
Abstract
Cytotoxic NK/CD8+ T cells interact with MHC-I ligands on tumor cells through either activating or inhibiting receptors. One of the inhibitory receptors is CD94/NKG2A. The NK/CD8+ T cell cytotoxic capability is lost when tumor-associated human leukocyte antigen, HLA-E, binds the CD94/NKG2A receptor, resulting in tumor progression and reduced survival. Failure of cancer patients to respond to natural killer (NK) cell therapies could be due to HLA-E overexpression in tumor tissues. Preventing the inhibitory receptor–ligand interaction by either receptor- or ligand-specific monoclonal antibodies (mAbs) is an innovative passive immunotherapeutic strategy for cancer. Since receptors and ligands can be monomeric or homo- or heterodimeric proteins, the efficacy of mAbs may rely on their ability to distinguish monospecific (private) functional epitopes from nonfunctional common (public) epitopes. We developed monospecific anti-HLA-E mAbs (e.g., TFL-033) that recognize only HLA-E-specific epitopes, but not epitopes shared with other HLA class-I loci as occurs with currently available polyreactive anti-HLA-E mAbs. Interestingly the amino acid sequences in the α1 and α2 helices of HLA-E, critical for the recognition of the mAb TFL-033, are strikingly the same sequences recognized by the CD94/NKG2A inhibitory receptors on NK/CD8+ cells. Such monospecific mAbs can block the CD94/NKG2A interaction with HLA-E to restore NK cell and CD8+ anticancer cell cytotoxicity. Furthermore, the HLA-E monospecific mAbs significantly promoted the proliferation of the CD4−/CD8+ T cells. These monospecific mAbs are also invaluable for the specific demonstration of HLA-E on tumor biopsies, potentially indicating those tumors most likely to respond to such therapy. Thus, they can be used to enhance passive immunotherapy once phased preclinical studies and clinical trials are completed. On principle, we postulate that NK cell passive immunotherapy should capitalize on both of these features of monospecific HLA-E mAbs, that is, the specific determination HLA-E expression on a particular tumor and the enhancement of NK cell/CD8+ cytotoxicity if HLA-E positive.
Introduction
Active specific and passive immunotherapies of human cancers aim to reprogram the metabolic profile of the tumors and their microenvironment during the progression of cancers. There is a need to identify and detect the specific biomarkers of the metabolic profile of a tumor or an immune cell on an individual patient basis. Biomarkers, whether composed of proteins or polysaccharides or lipids, can be monomeric, homodimeric, or heterodimeric. Monoclonal antibodies (mAbs) are indeed the most valuable tools for their specific immunodiagnosis. Furthermore, biomarkers may contain common (public or shared) epitopes in addition to specific and individual or unique epitopes. Therefore, it is critical to examine whether a mAb is genuinely monospecific for a biomarker, by demonstrating reaction only with private epitopes as opposed to shared epitopes.
Although cellular immunotherapy has successfully explored antitumor CD8+ T cell responses, it is increasingly realized that human cancers reprogram by developing strategies to escape T cell recognition. Counter strategies are needed to upregulate killer T cells as well as manipulate natural killer (NK) cells to attack the cancer cells. The NK cells share expression of cell surface antigens and effector molecules, such as CD2, CD7, CD90, perforin, granzyme A, and interferon-γ, with T lymphocytes on one hand, and the surface antigens such as CD11b and CD11c with myeloid cells on the other, based on specific genes required for T cell receptor (TCR) rearrangement.(1) The 112 genes for cell surface and signaling receptors and molecules are found to be significantly upregulated in NK cells,(2) and such upregulation may exhibit both inter- and intraindividual variations during various stages of tumorigenesis, metastasis, and infections. Therefore, personalized therapy should precisely monitor these variations in the molecular expression of such biomarkers. Indeed, precise monitoring requires well-defined and precise tools.
The NK cell receptors of the immunoglobulin (Ig) superfamily (e.g., killer cell Ig-like receptors, also known as KIRs and Ig-like transcripts) recognize human major histocompatibility complex (MHC) class-I molecules (human leukocyte antigens [HLAs] -Ia, -Ib, and MICA/MICB).(3–6) Some of these receptors transmit activating signals, whereas others mediate inhibition. One major class of genes controlling both activating and inhibitory NK cell receptor groups (NKGs) is Klrc1 [NKG2A], Klrc2 [NKG2C], and Klrk1 [NKG2D] and are involved in specific interactions with the MHC of tumor cells and virally infected cells. Each NKG2 subunit is a type II glycoprotein belonging to the C-type lectin superfamily, with an extracellular domain with transmembrane and cytoplasmic segments. The analogous murine Ly49 family receptors(5) and human KIR(3,4) both bind to cell surface HLA-Ia molecules on target cells. Natural killer cell group 2 (NKG2) receptors interact specifically with HLA-Ib and MICA/MICB as the homodimer (NKG2D) or as heterodimers (NKG2A, NKG2C), and such interactions are highly conserved across species. Most of the NKG2 isoforms (NKG2A, B, C, E, and H, but not NKG2D) form disulfide-linked heterodimers with an invariant chain, CD94.(7,8) NKG2A and NKG2B, which are alternatively spliced products from a single gene, have two immunoreceptor tyrosine-based inhibitory motifs in their cytoplasmic domains and form inhibitory receptors when complexed with CD94.(2,7) The specific ligands for CD94/NKG2A/C heterodimers are identified as nonclassical class-I molecules (HLA-Ib) in humans.(8–10)
The homodimer NKG2D interacts with MICA on tumor cell surface
NKG2D is a unique activating receptor of NK cells that share little similarity with the other isoforms of NKG2 receptors and does not associate with CD94. The receptor is on natural killer T cells (NKT) cells, subsets of γδ T cells,(2) activated macrophages,(11) and naive human CD8+ T cells. CD4+T cells can be induced to express it under certain pathological conditions, such as Crohn's disease, juvenile-onset lupus, and cytomegalovirus infection.(12) NKG2D recognizes highly polymorphic MHC loci MICA (60 allelic variants) and MICB (30 allelic variants), also known as “stress-induced MHC” on normal cells,(13,14) tumor,(15) and virally infected cells.(16) NKG2D–MICA interaction leads to tumor cell destruction, mediated by the release of perforin by the NK and other immune cells.(11) NKG2D is also to recognize other protein ligands, encoded by genes that encode functional proteins [in square brackets] in humans (RAET1E [ULBP4], RAET1G [ULBP5], RAET1H [ULBP2], RAET1I [ULBP1], RAET1L [ULBP6], and RAET1N [ULBP3]).(17)
The interaction of the activating NK cell receptor NKG2D with tumor cell surface MICA and MICB has been extensively studied(18–29) in gastrointestinal (GI) epithelium and several epithelial tumors. The extracellular transport of MICA and MICB is independent of the general peptide processing machinery that is required for the assembly of peptides for other antigen-presenting HLA-I molecules.(13–18) The MICA/B proteins share ∼30% identical amino acid residues throughout α1, α2, and α3 domains with other HLA class-I proteins. They possess seven to eight N-linked glycosylation sites, unique transmembrane and cytoplasmic tail sequences, and three extra cysteine residues in the α1 and α3 domains. Unlike HLA class-I proteins, they do not dimerize with β2-microglobulin (β2m).
Tumor cells may escape NKG2D–MICA-mediated immune attack by disulfide–isomerase-enabled proteolytic degrading and shedding of MICA.(18) Ferrari de Andrade et al.(19) have designed antibodies targeting the MICA α3 domain, the site of proteolytic cleavage for shedding, and found that these antibodies prevented loss of cell surface MICA and MICB in human cancer cells. Interestingly, these antibodies do not inhibit binding of the α1 and α2 domains with receptors and allowed continued NKG2D–MICA interactions to inhibit murine tumor progression in mouse models and reduced human melanoma metastases in a humanized mouse model. However, in some cancers at the advanced stage, the expression of ligands for NKG2D may promote tumor progression rather than regression. A recent report documents NKG2D activation-mediated tumor progression in a model of inflammation-driven liver cancer,(20) with the critical differentiating factor purported to be the surrounding inflammation. Hence, the nature of the microenvironment within and surrounding tumors may have the potential of altering the impact of NKG2D activation, and this aspect deserves attention while studying the impact of NKG2D interaction with MICA and other such ligands on the tumor cell surface.
