
Abstract
Homeostasis of the lacrimal functional unit is needed to ensure a well-regulated ocular immune response comprising innate and adaptive phases. When the ocular immune system is excessively stimulated and/or immunoregulatory mechanisms are disrupted, the balance between innate and adaptive phases is dysregulated and chronic ocular surface inflammation can result, leading to chronic dry eye disease (DED). According to the Tear Film and Ocular Surface Society Dry Eye Workshop II definition, DED is a multifactorial disorder of the ocular surface characterized by impairment and loss of tear homeostasis (hyperosmolarity), ocular discomfort or pain, and neurosensory abnormalities. Dysregulated ocular immune responses result in ocular surface damage, which is a further contributing factor to DED pathology. Several therapeutics are available to break the vicious circle of DED and prevent chronic disease and progression, including immunosuppressive agents (steroids) and immunomodulators (cyclosporine and lifitegrast). Given the chronic inflammatory nature of DED, each of these agents is commonly used in clinical practice. In this study, we review the immunopathology of DED and the molecular and cellular actions of current topical DED therapeutics to inform clinical decision making.
Introduction
The Tear Film and Ocular Surface Society International Dry Eye Workshop II report defines dry eye disease (DED) as “a multifactorial disease of the ocular surface characterized by a loss of homeostasis of the tear film and accompanied by ocular symptoms, in which tear film instability and hyperosmolarity, ocular surface inflammation and damage, and neurosensory abnormalities play etiological roles.” 1 DED is believed to be progressive in some patients.2,3 It can have significant impact on visual function, daily activities, social and physical functions, workplace productivity, and general quality of life. 4 Global prevalence estimates range from 5% to 50%, 5 and several reports find higher prevalence in women than in men.6–8 Additional risk factors include increasing age,6–8 systemic comorbidities such as diabetes and autoimmune disease,9,10 and therapeutic treatments for anxiety, depression, and sleep disorders.11–15 The pathologic processes of chronic inflammation and related biomarkers have been the focus of recent immunologic research to identify potential therapeutic targets. 16 The “vicious cycle of inflammation” has been proposed as a core driver in DED with the bidirectional interaction between the ocular sensory neurons and local immune system disrupting ocular homeostasis.16,17 Ocular surface sensory neurons in response to inflammation can provoke nerve impulse activity resulting in differences in sensations, tear flow, and blinking. 17 In addition, peripheral sensory neurons can illicit an immune response by releasing neuropeptides and immunomodulatory factors contributing to neurogenic inflammation. 17 While there are many factors involved in DED such as/including hyperosmolarity, tear film instability, and meibomian gland dysfunction (MGD),18,19 the purpose of this article is to explore the specific immunopathogenic processes and the impact of current pharmacological therapies.
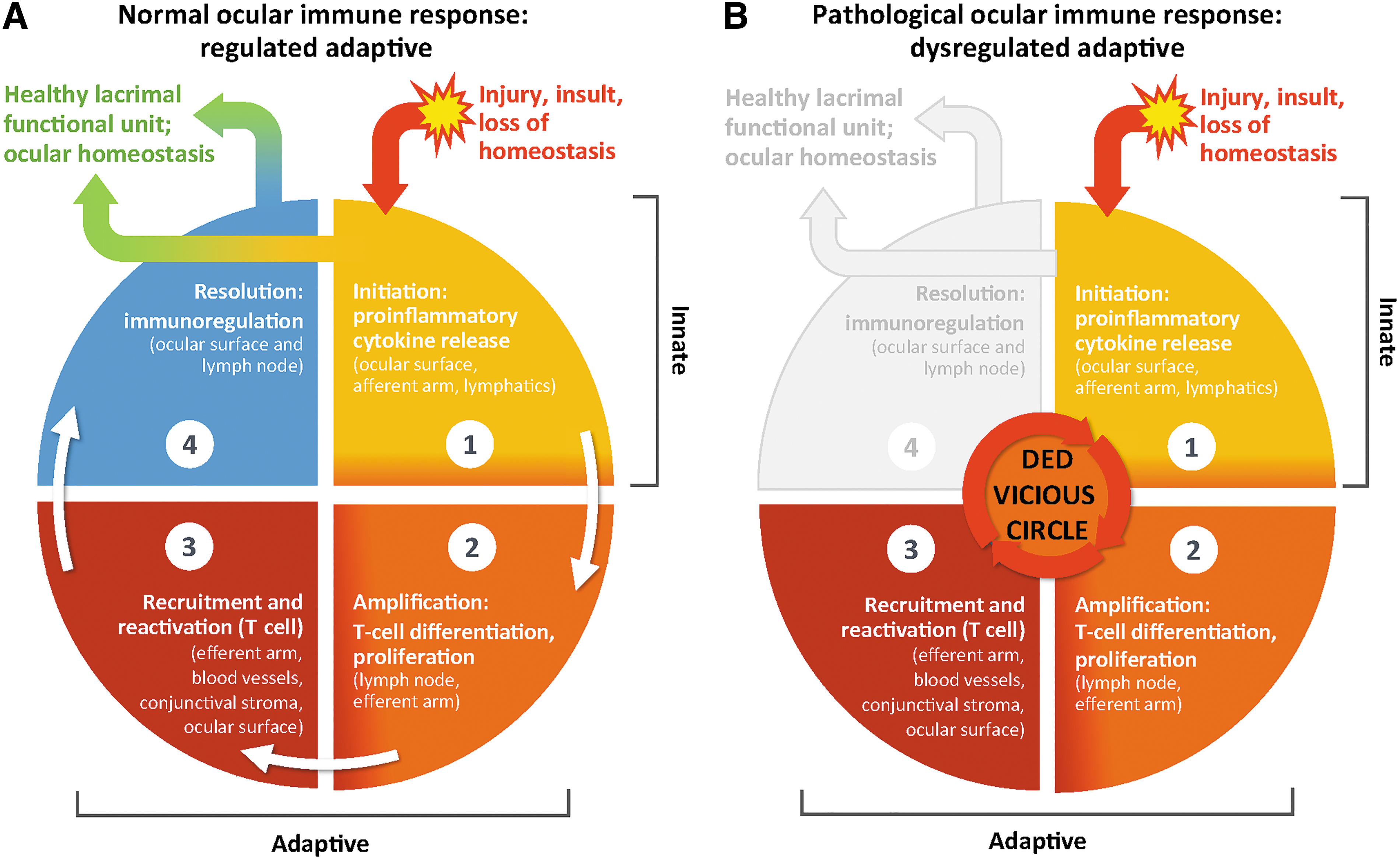
