
Abstract
Chaetomium globosum is a hydrophilic fungal species and a contaminant of water-damaged building materials in North America. Methods to detect Chaetomium species include subjective identification of ascospores, viable culture, or molecular-based detection methods. In this study, we describe the production and initial characterization of a monoclonal antibody (MAb) for C. globosum enolase. MAb 1C7, a murine IgG1 isotype MAb, was produced and reacted with recombinant C. globosum enolase (rCgEno) in an enzyme-linked immunosorbent assay and with a putative C. globosum enolase in a Western blot. Epitope mapping showed MAb 1C7 specific reactivity to an enolase decapeptide, LTYEELANLY, that is highly conserved within the fungal class Sordariomycetes. Cross-reactivity studies showed MAb 1C7 reactivity to C. atrobrunneum but not C. indicum. MAb 1C7 did not react with enolase from Aspergillus fumigatus, which is divergent in only two amino acids within this epitope. The results of this study suggest potential utility of MAb 1C7 in Western blot applications for the detection of Chaetomium and other Sordariomycetes species.
Introduction
W
Conventional methods to detect C. globosum in indoor environmental samples include the identification of round, oval, or flask-shaped perithecia (sexual fruiting structures) or darkly pigmented, lemon-shaped ascospores (spores) using viable or non-viable exposure assessment approaches. Although conventional methods are an important tool in fungal surveillance in the commercial and academic sectors, these approaches are often subjective, require several days for processing, and lack specificity, as many spores share similar morphological features. In contrast, monoclonal antibody (MAb)-based detection methods may provide a standardized approach to quantify the target organism(s) using rapid immunoassay platforms such as lateral flow assays, enzyme linked immunosorbent assays (ELISAs), or Western blot analysis. To date, MAbs have been developed for the detection of S. chartarum,(13) whereas for Chaetomium species, only polyclonal antibodies have been produced in rabbits.(5)
C. globosum produces a variety of intracellular and extracellular antigens as part of the organism's life cycle. Enolase, a 45–50 kDa enzyme that catalyzes the conversion of 2-phosphoglycerate to phosphoenolpyruvate in fungal glycolysis, is present within the cytosol and cell wall, and is secreted during hyphal growth of various fungi.(14,15) Based on these data, C. globosum enolase was selected as a candidate biomarker for the detection of C. globosum. In the present study, C. globosum enolase was cloned and a recombinant was expressed in Escherichia coli for the production of MAbs. Here we describe the development of MAbs against the recombinant enolase. The production of enolase MAbs may have unique application for the detection of this cellulolytic fungal species as well as other closely related Chaetomium species in the built environment.
Materials and Methods
Fungal cultures
Fungal isolates were acquired from the University of Alberta Microfungus Collection and Herbarium (UAMH), the United States Department of Agriculture, Agriculture Research Service Culture Collection (NRRL), and the American Type Culture Collection (ATCC, Manassas, VA). Isolates evaluated in this study included seven strains of C. globosum (Table 1). Additional fungal species, including C. indicum, C. atrobrunneum, and Aspergillus fumigatus, were also evaluated in cross-reactivity studies (Table 1). All Chaetomium isolates were maintained in short-term slant cultures at 4°C and stored at −70°C for long-term storage by suspending ascospores in a 25% glycerol solution.
Cloning of recombinant C. globosum enolase
The putative sequence for C. globosum enolase was obtained by performing a BLAST search with Chaetomium enolase as a search query. The search yielded several results for homologs present in different fungal divisions and included a 419 amino acid putative uncharacterized C. globosum protein (Q2HFP6). Sequences of putative uncharacterized proteins from C. globosum, Thielavia terrestris, Neurospora crassa, and A. fumigatus were analyzed using Clustal O multiple sequence alignment software and is shown in Figure 1.(16,17)

Sequence alignment of putative and characterized enolase derived from Chaetomium and other closely related fungal species. The full-length sequence of Chaetomium globosum enolase was identified by translation of the cDNA sequence and is identified as cDNA. Alignment was performed using Clustal Omega. Database accession numbers identify putative or enolase sequences. CHAGB, putative uncharacterized protein Chaetomium globosum; THITE, putative uncharacterized protein Thielavia terrestris; NEUCR, enolase Neurospora crassa; ASPFU, enolase Aspergillus fumigatus. Highlighted yellow represents the epitope of MAb 1C7 and homology between CHAGB, THITE, and NEUCR. Highlighted red boxes represent two amino acid substitutions in the ASPFU enolase sequence and correspond to glutamic acid (E) for glutamine (Q) and asparagine (N) for aspartic acid (D). Symbols correspond to (*) positions that have a single, fully conserved residue; (:) indicates conservation between groups of strongly similar properties; (.) indicates conservation between groups of weakly similar properties. The sequence analysis was performed on February 21, 2014.