In recent years, NKG2D–MICA ligand interactions in different cancers have been reviewed extensively.(21–25) The crystal structure of MICA shows restructured α1 and α2 helices, the regions interacting with NKG2D, and there is altered folding with a shallow remnant of a peptide-binding groove without a peptide.(26–28) The interaction between deglycosylated recombinant MICA and NKG2D shows that the NKG2D can bind to the presumptive peptide grooves of α1 and α2 domains at the top of the MICA platform, analogous to αβTCR recognition of MHC class-I proteins.(29) Since there are 60 allelic variants of MICA proteins, remarkable variations may occur in the α1 and α2 domains affecting binding of NKG2D with the MICA alleles.(30) However, it is also suggested that the NKG2D binding sites are on the underside of the MICA α1 and α2 domains, this region is comparable with the cryptic domain in classical and nonclassical HLA-I proteins masked by β2m. This domain in classical and nonclassical HLA-I loci contains mostly shared or public epitopes.(31)
Cancer-Associated HLA Class-I Molecules and NKG2A/CD94
As noted previously, NKG2 receptors different from NKG2D may mediate NK suppression of tumor killing rather than activation. Earlier studies reported that NK cells can kill autologous and allogeneic tumor cells without involving the MHC.(32) Lymphomas deficient in MHC antigens were rejected in vivo in contrast to MHC-expressing lymphomas.(33) Such loss of MHC expression failed to activate inhibitory NKG2 receptors (other than the activating NKG2D).
Similarly, Liao et al.(34) observed that the normal T cell blasts from MHC-I-deficient mutant mice are effectively targeted by NK cells in vitro, a finding that supports the speculation that HLA class-I expressed on regular T cell blasts may result in inhibition of NK-mediated cell killing. Furthermore, an inverse correlation was noted between in vitro NK-mediated cytolysis and the level of target cell HLA class-I expression.(35) However, no such correlation was noticed with target cell HLA class-II molecules.(35)
Moretta et al.(36) have studied the interaction between the CD94 receptor and HLA class-I. The cells expressed HLA-B7 protected target cells from NK lysis. The protection was abolished by CD94-specific mAbs (XA-185) but not by the mAbs conjugated with the soluble CD94 molecule. The CD94-specific mAbs (XA-185) did not react with the inhibitory NKG2A receptor, as it failed to stain COS-7 cell lines (fibroblast-like cell lines) transfected only with NKG2A but stained those transfected with both CD94 and NKG2A. In contrast, yet another mAb Z199, which does not recognize CD94, strongly reacted with COS cells cotransfected with CD94/NKG2A,(37) suggesting the occurrence of heterodimerization of NKG2A with CD94. Contrastingly, the same group documented the inhibitory role of CD94/NKG2A in a predominantly CD8+ fraction of human T lymphocytes.(38) In the presence of IL-15, the CD8+ T cells expressed CD94 de novo. CD94 expression commenced 4–6 days after addition of IL-15. Both CD4+ and CD8+ cells expressed CD94, but the simultaneous expression of NKG2A occurred only with CD8+ cells. Similar data were obtained in T cell populations activated in mixed lymphocyte cultures in the presence of IL-15. The expression of CD94/NKG2A diminished the allospecific cytolytic activity by mixed lymphocyte culture-derived T cell populations. Importantly, the cytolysis was reinstated by adding anti-CD94 mAbs, which masks the inhibitory NK cell CD94. These and similar observations(39) confirmed that the binding of CD94/NKG2A with HLA class-I on tumor cells protected them from cytotoxic killing by both NK cells and a subset of CD8+ T cells, although the precise mechanism remained to be clarified.
Tumor-Associated HLA-E as the Specific Ligand for CD94/NKG2A
HLA class-I molecules include highly polymorphic classical HLA class-Ia (HLA-A, -B, and -C alleles) and the least polymorphic nonclassical HLA class-Ib (HLA-E, -F, or -G). Literature increasingly documents that HLA-E may function as the specific ligand for the inhibitory NKG2 receptors. The fundamental structure of HLA-E is not much different from any one of the HLA class-Ia molecules (Fig. 1A). Interestingly, the HLA-E heavy chain shares several amino acid sequences with HLA class-Ia, despite several unique differences in the amino acid sequences or epitopes between HLA-E and HLA-Ia alleles (Tables 1 and 2). HLA-E functions by assembling with the leader peptide of HLA class-Ia/-Ib proteins as its cognate peptide.(40) The leader peptide is loaded at the N terminus of the newly synthesized HLA class-I molecule. The role of leader peptide is to ensure translocation of the newly synthesized HLA class-I heavy chain into the endoplasmic reticulum (ER) and promote the transportation of the HLA across the ER. Upon completion of the task, the leader peptide is cleaved from the heavy chain of HLA by a signal peptidase(41) and enters the cytoplasm. From there, the leader peptide itself must be transported across the ER membrane, which requires regular transporter associated with antigen processing (TAP) expression and function. Normal maturation and insertion of HLA-E into the cell surface membrane requires the leader peptide of HLA class-I molecules or leader peptides of HLA-F or HLA-G (but not the leader peptide of HLA-E itself) to bind the HLA-E peptide grove in the ER. A newly synthesized HLA-E matures and forms a stable complex only in the presence of the leader peptide of most HLA-A, -B, -C, and -G molecules, but not its own leader peptide.(40)
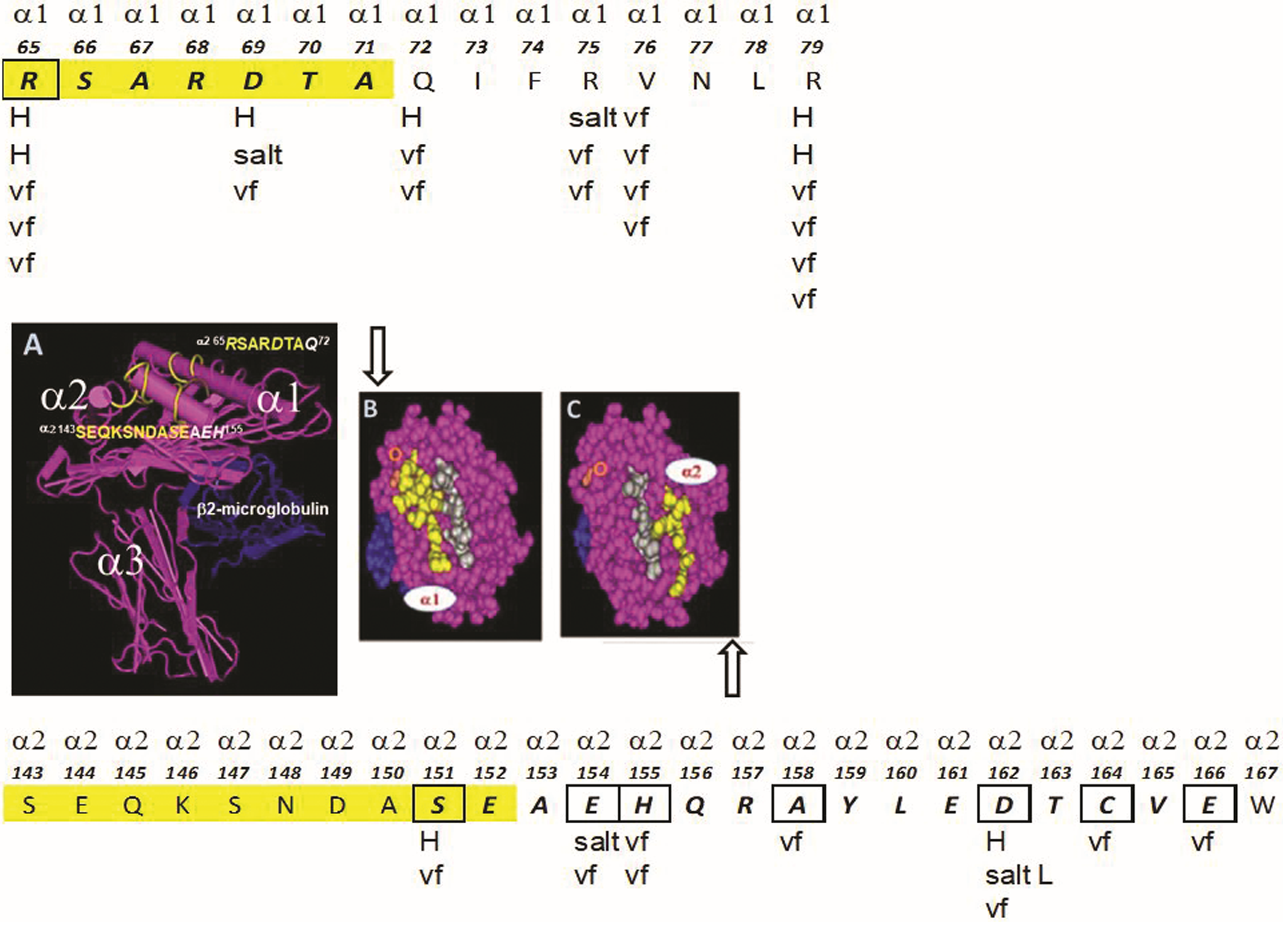
The structure of the nonclassical (HLA class-Ib) human leukocyte antigen HLA-E.