Over the past 15 years, a growing body of research on the role of inflammation in the pathogenesis of DED has led to the recognition of dysregulation of immune responses on the ocular surface. 20 An appreciation of the basic immunological factors associated with DED is essential for appropriate management of patients with the disease. 21 At the most basic level, there are 2 phases of immune responses coordinated to provide protection: innate and adaptive. Innate immune responses, which occur at the ocular surface, provide a first-line generalized defense. 22 Irritation, such as environmental or endogenous factors, or microbial stress on the ocular surface, activates signaling pathways that initiate an acute inflammatory response, including stimulation of the production of pro-inflammatory cytokines, matrix metalloproteinases (MMPs), and chemokines. The more specific adaptive immune response is then signaled to produce antigen-specific T cells in regional lymph nodes that migrate to the ocular surface to respond to the ocular stressors. DED in the chronic adaptive immunity phase is, in effect, a locoregional disease. The proliferation of T cells in the adaptive immunity phase and their amplified activation at the ocular surface cause damage that reinitiates the acute pro-inflammatory innate response, which, when accompanied by loss of immunoregulation, triggers a vicious circle 23 of pathological immune response.
Although the innate and adaptive immune responses differ and are triggered in distinct regions, key molecular interactions facilitating cellular migration are common to both. The interactions between 2 cell-surface factors—lymphocyte function-associated antigen 1 (LFA-1, an integrin protein) and its associated ligand, intercellular adhesion molecule 1 (ICAM-1)—are key to the proliferation and infiltration of immune cells. Basic knowledge of their roles in the dysregulation of ocular immunity is key to understanding current patient treatment strategies for DED.
The Ocular Surface Immune Response and DED
The ocular surface immune response involves both innate and adaptive mechanisms. 24 It occurs at the corneal surface, in ocular tissues and regional lymph nodes, and involves T helper (TH) cells, memory T cells, and regulatory T cells (Tregs). 25 It is a complex and tightly regulated process that is designed to protect and defend the ocular surface but, when dysregulated, can lead to DED.19,26–29
Specific insults or stress to the corneal surface triggers an innate immune response on the ocular surface that is maintained and regulated by the corneal epithelium. Mitogen-activated protein kinases (MAP-Ks)—specifically c-Jun N-terminal kinase (JNK), extracellular signal–related kinase (ERK), and p38—are activated and stimulate transcription nuclear factor kappa B (NF-κB), chemokines, and MMPs.30,31 A variety of immune cells reside at the ocular surface: natural killer immune cells, dendritic cells (which are the primary antigen-presenting cells [APCs]), macrophages, gamma delta (γδ) cells, and, to a limited extent, alpha beta (αβ) T cells (CD4+ and CD8+).26,32,33 During the innate response, specific immune cells at the ocular surface are activated to respond to the insult. 4 Cytokines and chemokines, specifically tumor necrosis factor alpha (TNF-α) and interleukin 1 and 6 (IL-1, IL-6), stimulate the maturation of APCs, while chemokine receptor 7 (CCR7) facilitates migration of mature APCs (mAPCs) in the afferent lymphatic vessels.33,34 These mAPCs are the primary immune cells that bridge the innate and adaptive immune responses. 33
In the adaptive immune response, mAPCs that have migrated to regional lymph nodes through the afferent arm facilitate the differentiation of naive T (TH0) cells into several types of mature T cells, such as (1) memory T cells, unique and specific to the antigen that caused the insult; (2) TH cells, which become the circulating effector T cells 35 ; and (3) Tregs, which modulate the immune response. 33 All of these T cells are generated in the lymph nodes and subsequently migrate to the site of inflammation, the conjunctiva, and the ocular surface.35–37 Although these have seemingly contradictory functions, they all play a role in a normal response, highlighting the multiplicities and complexities of the immune system.