C. globosum (NRRL 1870) was initially inoculated and grown on minimal agar media for the development of sporulating cultures. Ascospores derived from C. globosum were then inoculated in oatmeal broth media (50 mL) at a final concentration of 200 spores/mL. Cultures were grown for 3 days at room temperature (RT), after which the mycelium was harvested and washed thoroughly in phosphate buffered saline (PBS, pH 7.4) as previously described.(18)
A schematic of the cloning strategy used to generate recombinant C. globosum enolase (rCgEno) is shown in Figure 2. Total RNA was extracted from washed C. globosum mycelia using a phenol-free RNeasy Plant Mini Kit, according to manufacturer's instructions (Qiagen, Valencia, CA). In brief, hyphae, spores, or cells were macerated in liquid nitrogen, followed by a homogenization step in a lysis/binding buffer supplied by the manufacturer. Following homogenization, samples were briefly centrifuged to remove insoluble debris. The cleared lysate was then applied to an RNA-binding filter cartridge and washed, and the resulting purified RNA was eluted with an appropriate volume of pre-heated elution buffer. DNase was then added according to the manufacturer's instructions (Qiagen).
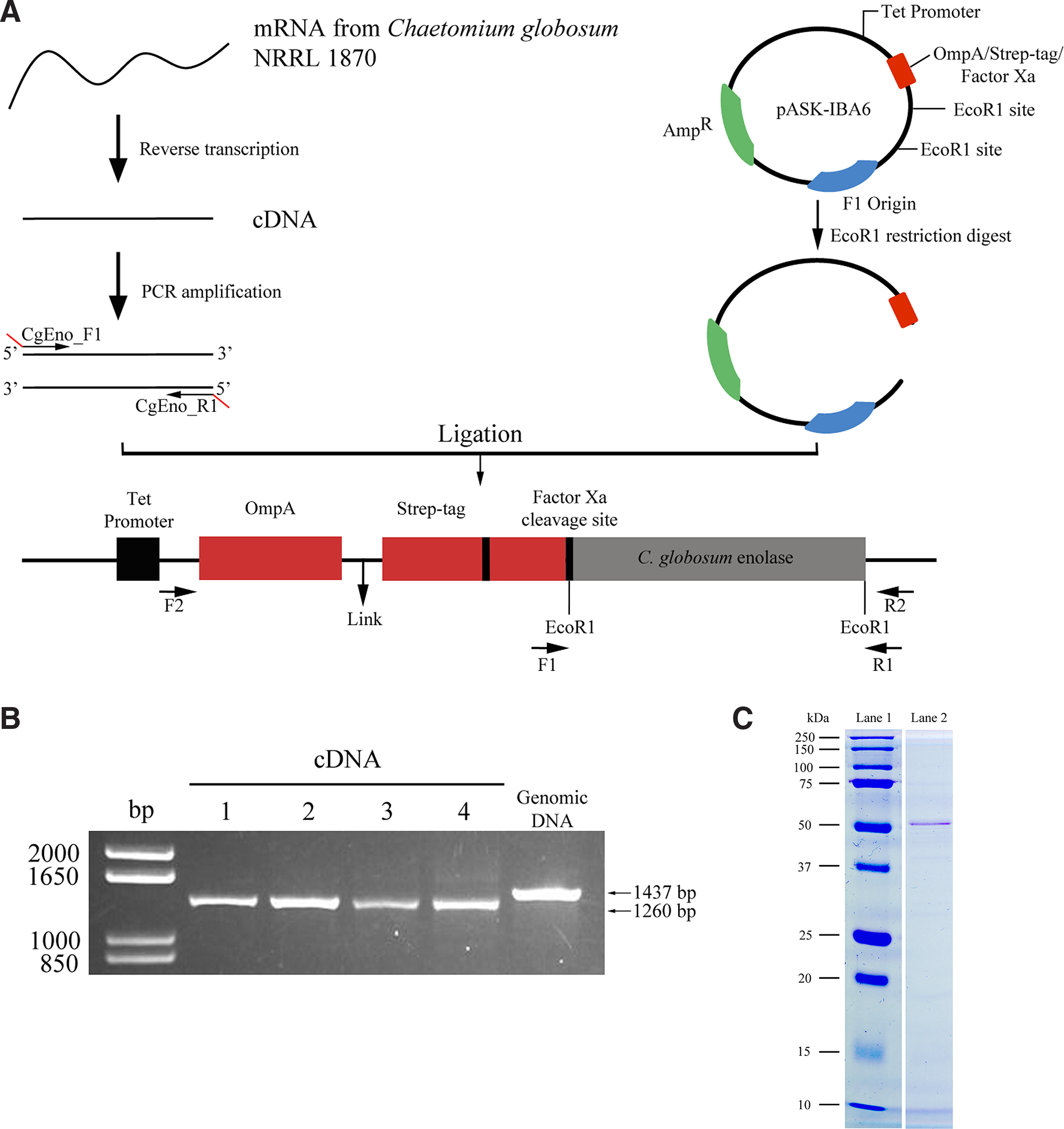
Cloning strategy for recombinant enolase in pASK-IBA6 vector and expression in Escherichia coli (
Reverse transcription of the purified total RNA into the complementary DNA (cDNA) was carried out using the RETROscript RT-PCR Kit (Ambion, Austin, TX). Utilizing a two-step method in combination with heat denaturation, the RNA template was primed with oligo dT primers at a final concentration of 5 mM. Reaction conditions were as follows: 3 min at 80°C, 60 minutes at 44°C, and 10 min at 92°C. Enolase-specific primers were designed using Primer3 software (
Enolase-pASK-IBA6 constructs were submitted to the University of Georgia Sequencing and Synthesis Facility for sequence analysis using the ABI PRISM® 3100 Genetic Analyzer. Both M13 Forward and T7 Promoter priming sites present on the pASK-IBA6 vector were used to sequence cDNA clones. Data analysis and sequence homology searches were performed using various programs available through the National Center for Biotechnology Information (NCBI) in order to confirm that the enolase gene was in-frame.
Expression and purification of recombinant C. globosum enolase
For expression of rCgEno, the pASK-IBA6 plasmid (IBA LifeSciences) containing an in-frame CgEno coding sequence was introduced into E. coli BL21 Star™ (DE3) One Shot cells (Invitrogen) (Fig. 2). This cell line has been engineered for the expression of recombinant proteins utilizing the tetA promoter. Following a similar transformation protocol as described above, rCgEno expression was induced using anhydrotetracycline. Briefly, the transformed cell line was cultured in Luria-Bertani medium (50 mL) supplemented with 100 μg/mL ampicillin overnight at 37°C at 200 RPM until cell growth reached the optical density (OD) of 0.5–0.8 at OD600. Anhydrotetracycline was then added at a concentration that was non-toxic to cells (200 ng/mL), and cell samples were further cultured overnight at 25°C.