Gene Sequence-Based (First Row of Numbers) and Secreted Heavy Chain-Based Amino Acid Sequences (Second Row of Numbers) of Nonclassical HLA-Ib, HLA-E Showing Private (Monospecific) and Public (Polyspecific) Epitopes in Boxes
Amino Acid Sequences (Epitopes) Shared with Classical HLA-Ia Loci (HLA-A, HLA-B, HAL-Cw, and Nonclassical HLA-Ib Loci (HLA-F and HLA-G)
HLA-E-restricted epitopes are shown in bold italics with (*).
HLA-E is expressed at the cell surface only if a suitable leader peptide from specific class-Ia or class-Ib alleles (other than HLA-E itself) associates as the cognate peptide of a complete HLA trimer. Some viral peptides (Table 3) can also fulfill this function. These leader peptides should be present in the ER at sufficient levels to ensure expression of a significant amount of trimeric HLA-E on the cell surface. Thus HLA-E is highly evolved to bind specifically to the class-I leader peptide.(41) The class-I leader peptide or microbial (cytomegalovirus, human immunodeficiency virus, Hep-C, and others) peptides may not be available when tumor cells and virally infected cells have downregulated HLA-I expression.(42) Furthermore, viruses can inhibit the function of TAP(43,44), which is required to transport leader peptides, and cleaved from nascent class-I molecules, from the cytoplasm into the ER. Under these circumstances, intact HLA-E may not be expressed on the cell surface, permitting NK lysis of the tumor cell or virally infected cell.(41)
Sources of Peptides and Their Sequences Presented by HLA-E During Interaction with CD94/NKG2
Sequences: A, alanine; F, phenylalanine; G, glycine; I, isoleucine; L, leucine; M, methionine; N, asparagine; P, proline; S, serine; R, arginine; T, threonine; tyrosine; V, valine.
CMV, cytomegalovirus; NKG2, natural killer cell group 2.
Two independent groups(10,45) have validated the finding that HLA-E functions as the specific ligand for CD94/NKG2A receptors. The absence of binding between CD94/NKG2A and classical HLA-Ia molecules was confirmed(46) by the direct binding of the soluble receptor and functional assays with CD94/NKG2A-positive NK cells. Braud et al.(10) constructed in vitro the phycoerythrin-labeled biotinylated tetrameric complexes of the heavy chain of HLA-E and β2m with a synthetic peptide (VMAPRTVLL) derived from the signal sequence shared by HLA-B allelic proteins. The peripheral blood mononuclear cells from nine regular donors stained for the HLA-E tetramer. An HLA-A2 tetramer with an Epstein–Barr virus peptide was used as a negative control. The HLA-E tetramer bound to 2%–11% of lymphocytes from different individuals, whereas the HLA-A2 tetramer bound to 0% and 0.8% of the lymphocytes. On average, an HLA-E tetramer bound to ∼57% (varied from 35% to 83% among individuals) of CD3−/CD56+ NK cells, while the HLA-E tetramers bound to 12%–60% of CD3+ T cells (some were CD56+). Most importantly, the HLA-E tetramer did not bind to the lymphocytes when the cells were pretreated with the anti-CD94 mAb HP3D9. The interaction between HLA-E tetramers and CD94 was further confirmed by staining a number of well-characterized CD94+ NK-cell clones with HLA-E tetramers and demonstrating that another anti-CD94 mAb DX22 completely abolished this binding. The HLA-A2 tetramer failed to stain the CD94+ NK cell clones.
Lee et al.(45) provided further experimental proof for the functional dynamics of the interaction between HLA-E and the NK inhibitory receptors CD94/NKG2A. The validity of their observation is based upon using two different HLA-I-deficient LCL 721.221 target cells, one expressing cytoplasmic but not cell surface HLA-E (HLA-E*0101 allele), and the other, LCL.221-AEH cells, that do express cell surface HLA-E (HLA-E*0101 allele). Besides, they used LCL 721.221 cells transfected with HLA-B*0702, B*2705, C*0401, C*0302, and B*5101 as controls. Using an effector NK cell line (CD94/NKG2A positive but negative for Ig superfamily or killer cell receptor), the effects of the presence or absence of HLA-E on the surface of LCL721.221 cells were examined. NK cell-mediated cytolysis was inhibited only when HLA-E was present on the surface of the target cells (LCL.221-AEH) but not when it is cytoplasmic (LCL 721.221). Total restoration of lysis was observed after the addition of various mAbs, including anti-HLA-E “specific” mAb 3D12 (IgG1), pan-HLA-I mAb HP-1F7 (IgG1), anti-CD94 mAb HP-3B1 (IgG2a), or anti-CD94/NKG2a mAb (Z199, IgG2b), but not with IgG1 negative control mAb. The results confirm that the inhibition of lysis was specific both for HLA-E, because protection was reversed by the 3D12 mAb, and for the CD94/NKG2A complex, because cytotoxicity was reconstituted by CD94 (HP-3B1) or CD94/NKG2A (Z199)-specific mAbs.
The results confirm the importance of HLA-E but highlight the fact that mAbs binding can abrogate the binding of NK-associated CD94/NKG2A to the extent of restoring the cytolytic capabilities of the NK cells. The observations had potential merit since a specific cell line devoid of HLA-I but incorporated explicitly with cell surface HLA-E was used. However, it is neither established whether the mAb 3D12 is capable of binding to the amino acid ligands of CD94/NKG2A nor whether mAb 3D12 is monospecific for the two well-known alleles of HLA-E (HLA-EG107 and HLA-ER107). Such in-depth analysis of monospecificity of anti-HLA-E mAbs is a prerequisite for using of anti-HLA-E mAbs for passive immunotherapy either in a preclinical model or for human trials.
We examined(47) the HLA-I binding affinity of mAb 3D12 using the Luminex single antigen bead (SAB) assay with beads coated with 30 HLA-A, 50 HLA-B, 16 HLA-Cw, 2 HLA-E, 1 HLA-F, and a couple of HLA-G alleles. We found that the mAb 3D12 recognized not only HLA-E but also several HLA-Ia alleles; hence, that the mAb 3D12 is not specific for HLA-E. Therefore, a mAb that is monospecific for HLA-E and at the same time capable of binding to the amino acid ligands of CD94/NKG2A on the HLA-E molecule will be potentially useful to restore cytotoxic killing of tumor cells as well as for the specific recognition of tumor cells expressing HLA-E on the cell surface, potentially being amenable to such therapy.
Specific Amino Acids on α1 and α2 Helices of HLA-E as Ligands for CD94/NKG2A
The disulfide-liked CD94/NKG2A dimers “sit across” the peptide-bound cleft of carboxy-terminal end of HLA-E, precisely interacting with α1 and α2 helices of HLA-E, respectively, as shown in Figure 1A. Petrie et al.(48) have critically analyzed the electrostatic surfaces of HLA-E, carrying the leader sequence peptide of HLA-G namely VMAPRTLFL in the α1 and α2 helical groove and CD94-NKG2A. They showed that a basic region on the α1 helix of HLA-E interacted with an acidic domain on CD94 and, contrariwise, an acidic region on the HLA-E α2 helix docked with a basic domain on NKG2A. This study is restricted to the leader sequence peptide of HLA-G (residues 3–11). However, several other leader peptides sequences of HLA-Ia are known to bind to the α1 and α2 helical groove of HLA-E (Table 3). Whether the strength of acid–base interaction of NKRs and HLA-E remains the same for all these leader peptides is not clear at present, although it is assumed to be similar. However, although all HLA-I leader sequence peptides tested bound to HLA-E and were recognized by CD94/NKG2A, amino acid variations in the leader sequences affected the stability of HLA-E. It appears that CD94/NKG2A recognition of HLA-E is controlled by the degree of stabilization of peptide with HLA-E and their cell surface expression, and both HLA-E heavy chain and cognate peptide form the ligand for CD94/NKG2A. However, a comparison of the CD94/NKG2A–HLA-E VMAPRTLFL complex with the unligated CD94-NKG2A(49) and HLA-E(50) revealed no significant conformational change in either HLA-E or CD94-NKG2A upon complex formation.