Memory T cells may survive in an inactive state for long periods and proliferate upon restimulation (possibly at the ocular surface).35,38 TH cells can be autoreactive—that is, they can react to self-antigens. 35 Once activated, they secrete pro-inflammatory factors that sustain the immune response.27,39 Loss of homeostatic mechanisms of the lacrimal functional unit can lead to dysregulation of the natural immune responses. 40 Myriad systemic conditions are associated with loss of homeostatic mechanisms, including hormonal abnormalities, systemic and topical medications, preexisting ocular stress, lifestyle habits, and environmental conditions.5,18,41,42
Inflammation at the ocular surface can be both a cause and a consequence of DED. 23 The resolution of inflammation at the ocular surface is usually controlled by immunoregulatory processes, such as goblet cell secretion of the immunoregulatory cytokine transforming growth factor beta (TGF-β) and programmed death-ligand 1 regulation of activated effector T cells.35,40 When homeostatic control mechanisms fail and immunoregulatory mechanisms such as these are suppressed or overwhelmed, the immune response becomes amplified, particularly the adaptive response. This results in increased mAPC activity and increased production and recruitment of CD4+ TH cells to the ocular surface. At the ocular surface, the dysregulated activity of effector T cells includes increased release of pro-inflammatory cytokines, causing further inflammation and damage.39,43 This, in turn, reinitiates the innate immune response, thus creating a vicious circle. 23 The resolution of inflammation by immunoregulatory processes is bypassed and/or inadequate to address the sustained inflammation (Fig. 1).35,40
Ocular surface immune response.
Immunopathogenesis of DED
Broken down stepwise, the immunopathogenesis of DED is a 4-part process of initiation, amplification, recruitment, and damage/self-perpetuation (Figs. 2 and 3). The initiation phase of the innate immune response at the ocular surface induces localized acute inflammation. 27 Specifically, initiation involves upregulation of MAP-K JNK, ERK, and p38 stimulation of NF-κB. Various pro-inflammatory mediators, including cytokines (TNF-α, IL-1, IL-6), various inflammatory chemokines (CCL3, CCL4), and T cell–attracting chemokines (CCL5, CXCL9, CXCL10), are released into the corneal and conjunctival epithelium (Fig. 2: step 1).30,31 The release of IL-1 and TNF-α induces the activation and maturation of APCs, which are predominantly antigen-presenting dendritic cells (Fig. 2: step 2). 34 Upregulated ICAM-1 on lymphatic endothelial cells facilitates mAPC adhesion (Fig. 2: step 3).44,45 The mAPCs then migrate, in a CCR7-mediated manner, through the efferent arm of the ocular anatomy to regional lymph nodes (Fig. 2: step 4).46–48 The adaptive immune response involves the creation and recruitment of effector T cells.49,50 This occurs in the following manner: in the lymph node, mAPCs engage with TH0 cells through an immune synapse (partly mediated by LFA-1:ICAM-1 interaction and also T cell receptor and major histocompatibility complex, TCR:MHC) to cause differentiation of TH0 cells into several subsets of TH cells: TH1, TH2,TH17, and Tregs. T cell differentiation is determined by mAPC expression of a balance of signaling factors, including IL-6, IL-12, IL-17, IL-23, TGF-β, and interferon gamma (IFN-γ) (Fig. 2: step 5). 51 The proportions and balance of the various cytokines help to drive differentiation of the effector T cells into specific subtypes.