rCgEno was then harvested from E. coli by centrifugation at 12,000 g for 15 min at 4°C. The cell pellet was resuspended in 10 mL of CelLytic B solution (Sigma, St. Louis, MO). Phenylmethylsulfonyl fluoride (100 μL) was added to inhibit endogenous protease activity. Avidin (100 μL, 2 mg/mL) was also added to the CelLytic B solution to remove endogenous biotin carboxyl carrier protein (BCCP) that would otherwise bind to the Strep-Tactin column (IBA LifeSciences) and interfere with the purification of rCgEno. The suspension was then centrifuged at 12,000 g for 15 min at 4°C to remove lysed E. coli and cellular debris. The supernatant was applied to an affinity column packed with 10 mL Strep-Tactin-sepharose (IBA LifeSciences), and unbound proteins were removed with washing buffer (100 mM TRIS 150 mM NaCl, 1 mM EDTA [pH 8.0]; IBA LifeSciences). rCgEno was then eluted with washing buffer containing 2.5 mM desthiobiotin [pH 8.0]; IBA LifeSciences) and the concentration of the purified rCgEno was determined using a NanoDrop ND1000 Spectrometer (Thermo Scientific, Wilmington, DE). rCgEno was then dialyzed in PBS (pH 7.4) and stored at −20°C until use in SDS-PAGE and immunoblotting studies.
SDS-PAGE
Purified rCgEno (1 μg/lane) was separated by SDS-PAGE on a 12% acrylamide separating gel and 4% stacking gel after denaturation in electrophoresis sample buffer according to the method of Laemmli.(19) SDS-PAGE was carried out using 100V constant voltage for 90 min, and the separated proteins were stained with Imperial™ protein stain (Thermo Scientific, Rockford, IL) as previously described.(20) Individual bands corresponding to rCgEno were observed at 49 kDa, and excised bands were submitted for mass spectrometry analysis.
Vertebrate animals
Female BALB/cJ mice (n=3) were acclimated for approximately 1 week prior to the rCgEno immunizations. The mice were housed in HEPA-filtered ventilated polycarbonate cages on autoclaved hardwood chip bedding. The temperature in the animal facility was maintained between 68°F and 72°F and the relative humidity between 36 and 57%. The light/dark cycle was maintained on 12-h intervals. Mice were provided NIH-31 modified 6% irradiated rodent diet (Harlan Laboratories, Madison, WI) and autoclaved tap water ad libitum. Sentinel mice housed in the animal quarters were free of viral pathogens, parasites, mycoplasmas, Helicobacter species, and cilia-associated respiratory Bacillus species. The NIOSH animal facility is an environmentally controlled barrier facility that is fully accredited by the Association for the Assessment and Accreditations of Laboratory Animal Care International. All animal procedures were performed under a National Institute for Occupational Safety and Health (NIOSH) Animal Care and Use Committee approved protocol 09-DS-M-007, amendment A-10-071.