According to Petrie et al.,(48) the “lock and key” engagement between HLA-E VMAPRTLFL and CD94-NKG2A exemplified the “innate characteristic” of this interaction. The binding of CD94-NKG2A heterodimer with HLA-E involves 8 salt linkages and 19 H bonds (Table 4 and Fig. 1B, C). Besides, several van der Wall interactions are found between the heterodimer and the HLA-E (Table 4). CD94 shows greater association with α1 helix than NKG2a with the α2 helix. The CD94-NKG2a “footprints” on HLA-E was further confirmed by studying structural data, and mutating the amino acids of CD94, NKG2a, and HLA-E involved in the binding.
Amino Acid Interactions Between NKG2A/CD94 and HLA-E with Leader Peptide from HLA-G
Modified from Petrie et al.(48)
The interaction between the structures of the CD94-NKG2A and HLA-EVMAPRTLIL was compared with the αβTCR–HLA-E complex(51) and that of NKG2D–MICA.(20,23) The position of the CD94-NKG2A footprint on HLA-E was like that HLA-E–restricted TCR (KK50.4); however, in the KK50.4TCR interaction with HLA-E, both heavy chains of the TCR were involved equally in the binding. Similarly, the proximity of the α1 and α2 helices of MICA enabled each chain of NKG2D homodimer to interact with both helices.(13,23) While comparing the HLA-E binding of the inhibitory receptor CD94-NKG2A with activating receptors CD94-NKG2C, differences in the binding affinities were observed. Both NKG2A and NKG2C show remarkable sequence differences and the binding affinity of NKG2C with HLA-E is found to be much lower than that of NKG2A.(50,52,53) All these studies fundamentally clarify a critical aspect of HLA-E function, namely that there are HLA-E-specific epitopes involved in the interaction with NK or TCRs, most importantly the HLA-E-specific amino acid sequences found in the α1 and α2 helices of HLA-E. Table 4 compares the unique amino acid sequences of HLA-E-specific epitopes with other HLA class-I molecules.
Background and the Central Hypothesis
HLA class-I molecules, which includes HLA-E, are heterodimers consisting of a heavy chain polypeptide with β2-β2m with or without a peptide in the grove. The heavy chains consist of α1, α2, and α3 helices. It is well known that HLA class-I loci include HLA-A, (with 3913 alleles and 2747 proteins), HLA-B (4765 alleles and 3465 proteins), HLA-Cw (3510 alleles and 2450 proteins), and least polymorphic HLA-E, HLA-F, and HLA-G. The genes are located on chromosome 6 (6p21.31). The heavy chain of HLA-I shares several commonly shared amino acid sequences (epitopes) and a very few specific amino acid sequences. Several shared sequences are cryptic in the heavy chain due to heterodimerization with β2m. When a trimeric HLA used as an immunogen either in animal models or when used as autologous or allogenic vaccines in patients, the shed immunogen rarely remains intact during immune recognition. Both the shed heavy chain and the β2m are recognized independently.
Consequently, upon immunizing HLA-E heavy chain in three mice, we obtained 258 mAbs. Although most of them were recognized as shared sequences and polyreactive, 31 mAbs were monospecific.(54) We hypothesize that only the monospecific anti-HLA-E mAbs that are monospecific for HLA-E are not only useful for specific recognition of HLA-E on tumor cells and tissues but also can serve to prevent tumor escape from NIK cell killing by blocking by the interaction of the inhibitory NK cell CD94-NKG2A with tumor expressed HLA-E. It is critical and essential that the therapeutic anti-HLA-E mAbs generated should recognize unique HLA-E-restricted epitopes but no other epitopes are public and shared by other HLA-Ia or HLA-Ib molecules.
Polyreactive HLA-E mAbs
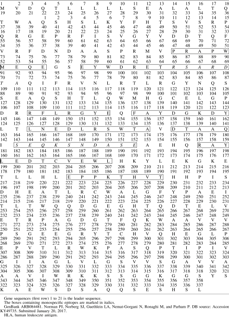
Table 5 summarizes the literature on the commercial anti-HLA-E mAbs used for identification of HLA-E on normal and tumor tissues, and on tissues were monitored during tumor progression and metastasis. However, the commercial mAbs such as MEM-E/02, MEM-E/06, MEM-E/07, and MEM-E/08,(55–60) 3D12,(61) and others listed in Table 5 failed to comply with the stringent criteria of monospecificity for HLA-E, based on the binding of these mAbs to other HLA class-I antigens. This finding is well illustrated in Table 6, which shows how the most commonly used anti-HLA-E mAb MEM-E/02 and mAb 3D12 react with HLA-A, HLA-B, and HLA-Cw molecules coated on a solid matrix in the form of SABs. Each bead is coated with one of the following HLA molecules: 31 HLA-A, 50 HLA-B, and 16 HLA-Cw. Two essential and specific publications(47,62) on these two mAbs clarify the notable findings in greater detail. The possible epitopes recognized by MEM-E/02 were tested at 1/1200 dilution of the mAb and by dosimetric peptide inhibition.(47) Since MEM-E/02 bound to several HLA-I molecules, we have selected the epitope present in the heavy chain of HLA-E, which are most commonly shared by several HLA-I molecules, as summarized in Table 2. They are 115QFAYDGKDY123, 137DTAAQI,142, and 126LNEDLRSWTA135. Interestingly, these shared peptides remain cryptic in intact trimeric native HLA-I (Fig. 2A), but will be exposed in β2m-free HLA heavy chain (Fig. 2B), well recognized as open conformers. Figure 2 illustrated in a prior publication(47) showed that the binding of MEM-E/02 to the HLA-Ia molecules coated on the LABScreen HLA-Ia SAB beadsets † is selectively inhibited by two of the shared peptides (DTAAQI [48%] and QFAYDGKDY [24%]) (Fig. 2C, D), but not inhibited by another commonly shared peptide (LNEDLRSWTA), which served as a potential control representing a shared but noninhibitory peptide. The inhibition is further confirmed by dosimetic analysis as shown in Figure 4 of the report.(47) Recently, Tremante et al.(60) tested the HLA-I binding of MEM-E/02 on Western blots using the purified heavy chains (NP40 lysates) from the cell lines that express HLA-A*11:01, HLA-B*35, HLA-Cw*04:01 (CJO), Cw*05:01 (221 C5), and HLA-Cw*07:01(221 C7).(60) Indeed, MEM-E/02 bound to A*11:01, B*35, Cw*04:01, Cw*05:01, and Cw*07:01 as shown in our report.(47) However, the authors contended that the MEM-E/02 binding to HLA-Ia antigens on Western blots is not as intense as the staining of HLA-E heavy chains. We have also reported(47) that MEM-E/06, E/07 and E/08 were also cross-reactive to several other HLA class-I molecules and hence none of the MEM categories of mAbs are HLA-E monospecific.
Structural variants of HLA class-I molecules.
Documention Cell Surface Expression of HLA-E on Human Cancers: Monoclonal Antibodies Used Include MEM-E/02, MEM-E/06, MEM-E/07, MEM-E/08, 3D12, 3H2679, and TFL-033
mAbs, monoclonal antibodies.
Anti-HLA-E mAbs Reported To Be “Specific” for HLA-E (MEM-E/02 and 3D12) Cross-React with Several HLA-Ia Molecules
The mAbs were diluted 1/100. Only those alleles with MFI above >1000 are presented. The mAbs were tested on Luminex SABs assay using regular LABSCreen beadsets (Lot-7) and with iBeads. The regular beadsets are coated with both β2m-associated, peptide–associated, or peptide-free heavy chain (β2aHC) with admixture of β2m-free heavy chains of HLA (β2fHC).(63)
β2m, β2-microglobulin; MFI, mean fluorescent intensity; SABs, single antigen beads.