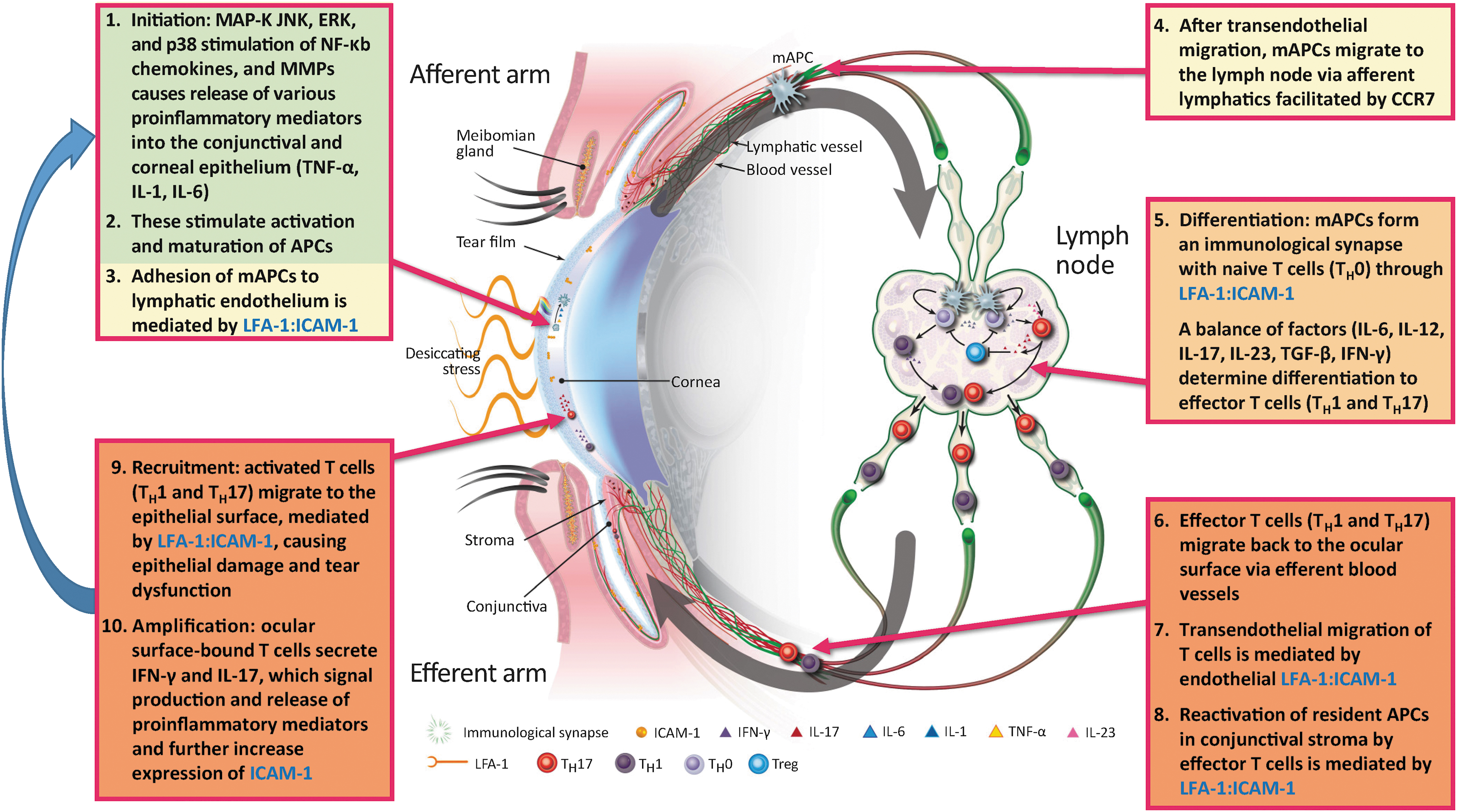
The immunoinflammatory pathway of dry eye disease. APC, antigen-presenting cell; CCR, chemokine receptor; ERK, extracellular signal–related kinase; ICAM-1, intercellular adhesion molecule 1; IFN-γ, interferon gamma; IL-, interleukin; JNK, c-Jun N-terminal kinase; LFA-1, lymphocyte function-associated antigen 1; mAPC, mature antigen-presenting cell; MAP-K, mitogen-activated protein kinase; MMP, matrix metalloproteinase; NF-κB, nuclear factor kappa B; TGF-β, transforming growth factor beta; TNF-α, tumor necrosis factor alpha; Treg, regulatory T cell. Adapted from Pflugfelder et al. 52
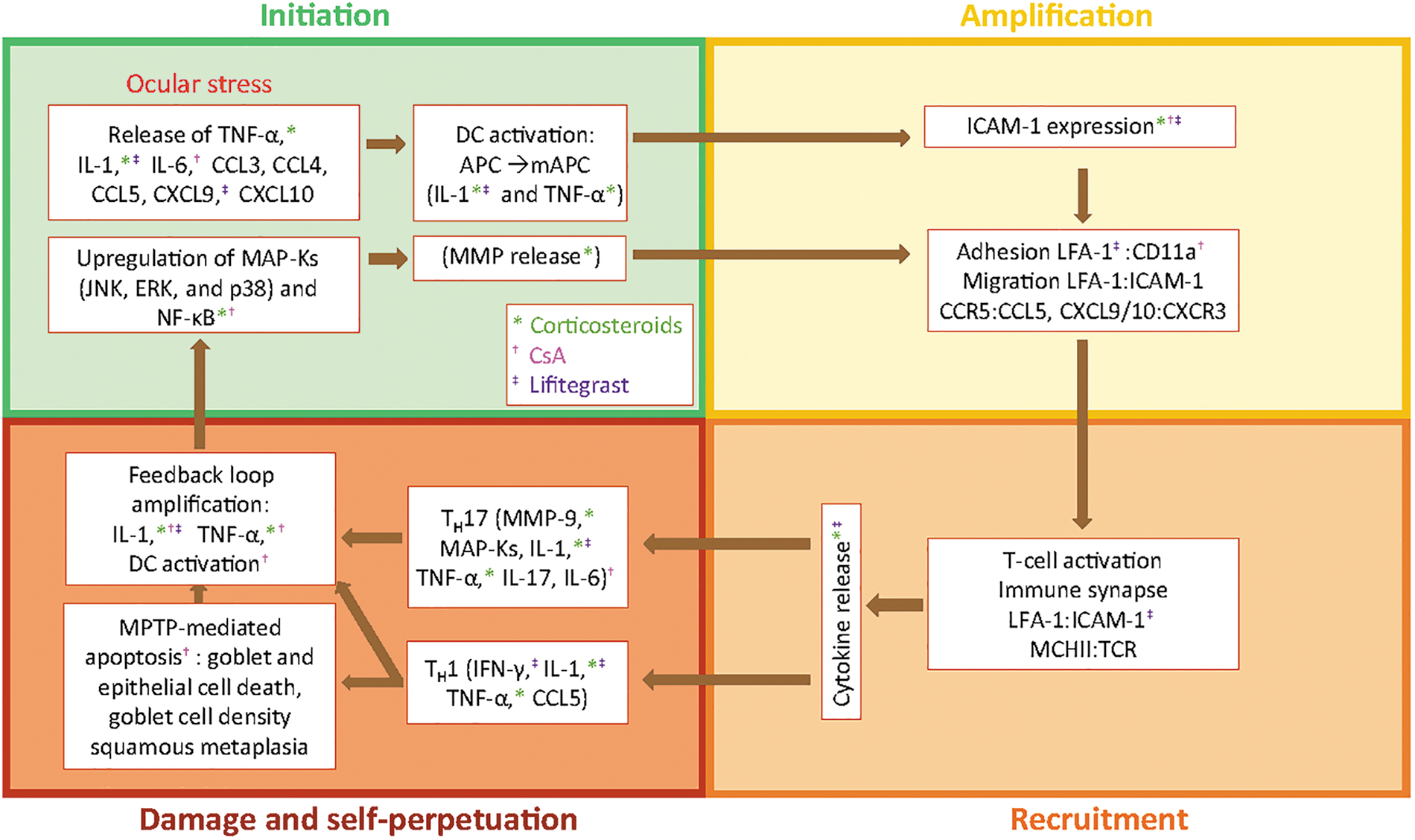
The phases of DED pathology and its processes, effectors, and actual treatments. Color-coded symbols denote specific DED treatment (corticosteroids, CsA, lifitegrast) modulation of DED effectors. APC, antigen-presenting cell; CCL, CXCL, chemokines; CCR, CXCR, chemokine receptors; CD11a, cluster of designation molecule 11a; CsA, cyclosporin A; DC, dendritic cell; DED, dry eye disease; ERK, extracellular signal–related kinase; ICAM-1, intercellular adhesion molecule 1; IFN-γ, interferon gamma; IL, interleukin; JNK, c-Jun N-terminal kinase; LFA-1, lymphocyte function-associated antigen 1; MAP-K, mitogen-activated protein kinase; mAPC, mature antigen-presenting cell; MHCII, major histocompatibility complex II; MMP, matrix metalloproteinase; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; NF-κB, nuclear factor kappa B; TCR, T cell receptor; TNF-α, tumor necrosis factor alpha.
