
Abstract
Aims:
To examine the efficacy of tacrolimus ophthalmic suspension 0.1% in treating severe allergic conjunctivitis.
Methods:
This was a multicenter, randomized, double-masked, placebo-controlled clinical trial. Fifty-six patients with severe allergic conjunctivitis in whom topical antiallergic agents and corticosteroids had been ineffective were randomized to tacrolimus or placebo treatment. Patients were treated either with tacrolimus or placebo twice-daily for 4 weeks. Severity of objective signs in palpebral and bulbar conjunctiva, limbus, and corneal involvement was assessed using 4 grades. Seven subjective symptoms were evaluated by visual analog scale (VAS) assessment. The primary efficacy endpoint was change in the total score of objective signs at the end of treatment. The secondary efficacy endpoints included change in the score for each objective sign and change in the VAS for each subjective symptom. Safety was assessed based on the severity and the incidence of adverse events.
Results:
Mean change from baseline in total score for objective signs was significantly greater in the tacrolimus (−5.6 ± 5.1) than in the placebo group (−0.1 ± 4.5; P < 0.001). Tacrolimus significantly improved giant papillae (P = 0.001) and corneal involvement (P = 0.005). Five subjective symptoms (itching, discharge, hyperemia, lacrimation, and foreign body sensation) were significantly better in the tacrolimus than in the placebo group. The most frequent treatment-related adverse event in the tacrolimus group was mild ocular irritation upon topical instillation, which was well-tolerated.
Conclusion:
Tacrolimus ophthalmic suspension 0.1% is effective in treating severe allergic conjunctivitis.
Introduction
S
Since the report of BenEzra and colleagues,4,5 the efficacy of topical cyclosporine against severe allergic ocular diseases has been documented by many investigators.6–11 However, high-concentration cyclosporine, in oil-based formulations such as maize, results in intense stinging sensation and blurred vision upon instillation, leading to poor compliance.8 To overcome these problems, 0.05% cyclosporine emulsion was used for AKC and VKC patients and shown to be effective,10,11 though it failed to exhibit sufficient clinical efficacy in some steroid-dependent cases.12
Tacrolimus is a nonsteroidal immunomodulator isolated from Streptomyces tsukubaensis.13 Because of its potent immunosuppressive effect,14 topical tacrolimus is expected to exhibit excellent therapeutic efficacy in suppressing abnormal immune responses related to allergic ocular diseases. Based on the efficacy of tacrolimus ointment in treating atopic dermatitis, it has been successfully used at concentrations of 0.02%–0.1% in ointment for various immune-mediated ocular surface disorders.15–20 However, the efficacy of tacrolimus eye drops has not been prospectively evaluated in a randomized controlled trial. We conducted a placebo-controlled trial to evaluate the efficacy of tacrolimus ophthalmic suspension 0.1% in patients with severe allergic conjunctivitis refractory to conventional treatment.
Methods
Study design
This was a multicenter, randomized, double-masked, parallel-group, placebo-controlled clinical trial performed to compare tacrolimus ophthalmic suspension 0.1% and vehicle control placebo (ophthalmic solution free of tacrolimus). Tacrolimus ophthalmic suspension 0.1% was formulated to be an isotonic aqueous suspension using polyvinyl alcohol as dispersive agents. It is preserved with benzalkonium chloride and submicron size of tacrolimus particle was suspended in it. Dose of tacrolimus was based on the results from dose-ranging study. In that trial, tacrolimus ophthalmic suspension 0.01%, 0.03%, and 0.1% were tested. As a result, 0.1% showed stronger improvement and similar safety profile compared with 0.01% and 0.03%. Therefore, we chose 0.1% as an optimal dose (Astellas Pharma Inc., Japan, 2003). Patients were enrolled at 14 investigational institutions (Appendix) between February and September 2004.
Patients
Male and female patients 6 years of age or older with severe allergic conjunctivitis (AKC and VKC) refractory to topical antiallergic agents and corticosteroids with moderate to severe giant papillae were eligible. Patients who received systemic administration or subconjunctival injection of corticosteroids, and/or ophthalmic or systemic administration of immunosuppressants within 2 weeks prior to the study; underwent cryosurgery or surgical excision of giant papillae within 4 weeks prior to the study; were receiving desensitization therapy or immune modulation therapy; had infectious eye disease; were pregnant, lactating, or planning pregnancy during the study period; exhibited drug hypersensitivity; were diabetic; or had cardiac, renal, hepatic, or pancreatic disease were excluded.
Before this study, the risks and benefits expected from participation were fully explained to all patients, and informed consent was obtained from each participant. This study was conducted in compliance with Good Clinical Practice and the Declaration of Helsinki. Before the study began, implementation of it was approved by the site Institutional Review Board.