Most interestingly, none of these MEM series antibodies bound to HLA-I antigens coated on a modified version of the regular beads called iBeads. The regular beadsets provided for the Luminex assay by the manufacturer (One Lambda, Inc., Canoga Park, CA) are coated not only with intact trimeric HLA class-I molecules but also contain free α-heavy chains of HLA class-I without β2m and/or the cognate peptide(63) (Fig. 2B). Realizing the existence of the monomeric and dimeric HLA-I variants in addition to intact trimeric HLA molecules on the beadsets, the manufacturer developed iBeads, provided as felix beads for in-house (at Terasaki Foundation Laboratory [TFL]) experimental use. Although the iBeads have been well investigated in clinical laboratories(63–70) and proven to be specific for containing only intact trimeric HLA-I molecules (Fig. 2A), their commercial production was abandoned as it was not cost-effective for commercial use. The iBeads carried only the intact trimeric HLA. These iBeads are produced by proprietary enzymatic treatment of regular HLA-Ia antigen coated microbeads to remove or reduce the amount of free heavy chains (also referred to as “denatured antigens”) by the manufacturers. Most interestingly, the mAb MEM-E/02 failed to bind to any one of the HLA molecules coated on the 97 different iBeads carrying HLA class-I alleles. This finding suggests that MEM-E/02 is not capable of reacting to intact cell surface HLA-E or HLA class-I molecules (Fig. 2A), but binds only to the heavy chain open conformers lacking association with β2m and/or peptide (Fig. 2B). We have further examined this issue with another mAb3D12,(62) considered to be specific for HLA-E,(61) by measuring the mean fluorescence intensity (MFI) with regular SABs. The results showed that 3D12 simulated MEM-E/02 in recognizing several HLA-B and HLA-C antigens. As observed with MEM-E/02, binding of 3D12 to HLA-E is inhibited by the public peptide sequences QFAYDGKDY and DTAAQI, shared by other class-Ia and class-Ib antigens. Furthermore, a decrease in binding of mAb 3D12 to HLA class-Ia SABs after heat treatment supports the contention that the epitope is located at the outside of the “thermodynamically stable” α-helix conformations of HLA-E. Therefore, we emphasize that reliable immunodiagnosis of HLA-E on histopathological studies on tissues, mainly derived from cancer patients, require proof of monospecificity of the mAb for HLA-E, by the dosimetric inhibition of the HLA-E mAbs with one or more of the HLA-E monospecific epitopes listed in Tables 1 and 2.
Monospecific HLA-E mAbs
Since the commercial anti-HLA-E mAbs, particularly those most commonly used on human cancer tissues such as MEM-E/02 and 3D12 (Table 5), do not meet the critical requirement of monospecificity of the mAb, they may not be considered reliable for specific immunorecognition of HLA-E on human cancer tissues. An imminent need is recognized to generate anti-HLA-E mAbs that can bind only to amino acid sequences (epitopes) unique or specific for HLA-E and to revalidate the expression of HLA-E on the cell surface of cancer cells. Therefore, we have generated(54) several hybridoma clones (n = 258) secreting anti-HLA-E mAbs using recombinant heavy chains of the two different alleles of HLA-E, namely HLA-EG107 and HLA-ER107. The glycine (G) at position 107 is also seen with HLA-F, HLA-G, and several other HLA-Ia alleles,(54) whereas arginine 107 is extremely rare among HLA-I, with two exceptions, HLA-A*01:01:01:02N and HLA-B*15:30. These mAbs were investigated for their specific affinity for HLA-E by measuring their MFI in Luminex SAB assays using beads coated with 31 HLA-A, 50 HLA-B, 16 HLA-Cs, one HLA-G, and one HLA-F antigen. Based on the analysis of the affinity of the antibodies to HLA-Ia and HLA-Ib molecules, the mAbs were categorized into eight groups as defined in Table 7. Group 1 of the eight groups constitutes mAbs that do not react with HLA class-Ia alleles or with HLA-F and HLA-G. However, they reacted to both HLA-EG and HLA-ER. The MFI of the isotypes of group 1 is compared to assess the potential strength of the mAbs (Fig. 3). The number of clones secreting the mAbs (MFI ≥1000) in general and the monospecific mAbs (MFI >1000) were much higher with HLA-ER107 (12 per mouse) than with HLA-EG107 (8 per mouse). The most common isotype is IgG1. Several clones showed remarkable proliferation in culture. The culture supernatants of five of these clones (TFL-033, TFL-034, TFL-073, TFL-074, and TFL-145) were purified using Protein-G columns tested for antibody titers (Table 8).(54) TFL-033 was selected for analysis of both ascites and culture supernatants. The titrimetric profile of TFL-033 is shown in Figure 4.
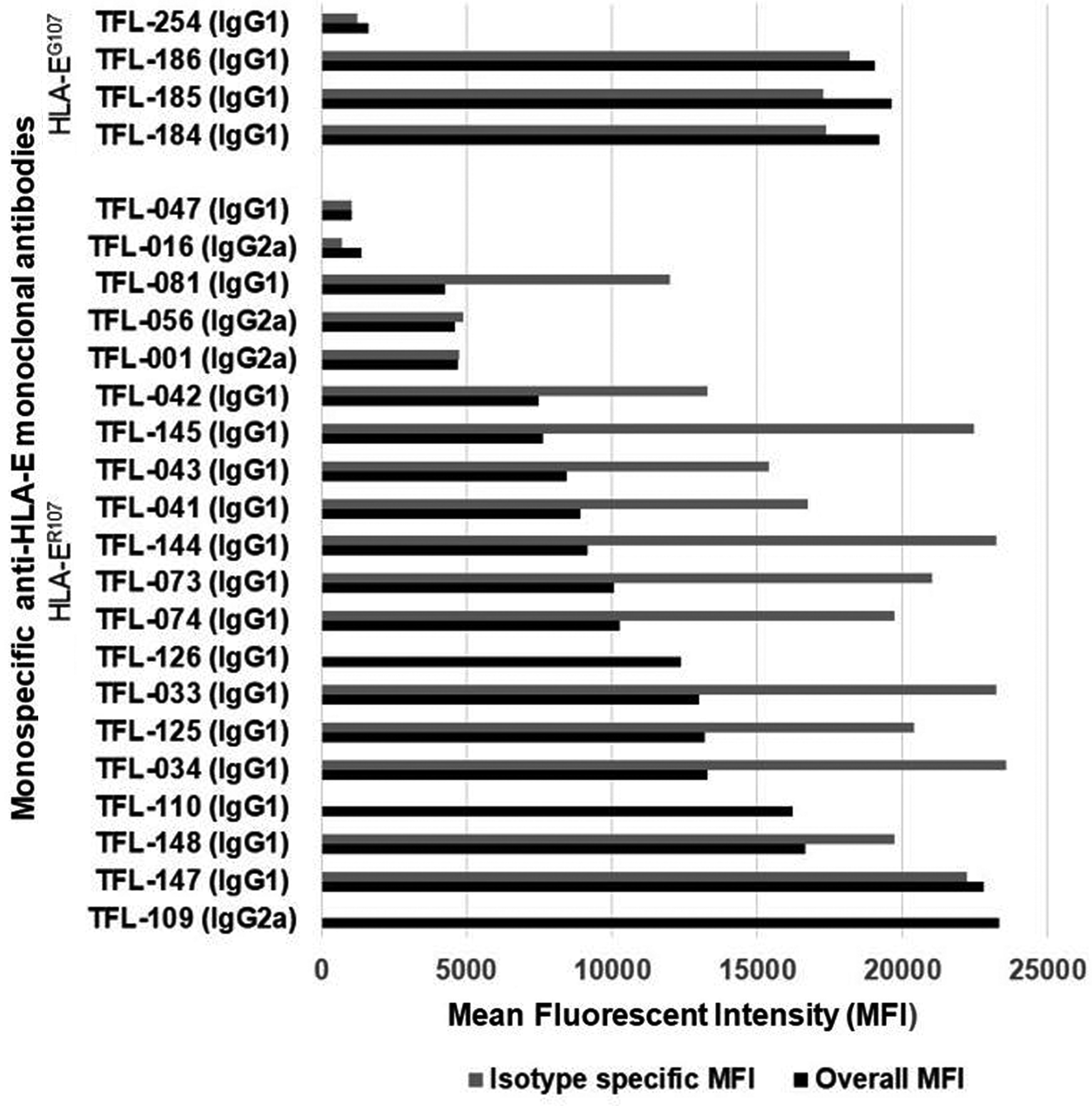
Several hybridoma clones secreting monospecific mAbs were generated by immunizing the heavy chains of either HLA-ER107 or HLA-EG107. The number of clones secreting the mAbs in general and the monospecific mAbs in particular were much higher with HLA-ER107 than with HLA-EG107. The most common isotype seems to be IgG1. The MFI of the culture supernatants as well the MFI of IgG isotypes is indicated in the figure. For detailed investigation, we have used TFL-033. Ig, immunoglobulin; MFI, mean fluorescent intensity.