Effector T cells then migrate through the efferent arm blood vessels to the conjunctival stroma (Fig. 2: steps 6 and 7).44,45,52–54 There, they reactivate resident mAPCs and are recruited to the ocular surface (Fig. 2: steps 8 and 9). 40 Specifically, TH1 and TH17 are the primary lymphocyte cells involved in the ocular surface damage and inflammation that is related to DED. 35 They release cytokines that alter the normal balance and cause epithelial damage and tear dysfunction.55,56 This, in turn, elicits an immune response and fuels the self-perpetuation cycle of DED pathogenesis (Fig. 2: step 10). 18 In nonpathogenic responses, the Tregs are responsible for dampening the effector response and regulating immunity. 55
T cell infiltration of the lacrimal gland and conjunctiva and amplification of the release of inflammatory cytokines are features of chronic inflammation in DED.23,27,40,57 Pro-inflammatory TH1 cells secrete the hallmark cytokine IFN-γ, which has been shown to promote conjunctival goblet cell loss and apoptosis of the ocular surface epithelium.37,58 In addition, IFN-γ is associated with conjunctival squamous epithelial hyperplasia. 37 TH17 cells secrete the hallmark cytokine IL-17 and promote production of the MMPs from corneal epithelial cells and fibroblasts.31,59,60 MMPs contribute to disruption of corneal epithelial barrier function, which contributes to further ocular insults. 26 IFN-γ and IL-17 also exert pathogenic effects by promoting production of pro-inflammatory cytokines, chemokines, MMPs, cell adhesion molecules, and prolymphangiogenic molecules. 40 These modulators lead to further damage of the ocular surface and thus amplify the cycle of inflammation.