Immunization of female BALB/cJ mice with recombinant C. globosum enolase
Three female BALB/cJ mice (Jackson Laboratory, Bar Harbor, ME), age 5–7 weeks, were immunized via intraperitoneal (IP) injection with a 50:50 (v/v) emulsion of 25 μg rCgEno and TiterMax® Gold Adjuvant (TiterMax, Norcross, GA). Five subsequent booster IP immunizations containing 5 μg rCgEno in sterile PBS were administered at biweekly intervals. The final boost was administered 3 days before the MAb fusion. Approximately, 100 μL of blood was collected from the tail of each mouse 7 days prior to the first immunization (pre-bleed) and 7 days after each subsequent booster immunization (post-bleed) to monitor the development of serum immunoglobulin G (IgG) responses.
Recombinant C. globosum enolase-antisera screening ELISA
Pre- and post-immunization mouse sera were screened using an indirect ELISA. Briefly, 96-well Nunc Immuno MaxiSorp microplates (Thermo Fisher Scientific) were coated with 2 μg/mL rCgEno in carbonate coating buffer (CCB, pH 9.6) and incubated at RT overnight. Wells were washed three times with PBS containing 0.05% Tween-20 (PBST) and then blocked for 1 h at RT with 200 μL PBST containing 3% non-fat dry skim milk powder (SMPBST). Murine IgG titers were evaluated by incubating duplicate wells for 1 h at 37°C with 100 μL/well of individual serum 2-fold serially diluted from 1:100 to 1:25,600 (v/v) in SMPBST. Wells were washed three times with PBST and bound murine IgG was detected by incubating wells for 1 h at 37°C with 100 μL/well alkaline phosphatase (AP) conjugated anti-mouse IgG (H+L) (Promega, Madison, WI) diluted 1:5000 (v/v) in SMPBST. Individual wells were washed three times with PBST and developed for 30 min at RT with 100 μL/well AP substrate and 0.5 mg/mL p-nitrophenyl phosphate-containing buffer (Sigma Aldrich, St. Louis, MO). The optical density was determined spectrophotometrically at 405 nm using an UltraMicroplate Reader (ELx800, Bio-Tek Instruments, Winooski, VT).
Splenocyte fusion and hybridoma screening
Mice were euthanized by CO2 asphyxiation 3 days following the final booster immunization with 5 μg rCgEno in sterile PBS. Individual spleens were aseptically removed from each mouse and single cell suspensions of the splenocytes were produced. Fusion of splenocytes with SP2/0-AG14 myeloma cells (ATCC# CRL-1581) was performed as previously described.(21,22) Hybridomas were selected by growing cells in Dulbecco's Modified Eagle Medium (DMEM, Life Technologies, Rockville, MD) supplemented with 1 mM sodium pyruvate, 100 U/mL penicillin, 100 mg/mL streptomycin, 0.292 mg/mL L-glutamine, 100 mM sodium hypoxanthine, 16 mM thymidine, 10% fetal calf serum (FCS, HyClone, Logan, UT), and 100 U/mL IL-6 (Boerhinger, Mannheim, Germany). DMEM was also supplemented with azaserine for selective propagation of hybridomas. After 10–14 days of growth, medium from individual wells with hybridoma cell growth was replenished with fresh DMEM.
The supernatant fluid from confluent hybridomas was diluted 1:2 (v/v) in SMPBST and tested using the rCgEno indirect ELISA. Hybridomas that were confirmed to produce rCgEno specific MAb were selected and grown in bulk, transferred to a 96-well tissue culture plate, and cloned twice by limiting dilution. Single positive clones were re-screened using the indirect ELISA, selected, grown in bulk, and purified according to the method of Kent.(23) Purified MAb was utilized in preliminary characterization studies. Hybridoma cell lines of individual clones were frozen in 10% dimethyl sulfoxide (Sigma Aldrich) and 10% fetal calf serum, stored at −80°C for 2 weeks, and then transferred to NIOSH liquid nitrogen facility for long-term storage.