Study procedure
Eligible patients were randomly allocated to the tacrolimus or placebo group by the minimization method with assignment factors of diagnosis of allergic conjunctivitis and participating medical institutions (implemented with Visual Basic Version 6.0, Microsoft). Patients were instructed to instill a single drop of either tacrolimus or placebo twice-daily for 4 weeks. Study drugs vials were covered by opaque film for masking. If topical corticosteroids, vasoconstrictors, and/or nonsteroidal anti-inflammatory agents were used before study entry, they were discontinued and switched to study drugs. Concomitant use of topical or systemic antiallergic agents was allowed, but only without dose change during the 4-weeks prior to the study and treatment period. Key codes for study drugs were kept secure by Astellas Pharma Inc. (Tokyo, Japan). These codes were broken after fixation of the database for this trial.
Objective signs and subjective symptoms were observed at baseline (before treatment) and 1, 2, and 4 weeks after treatment initiation. Ten objective signs for the palpebral conjunctiva (hyperemia, edema, follicles, papillae, and giant papillae), bulbar conjunctiva (hyperemia and chemosis), limbus (Trantas’ dot and edema), and corneal involvement were assessed using 4 grades (0 = Normal; 1+ = Mild; 2+ = Moderate; or 3+ = Severe, Table 1).21
G
The severity of subjective symptoms (itching, discharge, hyperemia, lacrimation, eye pain, foreign body sensation, and photophobia) was assessed by visual analog scale (VAS). The VAS scored 0 mm (none) to 80 mm (very severe). Ocular safety was assessed based on changes in visual acuity, IOP, pupil diameter, and clinical findings for the iris, lens, anterior chamber, and fundus. Laboratory examinations including hematological analysis, blood biochemical tests, and urinalysis were conducted.
The primary efficacy endpoint was change from baseline in total score for objective signs at end of treatment (at 4 weeks after treatment initiation or final observation if treatment was discontinued before week 4). Change from baseline in total score for objective signs at each study visit, change in score for each objective sign, and change in VAS score for symptoms were secondary efficacy endpoints. Safety was assessed based on the severity and incidence of adverse events.
Statistical analysis
The 2-sided 5% significance level was applied to statistical hypothesis testing, with confidence intervals (CIs) given as 2-sided 95% CIs.
Patients who met the eligibility criteria, had at least one dose of study drug, and had at least one efficacy assessment after baseline were included in the efficacy analysis. For each patient, the eye with higher total score for clinical findings was selected for efficacy assessment. For the primary endpoint, change from baseline in total score for objective clinical signs at end of treatment, the 2 groups were compared by 2-sample t-test. For the secondary endpoints, the 2 groups were compared by 2-sample t-test or Mann–Whitney U-test. Patients who had at least one dose of study drug and had safety assessment after baseline were included in the safety analysis. Safety assessments were performed based on the incidence of adverse events using Fisher’s exact test.
A sample size of 25 patients per group was calculated to detect a mean difference of 5.0 for the primary endpoint with 80% power and α = 0.05 (2-sided) assuming a standard deviation (SD) of 6.1.
Statistical analysis was performed with SAS Version 6.12 and Version 8.2 (SAS Institute, NY).
Results
Patient disposition and demographics
Of the 56 patients with severe allergic conjunctivitis (41 AKC and 15 VKC), 28 each were randomly allocated to the tacrolimus and placebo groups (21 AKC and 7 VKC for the tacrolimus group and 20 AKC and 8 VKC for the placebo group). All AKC patients had atopic dermatitis. Male patients predominated, accounting for 89.3% of patients (25/28) in both groups. Mean (±SD) age was 17.9 ± 9.1 years in the tacrolimus and 15.2 ± 8.1 years in the placebo group. Mean total score for objective clinical signs at baseline was 15.3 ± 4.9 in the tacrolimus and 17.1 ± 4.5 in the placebo group. All patients had received topical antiallergic agents (sodium cromoglicate, amlexanox, ketotifen fumarate, pemirolast potassium, ibudilast, and/or levocabastine hydrochloride) within 4 weeks prior to treatment, and continued to use them during the treatment period, except one patient in the tacrolimus group. Twenty-two patients (78.6%) each in the tacrolimus and placebo groups had been treated with corticosteroid eye drops including fluorometholone, betamethasone, or dexamethasone before the study.
Treatment with study drugs was discontinued for one patient in the tacrolimus group due to aggravation of allergic conjunctivitis with adverse event, while 5 patients in the placebo group were withdrawn from the study due to aggravation of allergic conjunctivitis and one patient due to an adverse event. All patients were included in safety and efficacy assessments.