Molecular Interactions Key to DED Pathogenesis
Integrins are cell-surface proteins expressed on a variety of cells. They are upregulated in response to insults and also play key roles in integrating signals among various cells.28,54,61 The integrin LFA-1 is a leukocyte cell-surface glycoprotein and a modulator of T cell activation and proliferation. 62 LFA-1 on T cells exists in an inactive or low-affinity binding state (bent conformation), but undergoes a conformational change to the high-affinity form of LFA-1 in response to inflammation. Specifically, this conformational change is due to TCR engagement in T cells and CXCR2 in neutrophils. 63 ICAM-1 is the natural ligand of LFA-1. During inflammation, acute-phase cytokines IL-1 and TNF-α induce upregulation of ICAM-1 expression on a variety of cells, including the vascular endothelium of patients with DED.36,44,64–66 Upregulation of ICAM-1 has several important functions, described further below (Fig. 2). 67
The LFA-1:ICAM-1 interaction has several important roles at key points in the immunoinflammatory pathway of DED (Fig. 2). 52 In general, binding of dendritic cells to the vascular endothelium through LFA-1:ICAM-1 interactions facilitates their migration to regional lymph nodes. 25 LFA-1:ICAM-1 interactions help to form the immune synapse between mAPCs and TH0 cells, leading to T cell differentiation. 61 The LFA-1:ICAM-1 interaction also aids the migration of activated T cells from blood vessels to the site of inflammation. 45 LFA-1:ICAM-1 interactions are thought to be necessary for the activation of effector T cells at the ocular surface through the immune synapse. 24 LFA-1:ICAM-1 interactions also play a role in the recruitment of T cells at the conjunctival epithelium and ocular surface. 4 Finally, the release of cytokines and chemokines is signaled through immune synapse/dual interactions between T cells and mAPCs using LFA-1:ICAM-1 and TCR:MHC, respectively.24,52–54 As the LFA-1:ICAM-1 interaction has a major role in ocular surface inflammation and the ocular immune response, treatment strategies focused on their association have emerged as therapeutic targets. 52 Whatever the precipitating cause of a patient's DED, treatment options are required that disrupt the chronic inflammatory process.20,21
Topical Ophthalmic Treatments for DED
Topical treatments for DED that are currently available in the United States and Canada include immunosuppressive agents (corticosteroids), immunomodulatory agents (cyclosporine ophthalmic emulsion 0.05% and cyclosporine ophthalmic solution 0.09%), and the recently available LFA-1 antagonist lifitegrast ophthalmic solution 5%. 68 The factors and processes involved in DED pathology, and the DED treatments that modulate these factors and processes, are summarized in Figure 3.
As potent inhibitors of multiple inflammatory mediators, topical corticosteroids are effective in interrupting the cycle of inflammation. 37 Suppression of NF-κB leads to suppression of acute-phase cytokines IL-1 and TNF-α, ICAM-1, MMPs, cytokines, chemokines, prostaglandins, and phospholipase A.65,69,70 Topical corticosteroids also reduce leukocyte infiltration of inflamed ocular tissues.71,72 However, known side effects (intraocular hypertension, cataracts, decreased wound healing, and predisposition to infection) limit their long-term use. 72 KPI-121, an investigational nanotechnology-based formulation of the corticosteroid loteprednol, has been evaluated in recently completed phase 3 clinical trials for patients with DED (NCT02793817 and NCT02813265); imminent publication of the results is expected. 73
Topical cyclosporin A (CsA) is indicated to increase tear production in patients with keratoconjunctivitis sicca. 74 CsA works by inhibiting the calcineurin–phosphatase pathway by intracellular complex formation with cyclophilin. 75 The clinical mechanism of action of CsA has not been fully elucidated, 74 but it includes increased natural tear production and increased goblet cell density.38,76 The scientific literature reports numerous molecular effects of CsA on DED immunopathophysiology, including inhibited T cell activation, 74 decreased cyclophilin-mediated gene transcription of IL-2 and IL-6, 39 and decreased epithelial and goblet cell apoptosis. 77
CsA inhibits the activation of T cells as measured by immunoactivation markers of human leukocyte antigen-D related and cluster of designation molecule 11a (CD11a) cell counts. 76 CD11a is a subunit of LFA-1.75,78 However, T cells that have already been activated can live for up to 164 days. 79
The original phase 3 clinical trial of CsA in patients with moderate-to-severe DED symptoms reported statistically significant improvements in subjective symptoms (P < 0.05), such as blurred vision, at 4 weeks. This was consistent with the improvements seen in objective signs: in corneal staining at 4 months and categorized Schirmer values (improved tear production) at 6 months. 80 The most common side effect of CsA treatment was ocular burning. 81 Other side effects included blurred vision, ocular itching, conjunctival hyperemia, discharge, foreign body sensation, and stinging.74,81 CsA ophthalmic solution 0.09%, recently approved in the United States, also showed increased tear production of ≥10 mm by Schirmer's test after 12 weeks of treatment compared with vehicle (16.8% vs. 8.6%, respectively). 82 The most common adverse event was mild, moderate, or severe instillation-site pain (the majority were mild). 82 Additional side effects included conjunctival hyperemia, blepharitis, eye irritation, headache, and urinary tract infection. 82 The exact molecular and cellular mechanism of action of this new CsA concentration and formulation remains to be elucidated.