Monoclonal antibody isotyping and quantification
An indirect ELISA was used to determine the isotype of selected rCgEno-specific MAbs. Briefly, 96-well Nunc Immuno MaxiSorp microplates (Thermo Fisher Scientific) were divided into five duplicate vertical wells coated with 100 μL of either 1 μg/mL AffiniPure goat anti-mouse IgG Fc subclass 1, 2a, 2b or 3 (Jackson ImmunoResearch Laboratories, West Grove, PA) in CCB and incubated overnight at RT. For murine IgM, duplicate wells were coated with 1 μg/mL AffiniPure goat anti-mouse IgM and μ chain-specific polyclonal antibody in CCB and incubated overnight at RT. Wells were washed three times with PBST and blocked for 1 h at RT with 200 μL SMPBST. Duplicate wells containing 100 μL/well purified MAb diluted 1:2 (v/v) in SMPBST were incubated for 1 h at 37°C. Isotype specific MAbs were included as isotype-specific positive controls. Wells were washed three times with PBST, and bound murine IgG isotype MAbs were detected by incubating wells with 100 μL/well AP-conjugated anti-mouse IgG (H+L) (Promega) diluted 1:5000 (v/v) in SMPBST for 1 h at 37°C. For IgM isotype detection, wells were incubated for 1 h at 37°C with 100 μL/well biotin-SP-conjugated AffiniPure goat anti-mouse IgG+IgM (H+L) (Jackson ImmunoResearch Laboratories) diluted 1:5000 (v/v) in SMPBST. IgM isotype wells were additionally washed and incubated for 1 h at 37°C with 100 μL/well AP-conjugated streptavidin (Jackson ImmunoResearch Laboratories) diluted 1:5000 (v/v) in SMPBST. Following secondary antibody incubations, individual wells were washed three times with PBST and developed for 30 min at RT with 100 μL/well AP substrate (Sigma Aldrich) and the OD was determined as previously described.
A modification of the indirect ELISA above was used to quantify the purified IgG1 isotype rCgEno-specific MAbs. Briefly, 96-well Nunc Immuno MaxiSorp microplates (Thermo Fisher Scientific) were coated with 100 μL of 1 μg/mL AffiniPure goat anti-mouse IgG Fc subclass 1 (Jackson ImmunoResearch Laboratories) in CCB and incubated overnight at RT. Wells were washed three times with PBST and blocked for 1 h at RT with 200 μL SMPBST. A standard curve for IgG1κ (Sigma Aldrich) was prepared in duplicate and two-fold serially diluted in 100 μL from 100–1.6 ng/mL. Duplicate wells containing 100 μL/well purified MAb were two-fold serially diluted from 1:100 to 1:51,200 (v/v) in SMPBST and incubated for 1 h at 37°C. Wells were washed three times with PBST and bound murine IgG1 isotype MAbs were detected by incubating wells with 100 μL/well AP-conjugated anti-mouse IgG (H+L) (Promega) diluted 1:5000 (v/v) in SMPBST for 1 h at 37°C. Following secondary antibody incubation, individual wells were washed three times with PBST and developed for 30 min at RT with 100 μL/well AP substrate; the OD was determined as previously described.
Fungal extraction
Fungal extracts derived from C. globosum, C. atrobrunneum, and C. indicum isolates were prepared separately according to a modification of the method of Hurkman and Tanaka.(24) Briefly, lyophilized spore, spore suspension, or hyphal material was macerated in a mortar and pestle in liquid nitrogen. The powdered fungal material was weighed and extracted at 4°C in a tissue extraction medium (0.1 M Trizma base [pH 8.8], 10 mM EDTA, 0.9 M sucrose, 0.4% β-mercaptoethanol) and Tris-buffered phenol (pH 8.0). The proteins were then precipitated in 0.1 M ammonium acetate in methanol. Previous studies conducted by our group have shown that this protein extraction protocol yields samples suitable for one-dimensional and two-dimensional electrophoresis. An extract of A. fumigatus was additionally prepared by collecting conidia from 10- to 14-day-old cultures by rolling approximately 1 g of 0.5 mm glass beads (BioSpec Products, Bartlesville, OK) over the plate. Glass beads with spores were collected into a 2 mL screw cap microcentrifuge tube and processed in a Mini Bead Beater (BioSpec Products) for 1 min to disrupt the outer cell walls of the spores. Spores were macerated in a mortar and pestal containing liquid N2 and suspended in cold PBS (pH 7.4), containing Complete Mini Protease Inhibitor Cocktail (Roche Applied Science, Indianapolis, IN). The protein concentrations were then determined according to the BCA method (Pierce Chemical, Rockford, IL) and the samples were stored at −20°C until further analysis.