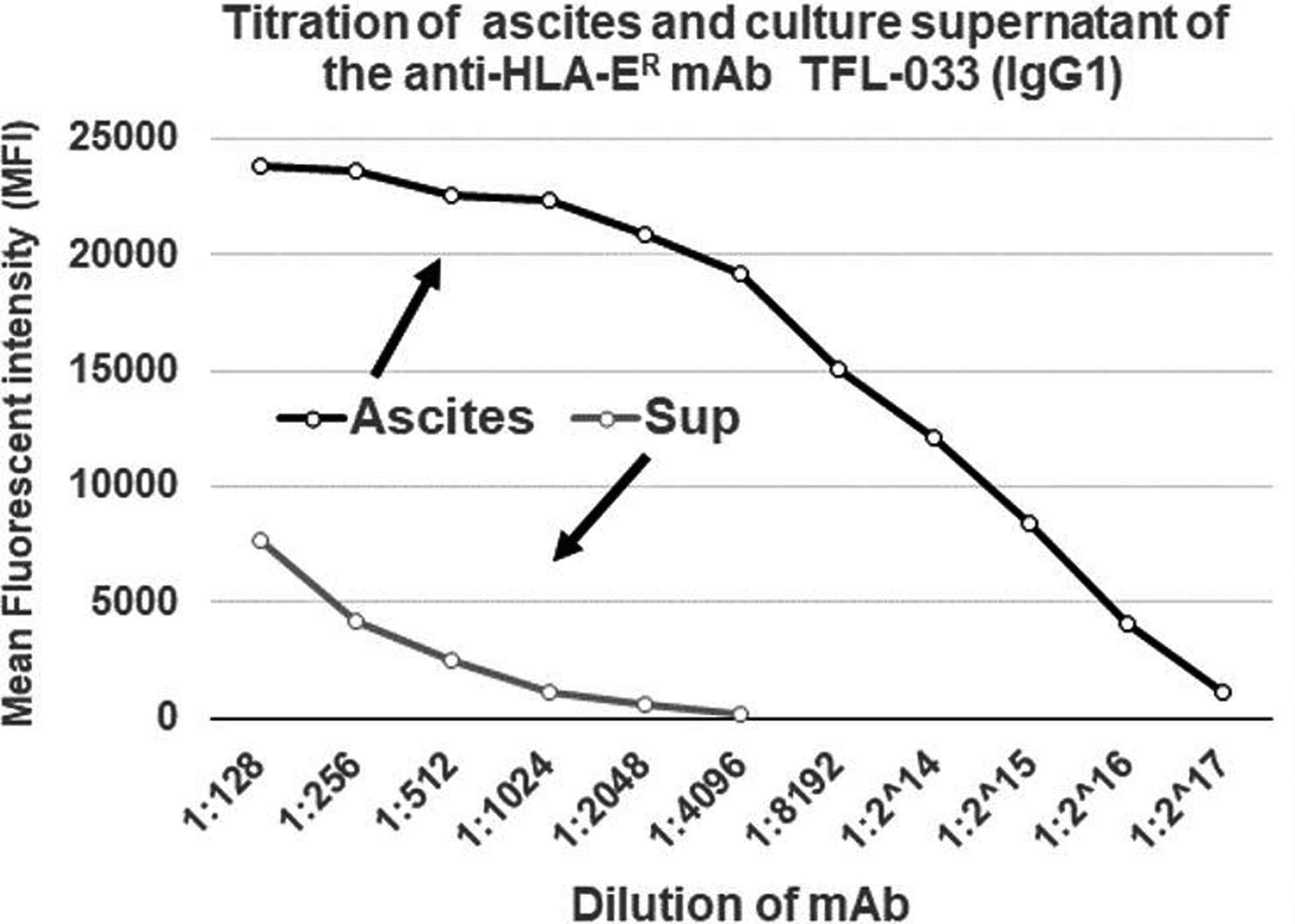
Titration of ascites and culture supernatant of the anti-HLA-ER mAb TFL-033 (IgG1). ER, endoplasmic reticulum.
HLA-E Heavy Chain Monomer, Upon Immunization in a Mouse Model, Elicits Eight Different Groups of Anti-HLA-E Antibodies; However, Only Group 1 Befits the Definition of HLA-E-Specific Monoclonal Antibody Because of Its Monospecificity
The monospecific monoclonal HLA-E antibodies recognize none of the other HLA-Ia or Ib molecules. TFL refers to Terasaki Foundation Laboratory in Los Angeles, where the antibodies were generated and characterized. The mAbs were monitored in Luminex SABs assay using regular LABSCreen beadsets.
Titers of Protein-G Purified Culture Supernatants of Monospecific Anti-HLA-ER
Titer of the mAbs measured after serial dilution of the immunogen used is the heavy chain of HLA-E.
Immunodiagnostic potential of a monospecific anti-HLA-E mAb
The purified fraction of the TFL mAbs was used for comparative staining with MEM-E/02 on tissues of human gastric cancer(71) and melanoma.(54) As noted earlier, HLA-E without a leader peptide in the grove does not give stability to the α-heavy chain/β2m heterodimer. However, as noted in our report,(54) of the eight groups of mAbs, only one group is truly specific for HLA-E, whereas others bind to epitopes shared with other HLA loci, such as HLA-A, HLA-B, HLA-Cw, HLA-F, and HLA-G. The mAb TFL-033-specific epitopes occur in α1 helix (which form the groove) and at adjacent naturally exposed domain on the helices, as illustrated in Table 9 and Figure 1A and B. The mAb TFL-033 binding to HLA-E coated on SABs was tested using such HLA-E monospecific peptides in a dosimetric manner, and the results are illustrated in detail in a figure [fig. 5 in Ravindranath et al.(54)]. The dosimetric inhibition of purified culture supernatants of TFL-033 was examined with two HLA-E-restricted peptides, 65RSARDTA71 and 143SEQKSNDASE152, at concentrations ranging from 4.4 to 0.27 mg/well. Although both peptides showed inhibition, the α2 helical peptide SEQKSNDASE showed higher inhibition than the other peptide. These findings suggest that TFL-033 has better specificity for recognition of intact trimeric HLA-E than most of the polyreactive commercial mAbs, which bind to β2m-free HLA-E. That is one of the reasons why MEM-E/02 did not bind to iBeads (the regular LABSCreen beadset enzymatically treated to eliminate monomeric and dimeric variants of HLA-I) but bound only to the regular LABScreen beads. Since human cancer cells can express both intact trimeric HLA and β2m-free HLA, there is a need to distinguish the different phenotypic expression of HLA-E on the surface of tumor lesions. Because of the unique peptide-binding affinities of TFL-033 and MEM-E/02, we used both mAbs to study this aspect in gastric cancer(71) and melanoma.(54) Only TFL-033 stained the cytoplasm of normal mucosa (Fig. 5) diffusely. The incidence and intensity of staining of the cell surface in early stages, poorly or undifferentiated and non-nodal lesions by TFL-033 are markedly higher than the staining by MEM-E/02. Of note, however, MEM-E/02 stained terminal stages of adenocarcinoma and lymph node metastatic lesions intensely, either owing to increased expression of β2m-free HLA-E with tumor progression or owing to expression of other β2m-free HLA-Ia molecules. Similar comparative observations made on human melanoma tissues(54) validate the hypothesis that monospecific anti-HLA-E mAbs reveal the presence of intact trimeric native HLA-E than the commercial polyreactive nonspecific HLA-E mAbs.