Lifitegrast is approved in North America for the treatment of the signs and symptoms of DED in adult patients. 83 It is an LFA-1 antagonist with a proposed mechanism of action of specifically blocking the binding of ICAM-1 to LFA-1 with high affinity (Fig. 4). 25 Lifitegrast has the potential to act on both afferent and efferent arms of the immunomodulatory pathway in DED. 84 In the afferent arm (to the lymph node), it may block LFA-1:ICAM-1 interaction between dendritic cells on the ocular surface and endothelial cells of lymphatic tissues, thereby inhibiting migration and homing of naive dendritic cells to draining lymph nodes or activation of resting T cells at the ocular surface. 25 In the efferent arm (from the lymph node), lifitegrast may inhibit migration of activated T cells into the conjunctiva, recruitment in conjunctival epithelium, and secondary activation in ocular tissues. 52 In experimental models, lifitegrast inhibited IFN-γ, IL-1β, IL-10, and macrophage inflammatory protein 1 alpha levels. 85
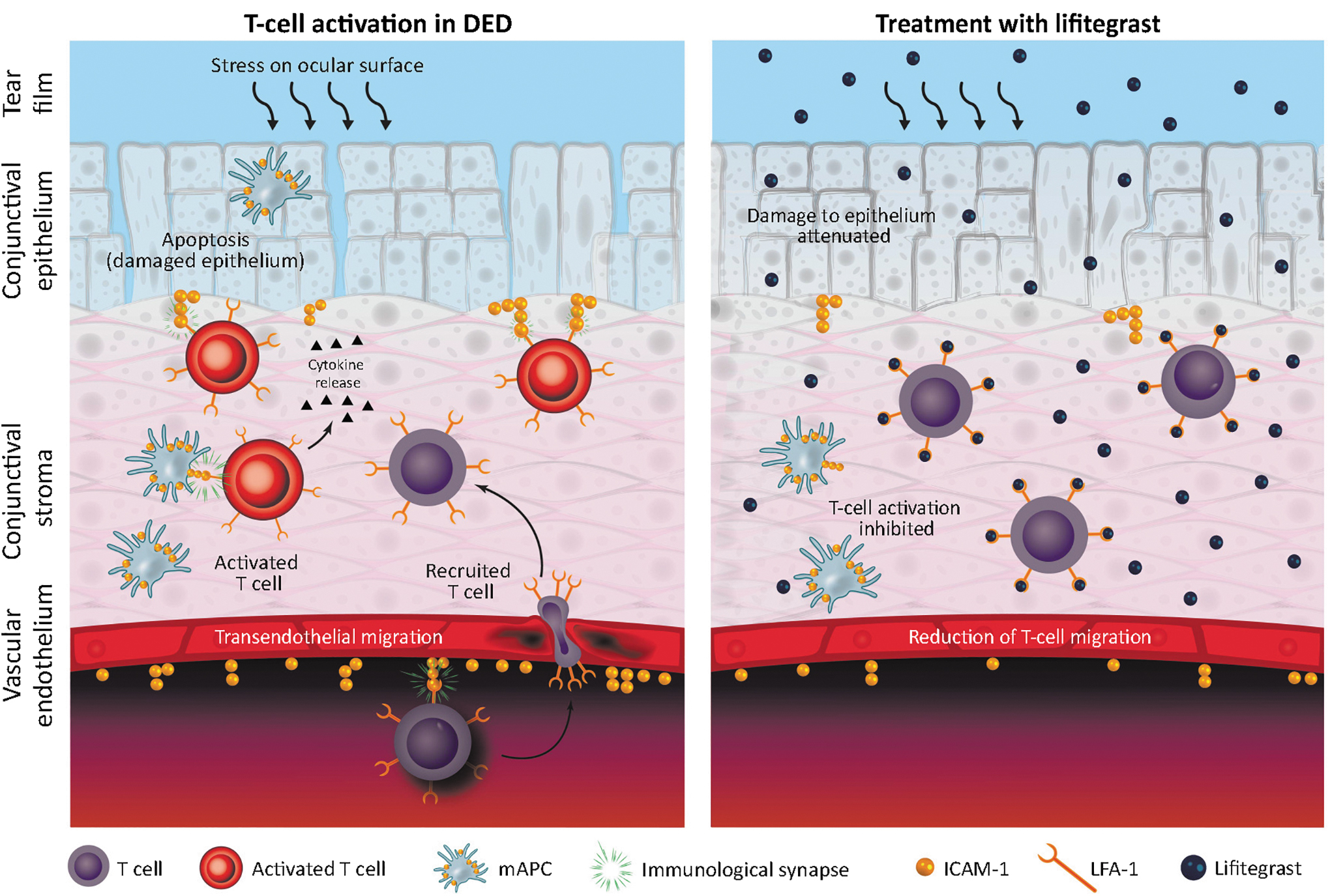
Effect of allosteric binding of lifitegrast to LFA-1 on LFA-1:ICAM-1 interactions. DED, dry eye disease; ICAM-1, intercellular adhesion molecule 1; LFA-1, lymphocyte function associated antigen 1; mAPC, mature antigen-presenting cell. Adapted from Perez et al. 25
A recent lifitegrast study in a murine model of desiccating stress provided evidence of lower expression of IFN-γ, CXCL9, and TH1-related genes compared with controls. 86 Mice treated with lifitegrast also had less corneal barrier disruption and greater conjunctival goblet cell density compared with controls. 86 Results of in vivo studies of lifitegrast in mice and dogs have demonstrated a potent dose-dependent inhibition of T cell activation, T cell recruitment, and release of cytokines that has been shown to correlate with clinical severity of DED.85,87–89 Likewise, in the chronic allergic eye disease murine model, an immune-mediated form of MGD is induced through a surprising TH17 and polymorphonuclear neutrophil response and has clinical parallels in humans. 90 Subsequent studies with lifitegrast treatment in this murine allergic eye disease model resulted in reduced severity of MGD and were associated with reduced Th17 cells and polymorphonuclear neutrophils in the conjunctiva (Saban DR. 2019; article in preparation). Clinical studies with lifitegrast have demonstrated improvements in signs and symptoms of DED.91–94 Combined results from phase 3 efficacy and safety studies (OPUS-1, 92 OPUS-2, 91 and OPUS-393) and a 1-year safety study (SONATA 94 ) demonstrated that lifitegrast alleviated symptoms of DED, with rapid onset of effect and improved signs of DED. The most common treatment-emergent adverse event was altered taste sensation (dysgeusia). 95