Western blot analysis
Western blot analysis was performed to screen the reactivity of MAb 1C7 to rCgEno as well as extracts derived from C. globosum strains, C. indicum, and C. atrobrunneum (Table 1). A. fumigatus was included as a negative control in Western blot experiments due to amino acid substitutions within the epitope of the A. fumigatus enolase sequence (Fig. 1; glutamic acid for glutamine and asparagine for aspartic acid). The rCgEno (0.05 μg/lane) and individual fungal extracts (20 μg/lane) were individually separated by SDS-PAGE as described above and transferred to a 0.2 μm nitrocellulose membrane (BioRad, Hercules, CA) overnight at 15 V as previously described.(20) The membranes were washed three times with Tris-buffered saline (TBS, Sigma Aldrich) containing 0.05% Tween-20 (TBST, Fisher Scientific). The membranes were blocked with 3% bovine serum albumin (BSA) in TBST for 1 h at RT. Membranes were then washed with TBST and incubated with 1 μg/mL of MAb 1C7 diluted in 3% BSA-TBST and incubated on a rocker for 1 h. Membranes were washed three times with TBST and incubated with AP-conjugated goat anti-mouse IgG antibody (H+L) diluted 1:5000 in 3% BSA-TBST for 1 h on a rocker. Following secondary antibody incubation, the membranes were washed three times with TBST, and immunoreactive proteins were visualized following 30 min incubation with One-Step nitroblue tetrazolium and bromocholor-indolyl phosphate (NBT/BCIP) substrate (Promega).
Epitope mapping
Epitope mapping was performed on synthetic peptide scans synthesized by Sigma Genosys (JPT Peptide Technologies, Berlin, Germany). For peptide scans, 216 peptides spanning the entire rCgEno sequence (including the Strep Tag) were synthesized as linear decapeptides overlapping by two amino acids. Peptides were covalently bound to a Whatman 50 cellulose support (PepSpots membrane) and the C- and N-termini of the peptides were acetylated for higher stability. The membranes were processed for epitope mapping following the manufacturer's instructions (JPT Peptide Technologies).
In brief, the PepSpots membrane was rinsed in methanol for 5 min, washed three times with TBS for 10 min, and blocked overnight at 4°C on a shaker with TBS containing 3% BSA. The membrane was incubated with 1 μg/mL of rCgEno MAb 1C7 for 3 h at RT on a shaker. The MAb 15B5 served as an IgG1 isotype control as it reacts with an epitope of the hemolytic protein terrelysin.(21) The membrane was washed in TBST three times for 5 min each and incubated with goat anti-mouse IgG horseradish peroxidase-conjugated antibody (Promega) diluted 1:50000 in blocking buffer for 1 h at RT on a shaker. The membrane was washed thoroughly in TBST three times for 5 min each and developed with ECL Western blotting substrate (Promega) as per the manufacturer's instructions. After a brief incubation, excess substrate was discarded and the membrane was exposed to CL-XPosure™ clear blue X-Ray film (Thermo Scientific) and developed on a SRX-101A tabletop processor (Konica Minolta, Ramsey, NJ). For regeneration, the PepSpots membrane was washed twice with water for 10 min each and then incubated with regeneration buffer I (62.5 mM TRIS containing 2% SDS [pH 6.7]; 100 mM 2-mercaptoethanol) at 50°C using four 30 min incubations. The membranes were washed three times for 20 min with PBS (10X), three times with TBST, and three times with TBS for 10 min at RT. The membrane was analyzed to ensure efficient removal of bound primary and secondary antibodies prior to analysis of new MAbs.
Results and Discussion
Enolase (2-phospho-D-glycerate hydro-lyase; EC 4.2.1.11) is a well-characterized prokaryotic and eukaryotic cytoplasmic enzyme that catalyzes the conversion of 2-phosphoglycerate to phosphoenolpyruvate in fungal glycolysis.(14,15) In baker's yeast (Saccharomyces cerevisiae), enolase may comprise up to 2% of the total protein concentration(25); due to its important role in glycolysis, this enzyme is expressed throughout the lifecycle of the organism.(15) Enolase has been localized microscopically in the fungal cell wall and the cytosol, and is secreted during hyphal growth. Enolase has been sequenced, cloned, and identified as an immunodominant antigen for several fungal species including S. cerevisiae,(26–28) Candida albicans,(25,27,29–32) Cladosporium herbarum,(15) Curvularia lunata,(33) Alternaria alternata,(15) Penicillium citrinum,(34) Rhodotourla mucilaginosa,(35) A. versicolor,(36) and A. fumigatus.(34) Although there is >83% identity between enolase derived from C. globosum (CHGG_00958) and other environmentally prevalent fungi (Fig. 1), these data suggest enough sequence divergence for enolase to function as a candidate biomarker antigen that would enable genus and even species-specific identification of C. globosum in the built environment. This has also been confirmed in IgE immunostaining studies where enolase sensitized subjects had variable responses to five different fungal enolases.(35)
Using a previously developed cloning and expression strategy,(18) the C. globosum enolase gene was cloned into the E. coli expression vector pASK-IBA6. The engineered vector was then transformed into E. coli and the rCgEno was expressed (Fig. 2A) and purified (Fig. 2B). The purified rCgEno was observed in SDS-PAGE analysis to correspond to the molecular weight (∼50 kDa) of putative C. globosum enolase (Fig. 2C). Mass spectrometry sequence analysis confirmed the purified recombinant protein to be C. globosum enolase. Although other recombinant C. globosum antigens have been previously published,(37) this is the first reported production of recombinant enolase derived from C. globosum.
