revealed a compact α-β fold reminiscent of cystatins (cysteine protease inhibitors) and amine-oxidases. 18 A disulfide bridge linked the two cysteine residues of the protein (C31-C107). Interestingly, these two cysteines and several other residues were conserved in the sequence of several proteins of unknown function belonging to the YebF family, which exhibited 25%–35% overall sequence identity with Cmi. 18 As previously observed for YebF, 43 the abundance of Cmi in membranes was dramatically reduced under circumstances in which the formation of the protein disulfide bridge was inhibited, after the production of Cmi in a DsbA oxidase defective strain or cysteine to alanine substitution. 18 The disulfide bond likely plays a role in Cmi folding and stabilization, but is not absolutely required for immunity. The role of 23 Cmi residues appearing as invariants or highly conserved in the YebF protein family was also analyzed by site-directed mutagenesis. Impaired Cmi immunity activity resulted from modification of the residues Y101, E113, and Y114, which are clustered in the C-terminal region of the protein, very close to the disulfide bond. The critical role of this region was also confirmed by showing that the deletion of the last four protein residues totally abolished ColM immunity. 18
In contrast to what was generally observed with other colicins, 13 all attempts to demonstrate direct interactions and significant binding affinity between ColM and its immunity protein have failed.19,31 No inhibition of the lipid II-degrading activity of ColM was observed in vitro in the presence of a great excess of Cmi, suggesting that the immunity protein did not interact efficiently with ColM or its lipid II substrate. 18 Further work is, thus, required to identify by which mechanism Cmi confers ColM immunity.
Genes encoding ColM-like proteins, PaeM, PflM, and PsyM, in the genomes of Pseudomonas spp. are immediately followed by another gene of unknown function that could code for an immunity protein. 4 The sizes of these gene products (140–150 amino-acid residues) and of Cmi (117 residues) are similar, but sequence alignments did not reveal a significant similarity (15%–25% identity). Whether these proteins are related and exert a similar immunity role in the corresponding species remains to be demonstrated.
Substrate Specificity and Antibacterial Spectrum
In vitro assays showed that ColM hydrolyzed lipid intermediates I and II with quite similar efficiencies, showing that the GlcNAc group was dispensable for activity. 14 C55-PP was not cleaved, or only very poorly, suggesting that the substitution of the β-phosphate group by the MurNAc residue was critical for activity. The nucleotide precursor UDP-MurNAc-pentapeptide was not a substrate either, indicating that the enzyme was specific for lipid-linked peptidoglycan precursors. 14 ColM accepted lipid II homologs with lower-size lipid moieties (data not shown), a quite expected finding, in fact, when considering that this part of the substrate might be embedded in the membrane and not really accessible to the enzyme.
The fact that lipid II is essential, found in all types of bacteria, and specific of the bacterial world clearly opened the way toward a potential exploitation of ColM, and its orthologs, as broad-spectrum antibacterial agents. Two prerequisites should be fulfilled for ColM-mediated killing of organisms other than E. coli: (i) ColM should get access to lipid II at the outer side of the cytoplasmic membrane in the absence of the cognate transporter; (ii) ColM should hydrolyze heterologous lipids II despite interspecies structural variations. We have investigated the latter prerequisite by preparing various forms of lipid II from L-lysine-containing UDP-MurNAc-pentapeptide. The specific lateral chains of Enterococcus faecalis (di-L-alanine), Enterococcus faecium (D-isoasparagine), and Staphylococcus aureus (pentaglycine)7,8,42 (Fig. 2) were introduced onto L-Lys at position 3 by chemical synthesis, and the branched nucleotides thus obtained were converted into the corresponding lipids II by using MraY and MurG enzymes. Intermediate forms with one L-Ala, or one or three Gly, which exist in the E. faecalis or S. aureus peptidoglycan biosynthesis pathway,1,8 were also prepared. All of the branched lipids thus generated (Fig. 2) were hydrolyzed by ColM at approximately the same rate as the meso-diaminopimelate-containing lipid II found in Gram-negative bacteria. 33 This result established that the catalytic efficiency of ColM was not affected by the presence of a side chain, even bulky (L-Ala2, Gly5), at position 3 of the peptide stem. Thus, the diversity of peptidoglycan composition among bacterial species 44 does not constitute an impediment to the exploitation of ColM as a candidate for the development of nonconventional antibacterial agents.
Conclusion
The recent discovery of the specific mode of action of ColM has clearly renewed the interest for this particular colicin. Indeed, various aspects of ColM biology have been (re)investigated in detail since the past 5 years, with particular attention being paid to the functional characterization of this toxin using complementary biochemical and structural approaches. Although a great deal of information was thus generated, many questions still remain unanswered. A map of the ColM active center was tentatively drawn, which was based on the identification of several residues essential for enzyme activity. However, no crystal structure of this protein in complex with its lipid II substrate or analogs thereof was obtained, and the catalytic mechanism remains to be established. In addition, the mode of action of the bifunctional chaperone/PPiase FkpA protein, recently demonstrated to be essential for ColM activation, remains quite unclear. The X-ray structure of the immunity protein Cmi is now available, but the mechanism by which this protein protects cells from ColM is unknown.
Colicin M is unique among the colicins in that it interferes with peptidoglycan biosynthesis and provokes the lysis of susceptible cells. Pesticin, a bacteriocin produced by Yersinia pestis, which has a similar general structural organization and narrow antibacterial spectrum as colicins, was earlier shown to also target this cell-wall component. However, in contrast to ColM, pesticin does not interfere with the biosynthesis of this polymer but acts as a peptidoglycan hydrolase (muramidase).17,45
Colicins are known to exhibit a narrow range of killing activity, because they parasitize receptors and translocation machineries that are species specific. The N-terminal domains of E. coli and Pseudomonas “ColM orthologs” do not exhibit sequence similarity, likely reflecting the import specificities of these bacterial genera. These toxins are, therefore, expected to play a role restricted to modulation of the population dynamics among clones of the same species, but the real advantage their expression confers to the producing strains in terms of competition, growth, and virulence remains to be determined. As this is the case for bacteriophages, colicins have often been suggested as potential alternative agents that are recruited in the battle against pathogenic strains. Owing to its particular mode of action and capability to hydrolyze lipid II of different structures representative of peptidoglycan variability in the bacterial world, ColM could be considered an interesting candidate for such application.
Footnotes
Acknowledgments
This work was supported by grants from the Agence Nationale de la Recherche (PEPGLYCOL project, ANR-07-MIME-020), the European Community (FP6, COBRA project, LSHM-CT-2003-503-335), and the Centre National de la Recherche Scientifique.
Disclosure Statement
All authors report no conflicts of interest relevant to this article.