Additional anti-inflammatory therapies for the treatment of DED are under investigation. RGN-259 ophthalmic solution is a synthetic copy of the naturally occurring 43 amino acid protein thymosin β4 (Tβ4), which is the major constituent protein of trauma response and wound repair cells such as platelets, macrophages, and polymorphonuclear cells. 96 Tβ4 was found to promote corneal epithelial cell migration, decrease inflammation, and accelerate epithelialization in a murine-controlled adverse environment model of DED. 97 In a phase 2 trial with RGN-259, the combined primary end point of signs and symptoms of DED was not met. 97 However, multiple secondary end points showed improvements with RGN-259 in the phase 2 trial and a recently completed phase 3 trial (NCT02974907).97–100
Conclusion
DED is a multifactorial disease that is characterized by a sustained inflammatory response on the ocular surface that, if left untreated, can lead to chronic disease. Whether DED can be considered “progressive”—that is, whether any clinical characteristics of DED will progress without treatment—remains to be determined. Progress in discerning the underlying immunopathology of DED has led to advances in treatment that target specific inflammatory effectors/pathways. Such treatments are needed to break the cycle of DED and prevent chronic disease and progression.
Method of Literature Search
PubMed searches were performed and included articles from 1987 through 2018. Terms related to dry eye (“dry eye,” “dry eye disease,” “keratoconjunctivitis sicca,” “DED”) and the ocular immune response (“immune response,” “immunopathology,” “pathophysiology,” “inflammation,” “ocular surface immune response”) were included. Related searches on specific ocular immune effectors, including “T cell,” “dendritic cell,” “goblet cell,” “integrin protein,” “adhesion molecule,” “lymphocyte function-associated antigen 1,” “LFA-1,” “intercellular adhesion molecule 1,” “ICAM-1,” and specific topics regarding DED prevalence and DED-associated comorbidities were also conducted in PubMed and Google. English translations of abstracts were reviewed from all languages. Articles were read from English sources. From the abstracts, articles were reviewed that addressed areas of ocular surface pathology, immunology, inflammation, and associated treatments of DED.
Footnotes
Acknowledgments
The authors thank Shirley Louise-May, PhD, of Excel Scientific Solutions, who provided medical writing assistance, supported by Takeda and Novartis. Takeda and Novartis supported the preparation of this article and reviewed the final draft for medical and scientific accuracy.
Author Disclosure Statement
L.M.P. has been a consultant and speaker's bureau member for Allergan, Lumenis, Shire, Sun, and TearLab. V.L.P. has been a consultant and speaker's bureau member for Alcon, EyeGate, Mallinckrodt, Trefoil, and Shire. D.R.S. has received an investigator-initiated research grant from Shire. M.C.L. has been a consultant for Health Advances, Novartis, and Shire and has received research funding from CooperVision, Inc., Essilor USA, Johnson & Johnson, Leo Lens, Inc., Orinda Pharma, Verily Life Science, and Viewpoint Pharmaceuticals. P.N. declares no conflicts of interest.
Funding Information
Takeda and Novartis funded medical writing support for the preparation of this article and reviewed the final draft for medical and scientific accuracy.