Immunization of three female BALB/cJ mice with rCgEno and TiterMax Gold adjuvant resulted in an elevated IgG antibody response. Following the fusion of murine splenocytes with SP2/0-AG14 myeloma cells, the resulting hybridomas were screened using an indirect ELISA for the secretion of rCgEno-specific antibodies. Three hybridomas (1C7, 21D5, and 30C3) were initially identified and exhibited variable reactivity to immobilized rCgEno in the indirect screening ELISA. Two clones that produced IgG1 isotype MAbs survived secondary screening after subcloning and were further assessed in SDS-PAGE and Western blot studies. Only 1C7 clones bound to both immobilized rCgEno (Fig. 3) and to putative enolase derived from C. globosum spore extracts (Fig. 4). Based on these preliminary results, clone 1C7 was selected for further characterization.
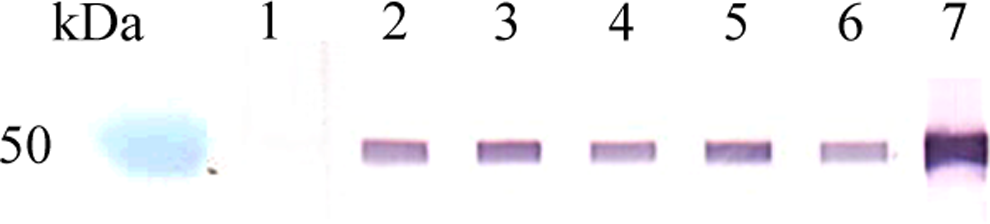
Western blot analysis of IgG1 isotype MAb 1C7 clone reactivity to recombinant C. globosum enolase. Lanes 2–6 represent multiple MAb 1C7 clones that were selected and purified following the limiting dilution steps. Lane kDa, MW markers; lane 1, 3% SMPBST; lane 2, 1C7-4D4-1D4; lane 3, 1C7-3E3-1C4; lane 4, 1C7-3E3-1D4; lane 5, 1C7-3D4-1C4; lane 6, 1C7-3D4-1D4; lane 7, rCgEno murine polyclonal antibody.

Cross-reactivity analysis of IgG1 isotype MAb 1C7 reactivity to enolase derived from Chaetomium species and A. fumigatus spore extracts. Lane kDa, MW markers; lane 1, C. globosum (UAMH 9683); lane 2, C. globosum (UAMH 841); lane 3, C. indicum (UAMH 7031); lane 4, C. atrobrunneum (ATCC 64497); lane 5, A. fumigatus (ATCC 13073); lane 6, rCgEno.
Western blot studies were designed to evaluate the reactivity of MAb 1C7 to C. globosum protein extract fractions including spore wash, homogenized spore, and mycelial extracts. Highest MAb 1C7 reactivity was observed in a ∼50 kDa band in the homogenized spore extract that corresponded to rCgEno (data not shown). This result supports previous studies that have demonstrated enolase to be associated with the fungal cell wall(38,39) and the cytosolic fraction.(40) Reduced MAb 1C7 reactivity was observed in the C. globosum spore wash and mycelial extracts; however, reactivity was restricted to several lower molecular weight protein bands (∼35–45 kDa) that could correspond to truncated enolase isoforms (data not shown). In protozoa, such as Plasmodium, multiple enolase isoforms have been described to represent degradation products or originate following phosphorylation or proteolytic cleavage.(41)
Epitope mapping of MAb 1C7 demonstrated the MAb to bind to the decapeptide at spot 139 (Fig. 5), which corresponded to amino acids 275-284, LTYEELANLY (Fig. 1). SPOTscan of the membrane using the isotype control MAb 15B5 and secondary antibodies resulted in non-specific MAb reactivity observed at spots 172 and 206 (Fig. 5). BLAST analysis of the peptide sequence LTYEELANLY revealed 100% sequence homology to 16 deposited fungal sequences (Table 2). An additional two fungal sequences and one bacterial sequence with 90% homology to the queried decapeptide were also identified (Table 2). All identified sequences in the BLAST query corresponded to enolase or putative proteins with a glycolytic function that belonged to fungi placed in the class Sordariomycetes. These data correspond to MAb 1C7 Chaetomium species reactivity shown in Figure 4. Greatest MAb 1C7 reactivity was observed to C. globosum UAMH strains 9683 and 841 as well as a putative enolase derived from C. atrobrunneum. Interestingly, MAb 1C7 did not react with C. indicum or A. fumigatus (Fig. 4). Divergence with as few as two amino acids in the A. fumigatus extract resulted in the loss of MAb reactivity (Fig. 1). Although there is no sequence data available for C. indicum, the results suggest that C. indicum may not share 100% homology with amino acids 275-284 as observed for C. thermophilum (Table 2). These combined datasets demonstrated that MAb 1C7 is specific for a highly conserved enolase epitope that is restricted within the fungal class Sordariomycetes. In addition to reacting with C. globosum and C. atrobrunneum, MAb 1C7 may additionally react with other closely related species within the fungal orders Sordariales and Hypocreales.