Outcome measures
Total score for objective signs in the tacrolimus group steadily decreased from weeks 1 through 4. Mean change from baseline (±SD) in total score for objective signs at end of treatment was −5.6 ± 5.1 in the tacrolimus and −0.1 ± 4.5 in the placebo group, and significant improvement in the former (P < 0.001) was shown in the primary analysis. The difference between 2 groups was −5.5, and CI ranged from −8.1 to −2.9. The mean changes from baseline in AKC and VKC patients in the tacrolimus group were lower than in the placebo group (Fig. 1).
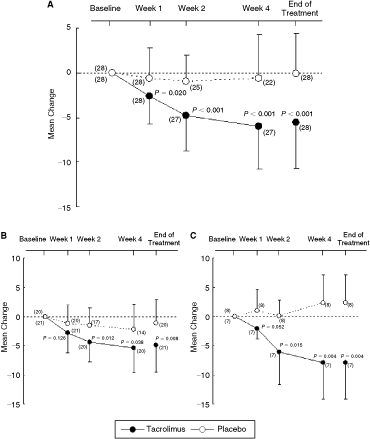
(
Figure 2 shows changes in distribution of severity of giant papillae, corneal involvement, palpebral conjunctival hyperemia, and bulbar conjunctival hyperemia. Treatment with tacrolimus yielded significantly greater improvement in giant papillae from week 1 through week 4. The proportions of patients with no or mild giant papillae were 60.7% (17/28) in the tacrolimus and 10.7% (3/28) in the placebo group at end of treatment. Corneal involvement was also significantly improved in the tacrolimus group compared with the placebo group from week 1 through week 4. No corneal epithelial disturbance was observed in 40.0% (10/25) of patients in the tacrolimus and 7.7% (2/26) in the placebo group at end of treatment. Representative photographs of changes in giant papillae and corneal involvement with tacrolimus are shown in Figure 3. Palpebral and bulbar conjunctival hyperemias were improved with time from week 1 to week 4 as giant papillae and corneal involvement. Palpebral conjunctival edema (P = 0.006), follicles (P = 0.001), and limbal edema (P = 0.033) were also significantly improved in the tacrolimus group compared with the placebo group at week 4.
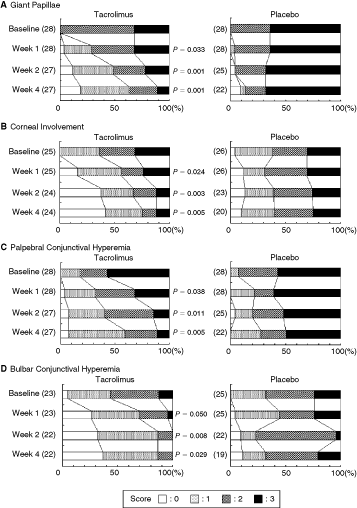
Distributions of scores for giant papillae, corneal involvement, palpebral conjunctival hyperemia, and bulbar conjunctival hyperemia. Significance of differences was evaluated by Mann–Whitney U-test for change in score compared with placebo group. ( ): patient number.
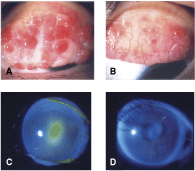
(
On VAS assessment, 5 of 7 symptoms (itching, discharge, hyperemia, lacrimation, and foreign body sensation) were significantly improved in the tacrolimus group compared with the placebo group at end of treatment (Fig. 4).
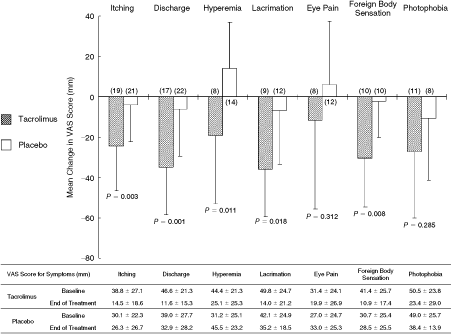
Visual analog scale (VAS) for each subjective symptom. The figure shows mean (SD) change from baseline in each symptom at end of treatment. Significance of differences was evaluated by 2-sample t-test compared with placebo group. Range for each symptom: 0–80 mm. ( ): patient number. Vertical bars: standard deviation.
In assessment of visual acuity, IOP, pupil diameter, and clinical findings for the iris, lens, anterior chamber, and fundus, hematological analysis, blood biochemical tests, and urinalysis, no abnormal change related to test drugs was observed. No serious adverse events were reported during the study. The incidence of treatment-related adverse events was significantly higher (P = 0.044) in the tacrolimus group (46.4% [13/28]) than in the placebo group (17.9% [5/28]). The most frequent treatment-related adverse event in the tacrolimus group was mild ocular irritation (42.9% [12/28]). As ocular infections, there was one case of suspected herpetic keratitis in the tacrolimus group and one of hordeolum in the placebo group. In the tacrolimus group, one case of throat irritation was observed.