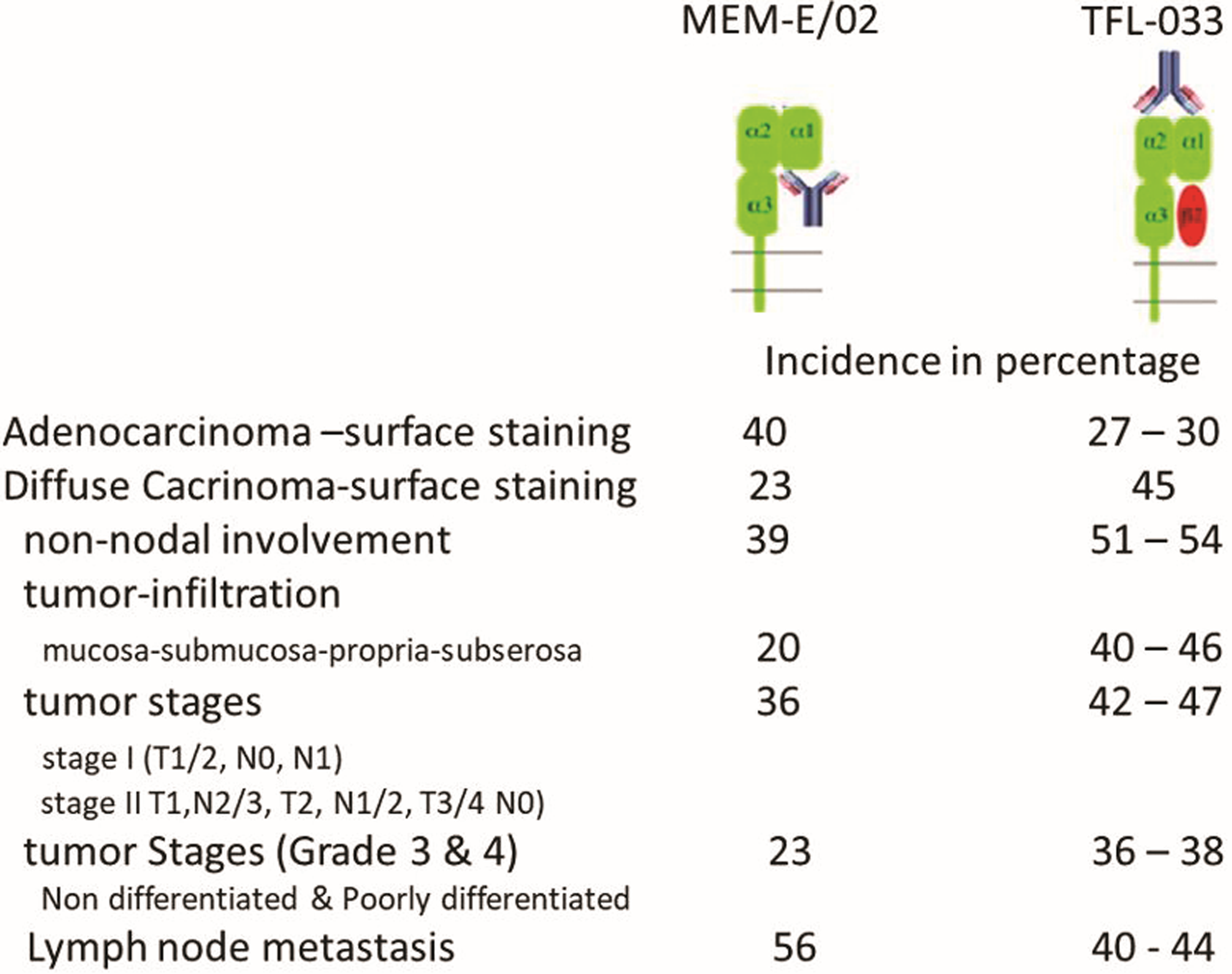
Comparative staining of gastric adenocarcinoma and diffuse carcinoma with trimeric HLA-E reactive monospecific mAb TFL-033 and β2m-free heavy chain of HLA-E and HLA-I polyreactive mAb MEM-E/02. Please note that intense staining or higher incidence of staining with MEM-E/02 does not imply it is due to intact, trimeric, or dimeric HLA-E but due to the monomeric heavy chain of HLA-I including HLA-E. In adenocarcinoma, MEM-E/02 stained 12 of 30 tissue samples (40%) and the incidence is higher than that of TFL-033 (27%–30%). In diffuse carcinoma, TFL-033 stained 18 of 40 tissue samples (45%). The incidence is higher than that of MEM-E/02 (23%). The node-negative was significantly more positive staining with TFL-033 than with the node-positive. Metastatic cancer included peritoneal (n = 5), liver (n = 3), ovarian (n = 5), and lymph node (n = 27) metastases. With the mAb MEM-E/02, 21 of the 40 metastatic carcinomas stained intensely (53%), higher than that of TFL-033 (38%–40%). With lymph node metastasis, the incidence of staining by TFL-033 was 40%, whereas that of MEM-E/02 is 56.6%. [Source: Sasaki et al.(71)].
Comparison of the HLA-E Monospecific Amino Acid Sequences (Epitopes) of Three Epitopes (One 6-mer [#1], One 9-mer [#2], and One 5-mer [#3])
Since each locus has >1000 alleles, due to space constraints, few alleles for each locus were selected. Qa-I, a murine equivalent of hu HLA-E is also compared. Peptide #1 and peptide #2 were selected for diametric inhibition of one of the monospecific mAbs [TFL-033, see fig. 5 in Ravindranath et al.(54)]. The term monospecificity implies the binding of the mAbs to a unique epitope of HLA-E (Table 4).
The bold letters of amino acids of alleles other than HLA-E * 01010101 indicate the corresponding position of the amino acids found in HLA-E. The lack of repetition of the entire sequence of amino acids found in HLA-E establish that the amino acid sequence is HLA-E restricted.
Potential of monospecific anti-HLA-E mAb to avert CD94/NKG2A interaction
The binding domain of the monospecific anti-HLA-E mAb (e.g., TFL-033) and CD94 and NKG2A on the α1 and α2 helices of HLA-E is the same. Both TFL-003 and the CD94/NKG2A heterodimer share some of the specific amino acids. However, we have not done peptide inhibition studies on all the monospecific anti-HLA-E mAbs. It is reasonable to infer that all of them may bind to HLA-E-specific epitopes, which could exist on the α1 and a2 helices of HLA = E. Possibly some mAbs may share more amino acids in the binding domain with the NK-inhibitory receptors, CD94-NKG2A, that react with HLA-E-specific epitopes on the α1 and α2 helices. Figure 1A illustrates the exact amino acid (in box) and its location in the epitope in the α1 helix, with either TFL-033 or CD94. Figure 1B documents the exact amino acid (in box) and its location in the epitope in the α2 helix that reacts with both TFL-033 and NKG2A. The nature of the interaction of CD94 or NKG2A with their respective binding domains on the helices is further clarified in Figure 1A and B as well as in Table 4. Two critical issues should be addressed in this context. CD94 or NKG2A bind to more than one amino acid in their binding domain, but TFL-033 binds to only one of those amino acids recognized by CD94 or NKG2A. Can such binding to a single amino acid in each helix by TFL-033 suffice to block the interaction of CD94 or NKG2A? It should be noted that for peptide inhibition studies on TFL-033, we have employed only the amino acids specific for HLA-E found on each helix. We did not test the whole amino acid sequence recognized by CD94 or NKG2A. Probably it would have provided better inhibition of TFL-033 binding to HLA-E on beads. Second, if a mAb binds to that epitope, it is sure to create stearic hindrance to block the binding of CD94 and NKG2A. Possibly other monospecific mAbs (n = 24) may differ in their efficacy to block binding of CD04/NKG2A binding based on the amino acid sequences recognized by the mAbs. Examining the nature of amino acids that are recognized by TFL-033, it may be noted in Figure 1A that Arginine [R] at position-65 constitutes the part of the epitope recognized by mAb TFL-033. The interaction of CD94 with R-65 involves to hydrogen bonding and three van der Waal forces (see Table 4 for details). Monospecific mAb binding can cause hindrance for this powerful interaction between R65 and CD94.
Similarly, serine [S] at position 152 recognized by the mAb involves both H bonding and van der Waal forces of interaction between NKG2A and S-152. However, it may be that the molecular size and configuration of the IgG1 mAbs that have spread and bound on the helices of HLA-E are sufficient to prevent the interaction between HLA-E and the inhibitory receptors. On blocking the binding of the inhibitory receptors on NK/CD8+ T cells, the antitumor cytotoxic functions of these immune cells are restored.
Another important issue is whether TFL-033 is capable of binding to the epitopes on the two helices when a peptide is present in the grove. The fact that CD94 and NKG2A involve in a variety of H bonding with salt linkage and multiple van der Waal force bindings with a single amino acid (see Table 4 for details) even in the presence of a peptide in the grove suggests that TFL-033 would also bind. As indicated in Figure 1A and B, the mAb binding sites are exposed at the periphery of grove peptide binding sites.
Therefore, a humanized or human monospecific anti-HLA-E mAb mimicking TFL-033 is bound to be a valuable agent for passive immunotherapy of human cancers. It is well known that the therapy benefits not all of the patients receiving the current NK immunotherapy protocols. Failure of patients to respond to NK therapy could be due to the level of expression of HLA-E on the tumor cell surface. If highly expressed, NK cells may be ineffective. The report provides hope that a combinational therapy admixed with anti-HLA-E monospecific mAb can be beneficial for these patients.