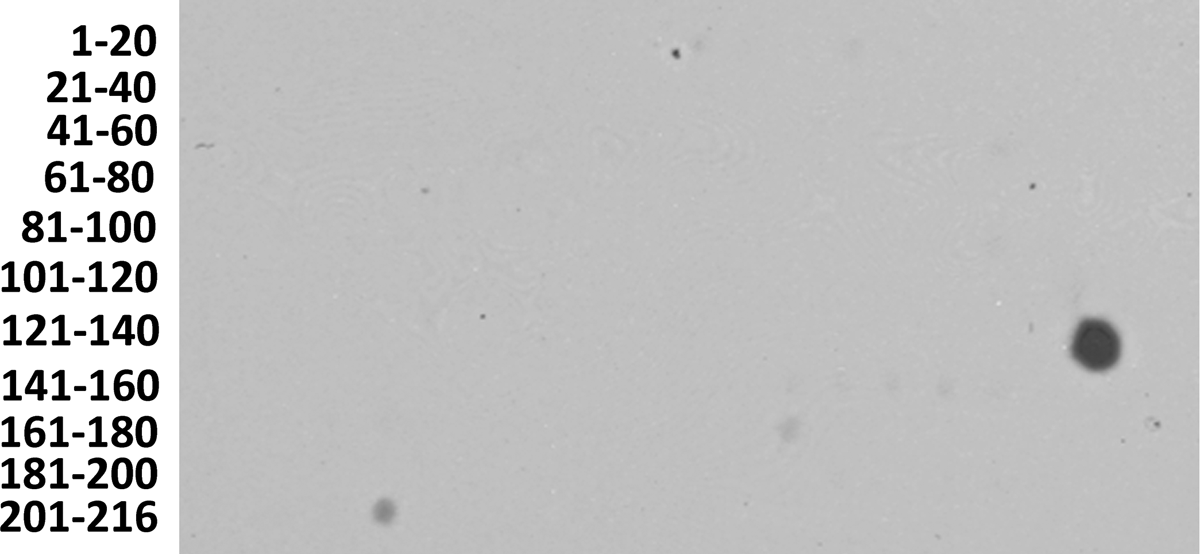
Epitope mapping of MAb 1C7 reactivity. Each spot represents a decapeptide of rCgEno sequence. Decapeptides were sequential with an overlap of two amino acids. In addition to the rCgEno sequence, the N-terminal purification Strep-tag II and the Factor Xa cleavage site are included.
90% homology with the bacterial species, Beijerinckia indica subsp. indica was additionally observed.
Putative uncharacterized protein.
ND, not determined.
C. globsoum enolase was selected as a candidate biomarker due to its elevated expression in several fungal species.(42) Targeting a highly expressed protein such as enolase was hypothesized to result in improved immunoassay sensitivity compared to other fungal antigens, such as terrelysin that may only be expressed during a specific growth stage of a fungal species.(21) Although the production of the IgG1 isotype MAb 1C7 is an exciting development for the detection of C. globosum and other members of Sordariomycetes, the results of these preliminary immunoassay studies demonstrated that MAb 1C7 could not bind to native rCgEno or putative C. globosum enolase in an inhibition ELISA format (data not shown). These results suggest that MAb 1C7 may only bind to a reduced form of C. globosum enolase and would limit the use of this MAb to Western blot applications. However, this antibody may prove useful for the detection of enolase derived from Chaetomium species and other members of Sordariomycetes in reduced extracts derived from contaminated indoor environments.
Footnotes
Acknowledgments
This study was supported in part by an interagency agreement with NIEHS IAA# AES 12007-00100000.
Author Disclosure Statement
The findings and conclusions in this report are those of the authors and do not necessarily represent the views of the National Institute for Occupational Safety and Health.