Discussion
The efficacy of topical tacrolimus treatment for severe allergic ocular diseases has recently been intensively studied by many investigators. For example, Vichyanond and coauthors reported marked clinical responses in 10 VKC patients with 0.1% tacrolimus ointment.15 Joseph and coauthors reported that one patient with severe AKC responded dramatically to 0.03% tacrolimus ointment.16 Miyazaki and coauthors reported that 1 VKC and 5 AKC patients who had been refractory to conventional treatment were successfully treated with 0.02% tacrolimus ointment.19 Nevertheless, the efficacy of tacrolimus requires evaluation in larger numbers of patients in a randomized, controlled trial.
Consistent with previous reports, we found that tacrolimus ophthalmic suspension 0.1% twice-daily yielded marked improvement in objective signs in severe allergic conjunctivitis refractory to conventional treatment. Improvement was observed as early as 1 week after initiation of treatment and continued up to week 4. Similar findings were observed in the subgroups of AKC and VKC patients. Tacrolimus also improved symptoms; the 2 most frequent symptoms, itching and discharge, were effectively suppressed by it. These findings verified the excellent efficacy of tacrolimus ophthalmic suspension in treating severe allergic conjunctivitis.
Corneal scarring due to severe, prolonged epithelial disturbance such as shield ulcer is known to impair visual acuity in patients with severe allergic conjunctivitis.1–3 In patients with conjunctival proliferative change, suppression of inflammation of giant papillae is essential for successful management, since corneal epithelial disturbance results from cytotoxic effects of inflammatory molecules released from lesions at the inflamed palpebral conjunctiva. With topical tacrolimus, giant papillae became less inflamed and flattened, with profound decrease in ropy discharge, finally resulting in inflamed flat giant papillae in about 60% of patients and disappearance of giant papillae in 20% of patients. Improvement of corneal involvement was rapid as well, and synchronized with suppression of inflammatory response of giant papillae. Tacrolimus ophthalmic suspension significantly improved the score for corneal epithelial disturbance including shield ulcers without affecting visual acuity. However, moderate to severe giant papillae (score ≥2+) persisted in some patients even after treatment with topical tacrolimus for 4 weeks, suggesting that longer treatment might have been needed to improve efficacy.
One of the expected advantages of nonsteroidal immunomodulator is safe tapering or discontinuation topical corticosteroids. Daniell and colleagues reported that low-concentration topical cyclosporine was not beneficial as a steroid-sparing agent in steroid-dependent allergic conjunctivitis.12 In the present study, patients treated with topical corticosteroids with insufficient effects could be switched to topical tacrolimus without rebound phenomenon. These findings suggest the usefulness of topical tacrolimus 0.1% as a steroid-sparing substitute for treating severe allergic conjunctivitis due to its potent immunosuppressive effect, which is up to 100 times stronger than that of cyclosporine.14 To reach more objective conclusions regarding steroid-sparing effects of topical tacrolimus, better planned clinical studies are needed.
Tacrolimus ophthalmic suspension 0.1% was well-tolerated. The most frequent tacrolimus-related adverse event was ocular irritation. In the tacrolimus group, ocular irritation occurred in 12 of 28 patients (42.9%), but no severe ocular irritation was observed. Opportunistic infection can often occur with use of immunosuppressants or immunomodulators. In the tacrolimus group, one AKC patient developed corneal lesion suspected to be herpes simplex virus infection, although precise virological diagnosis was not performed. Therefore, close follow-up of patients receiving topical tacrolimus, particularly those with atopic dermatitis, is thus always needed, since they are susceptible to herpes simplex virus infection. On the other hand, the incidence of neither herpes simplex virus nor varicella zoster infection was found to be increased with use of tacrolimus ointment in patients with atopic dermatitis in long-term follow-up studies.22,23 Whether topically instilled tacrolimus elicits reactivation of HSV requires further observation.
In conclusion, tacrolimus ophthalmic suspension 0.1% was proven effective in improving objective clinical signs and subjective symptoms of severe allergic conjunctivitis refractory to topical antiallergic agents and corticosteroids compared with placebo. Despite mild irritation upon topical instillation, tacrolimus ophthalmic suspension 0.1% is considered feasible for treatment of severe allergic conjunctivitis.
Footnotes
Acknowledgment
This study was supported by Astellas Pharma Inc., Japan, and Senju Pharmaceutical Co., Ltd., Japan.
Author Disclosure Statement
Dr. Ohashi was contracted with Astellas Pharma Inc. and Senju Pharmaceutical Co., Ltd. as medical adviser for this clinical trial. Dr. Kumagai and Dr. Nakagawa were contracted with Astellas Pharma Inc. as medical adviser. Dr. Hayashi was contracted with Astellas Pharma Inc. as biostatistical advisor. Other authors have no commercial or proprietary interest in the products or the company mentioned in this article.