Monospecific Anti-HLA-E mAbs Promotes Proliferation of CD8+ T Cells
The monospecific anti-HLA-E mAbs had the potential to augment the proliferation of nonactivated and activated CD3+/CD8+ cytotoxic T cells and CD3−/CD8+ NK cells or NKT cells.(54) Almost all monospecific mAbs augmented the proliferation of phytohemagglutinin (PHA)-untreated T lymphoblasts (TFL-033s at 1/30 (p < 0.02) and 1/150 (p = 0.001), TFL-034 at 1/10 (p = 0.005) and 1/50 (p = 0.002), TFL-073 at 1/50 (p = 0.001), TFL-074 at 1/10 (p = 0.006) (Table 10). The specific increase of “PHA-untreated” T lymphoblasts points out the potential of the monospecific anti-HLA-E mAbs in augmenting CD8+ T lymphoblasts. In addition, a significant increase in the blastogenesis of CD8+ T lymphoblasts was observed among the PHA-treated T lymphoblasts with TFL-033 at 1/30 (p < 0.02) and 1/150 (p = 0.003), with TFL-034 at 1/50 (p < 0.01), and with TFL-145 at 1/100 (p = 0.05). None of the other CD3+ T cells (CD4+/CD8−, CD4−/CD8−, and CD4+/Cd8+) PHA untreated or PHA treated showed any change in the number of lymphoblasts. We have also used a polyreactive anti-HLA-E mAb TFL-007 as control, decreasing the population of untreated CD3+ and CD8+ T lymphoblasts (Table 10). This unique property of augmenting the number of CD8+ T lymphocytes and not other lymphocytes (Table 10) by monospecific but not by polyreactive anti-HLA-E mAbs strongly clarifies the potential of the monospecific anti-HLA-E mAbs for passive immunotherapy of cancer. Immunodiagnostic observations and peptide inhibition studies reported earlier point out that monospecific anti-HLA-E TFL mAbs are indeed capable of binding to HLA-E molecules expressed on the CD8+ T and NK cells. HLA-E is a component of unspecified HLA class-I antigens earlier identified at low levels on inactivated CD8+ cells and upregulated(72,73) or heavily clustered(74) in activated (by PHA or IFNγ) CD8+ cells. Both blastic transformation and proliferation of immune cells induces transient expression of cell-surface molecules. These molecules include IL-2R, Fc receptors for IgG (FcγRI/CD64, FcγRII/ CD32 and FcγRIII/CD16, IgE (FcɛRII)/CD23), insulin receptors; insulin-like growth factor 1R and IL-2R, alpha-fetoprotein and transferrin receptors, a non-disulphide-linked heterodimer of polypeptide chains 33 kDa and 38 kDa called ‘Me14/D12’, MICA, and HLA class II antigens HLA-DR, -DP and -DQ [for detailed information and references(75)]. Most importantly, over-expression of β2m-free heavy chains of HLA class I are documented(76–78)). It is anticipated that the binding of the monospecific anti-HLA-E mAb onto α1 and α2 helices of the overexpressed and clustered open conformers of HLA-E(74) in activated normal human CD8+ T and NKT cells may induce phosphorylation, promoting proliferation of both nonactivated and PHA-activated CD8+ T and NKT cells. A model illustrating the hypothesis is presented in our previous report.(54) Since NKT cells are devoid of CD3 molecules, it is envisaged that the TFL mAb-mediated activation of CD8+ T cells and NKT cells may be independent of CD3 molecules or may involve different cell surface receptors.
Experimental Analysis of Proliferation of CD3+/CD8+ T Cells with Anti-HLA-E Monospecific mAbs (TFL-033, TFL-034, TFL-073, TFL-074, TFL-145) and an Anti-HLA-E Polyreactive mAb (TFL-007) as the Control
Peripheral blood lymphocytes were obtained from a normal healthy volunteer. Both PHA-untreated and PHA-treated CD4 − /CD8+ cells but not CD4+/CD8 − or CD4+/CD8+ or CD4 − /CD8 − cells are induced proliferation only by anti-HLA-E monospecific mAbs. The experimental protocols are described in detail elsewhere.(125) Paired test was performed between No mAb values versus TFL-mAb corresponding values. Significant values in the column of CD4 − /CD8+ are shown in bold.
Ig, immunoglobulin; mAb, monoclonal antibody; NS, nonsignificant.
Conclusions
HLA-E is one of the MHC class-I antigens with structural configuration identical to most of the other classical HLA-Ia and nonclassical HLA-Ib molecules. Using polyreactive anti-HLA-E mAbs, enumerable studies suggest overexpression of HLE-E on the cell surface of several human cancers. We have confirmed the overexpression of HLA-E on the cell surface of gastric cancers and melanoma, using monospecific anti-HLA-E mAbs.(54,71) Tumor-associated HLA-E, upon binding to the inhibitory receptors (CD94/NKG2A) on NK cells or cytotoxic T cells (CD8+), inactivates the ability of such cells to kill tumor cells. Possibly for this reason, the efficacy or outcome of NK and CD8+ T cell-mediated cancer treatment is not always as anticipated. We have developed two categories of mouse HLA-E mAbs, monospecific and polyreactive HLA-E mAbs.(54) Monospecific HLA-E mAbs bind to HLA-E only, but not to any other HLA-Ia or HLA-Ib molecules. In contrast, polyreactive HLA-E mAbs cross react with three or more of the other loci of HLA class-I, namely HLA-A, HLA-B, HLA-C, HLA-F, and HLA-G, which is similar to commercially available mAbs (such as MEM-E0/2, MEM- E0/6, MEM-E0/7, MEM-E0/8 and 3D12), Therefore, the monospecific anti-HLA-E mAbs are better able to confirm specific HLA-E expression on tumor cells and biopsies obtained from cancer patients. Furthermore, peptides from the amino acid sequences located on the α1 and α2 helices of HLA-E specifically inhibited the binding of the monospecific mAbs. Interestingly, the same sequences are also associated with the interaction of HLA-E with the inhibitory receptors (NKG2A and CD94) of NK and CD8+ cells. Since both the monospecific mAbs such as TFL-033 and inhibitory receptors NKG2A/CD94 on NK and cytotoxic T cells bind to the same sequence of the peptides located in the HLA-E α1 and α2 helices, these HLA-E-specific mAbs may have the potential to block the inhibitory pathway responsible for the poor outcome of the NK and CD8+ T cell-based cytotoxic killing of cancer cells in some cases. Furthermore, these anti-HLA-E monospecific but not polyreactive mAbs (such as TFL-007) have the potential to stimulate the blastogenesis and proliferation of the cytotoxic CD8+ T cells.(54) Therefore, the anti-HLA-E monospecific HLA-E mAbs have dual antitumor potential, both to release NK and CD8+ T cells from their inactive state caused by binding of CD94.NKG2A with HLA-E and to simultaneously augment proliferation of the cytotoxic CD3+/CD4−/CD8+ T lymphocytes.
Thus, the monospecific anti-HLA-E mAbs can enhance the immune cell-based therapies paving the way for a better clinical benefit. In addition, these monospecific anti-HLA-E mAbs may also represent a novel prognostic tool in patients with GI tumors or malignant melanoma and other metastasized cancers who plan to undergo immune cell (NK/NKT/CD8+ T cell)-based therapies by identifying which tumors/patients are most likely to respond. Preclinical and clinical trials are required to define the proper role of this technology in the prognostication and treatment of these common cancers. In principle, we suggest that anticancer NK immunotherapy requires determination of the degree of expression of HLA-E on patients' primary or metastatic tumors, with higher expression indicating those most likely to respond to monospecific anti-HLA-E mAb enhancement of cytotoxicity. Furthermore, tumor biopsies can be monitored before and during the course of therapy using these monospecific HLA-E mAbs.
Footnotes
Acknowledgments
M.H.R. thanks Terasaki Family Foundation for grant support for this investigation and the entire project was carried out at Terasaki Foundation Laboratory (TFL), Los Angeles, CA. The authors dedicate this project to Late Professor Paul Ichiro Terasaki, who has ardently supported this investigation and served as a coinventor with MHR for a U.S. patent application on anti-HLA-E monospecific mAb. We sincerely thank TFL-research associates Mr. Tho Pham and Mr. Vadim Jucaud for carrying out Luminex SAB assays and flow cytometric analysis, Ms. Judy Hopfeld for providing iBeads and Dr. Cecilia M. Freitas and Mr. Satoru Kawakita for immunostaining of melanoma and gastric carcinoma microarrays, which enabled specific localization of HLA-E on cancer cells. We thank Professor Dr. Soldano Ferrone, Department of Surgery, Massachusetts General Hospital, Harvard Medical School, Boston, for suggesting improvements in the article, continued support, and encouragements. M.H.R. wishes to express his sincere thanks to Professor Mark Terasaki, the first son of Professor Paul Ichiro Terasaki, for his continued support and encouragement during the preparation of this review.
Author Disclosure Statement
No competing financial interests exist.