
Abstract
Background:
Oral and inhalation-based cannabidiol (CBD) administration has been clinically evaluated for various therapeutic indications, alongside widespread off-label use. However, the long-term exposure kinetics and varied bioavailability have not been fully characterized.
Methods:
Human CBD plasma concentration–time profiles from six studies evaluating the oral administration of Epidiolex® and three studies evaluating inhalation-based delivery were obtained. A four-compartment pharmacokinetic (PK) model with Weibull-based oral absorption kinetics was employed to describe the long-term PKs of CBD. Furthermore, a Cedergreen–Ritz–Streibig model was applied to evaluate nonmonotonic oral bioavailability.
Results:
CBD was extensively distributed into tissue compartments with varied kinetics resulting in a long plasma terminal elimination half-life of >134 h in humans. For once-a-day oral dosing, the plasma trough concentrations require >70 days to reach a steady state. The oral bioavailability of CBD for different doses administered in fasted state follows a nonmonotonic pattern with an inverted U-shaped profile. Oral administration of CBD under fed state or subjects with hepatic impairment yields higher oral bioavailability with varied exposure. In contrast, inhalation-based delivery of CBD, while delivering a similar systemic delivered dose compared with oral dosing due to high device losses, bypasses first-pass metabolism and can be efficient.
Conclusion:
CBD PKs vary across different doses due to nonmonotonic oral bioavailability, and inhalation-based delivery could minimize such variability in humans. The delayed attainment of steady state and prolonged terminal half-life, resulting from differential but extensive tissue distribution, needs to be considered when dosing CBD in the long term. These fundamental findings are critical for establishing dose–exposure relationship for further clinical evaluation of novel CBD-based therapies.
Introduction
Epidiolex®, an oral solution of cannabidiol (CBD) in sesame oil, has been approved by the U.S. Food and Drug Administration for the treatment of seizures associated with Lennox–Gastaut syndrome or Dravet syndrome in patients 2 years or older.1,2 The human pharmacokinetics (PKs) of CBD have been evaluated in single and multiple dose clinical studies. 1 Long-term administration of CBD has been shown to be safe with mild-to-moderate side effects. 2 A starting dose of 2.5 mg/kg taken twice a day (5 mg/[kg day]) followed by a dose titration of ≤10 mg/kg taken twice a day (20 mg/[kg day]) is recommended based on tolerability and clinical response. 2
CBD is a highly lipophilic compound with low solubility. 3 The oral bioavailability of CBD is poor due to partial absorption from gastrointestinal tract and extensive first-pass metabolism. Although the absolute bioavailability has not been determined, 2 the oral bioavailability can significantly vary based on the food intake, with a high-fat meal increasing the maximum concentration (Cmax) by fivefold and area under curve (AUC) by fourfold. 2 CBD is primarily metabolized in the liver by different cytochrome P450 (CYP) enzymes such as CYP2C19 and CYP3A4.1,4
CBD can also undergo glucuronidation by several Uridine 5'-diphospho-glucuronosyltransferase (UGT) isoforms, such as UGT1A9, UGT2B7, and UGT2B17. 1 It is estimated that ∼70–75% of the orally absorbed dose is cleared by the liver during the first-pass metabolism, 5 thereby generating different metabolites. In vitro studies have reported that CBD reversibly inhibits CYP1A2, CYP2B6, CYP2C8, CYP2C19, and CYP3A4 in a time-dependent manner. 2 Furthermore, CBD is highly protein bound (>94%) with a large volume of distribution (>21,000 L).1,2 The terminal elimination half-life of single oral dose is estimated to be ∼14–17 h, and with multiple oral dosing, the estimated terminal elimination half-life is 56–61 h.1,2
The medical use of CBD is receiving increased attention due to its potential therapeutic benefits. Clinical evidence supports the use of CBD to treat anxiety, pain, psychosis, schizophrenia, and other therapeutic indications. 6 The PK of CBD administered orally is associated with high variability potentially due to differential rates of dissolution, interindividual differences in metabolism, body mass index, fasting and fed states, etc. 7 In addition to oral, inhalation-based delivery is a common administration route for off-label use. CBD has high permeability, and inhalation of aerosolized CBD results in rapid systemic delivery with a shorter Tmax (time to reach Cmax). 8 Inhaled CBD bypasses the first-pass metabolism, leading to low levels of metabolite generation. 8
Compartmental PK models are simplified nonmechanistic models that enable the calculation of PK parameters, prediction of PK at various doses, dosing regimen selection, and evaluation of population-level variability in PKs. 9 They typically consist of one or two physiological components, that is, central compartment representing the plasma and rapidly perfused tissues, and optional peripheral compartments representing tissues with differential perfusion and retention capacities.
The number of peripheral compartments typically depends on the concentration–time profiles. 9 Most concentration–time profiles are commonly described using one, two, or three compartmental PK models. 9 Earlier studies have successfully modeled CBD PKs using a three-compartmental PK model7,10,11 but did not describe long-term PKs resulting from multiple dosing regimens. Obtaining an understanding of long-term exposure kinetics is critical to improving the efficacy and safety of CBD. This study aims to describe the long-term PKs and bioavailability of CBD administered by different routes.
Methods
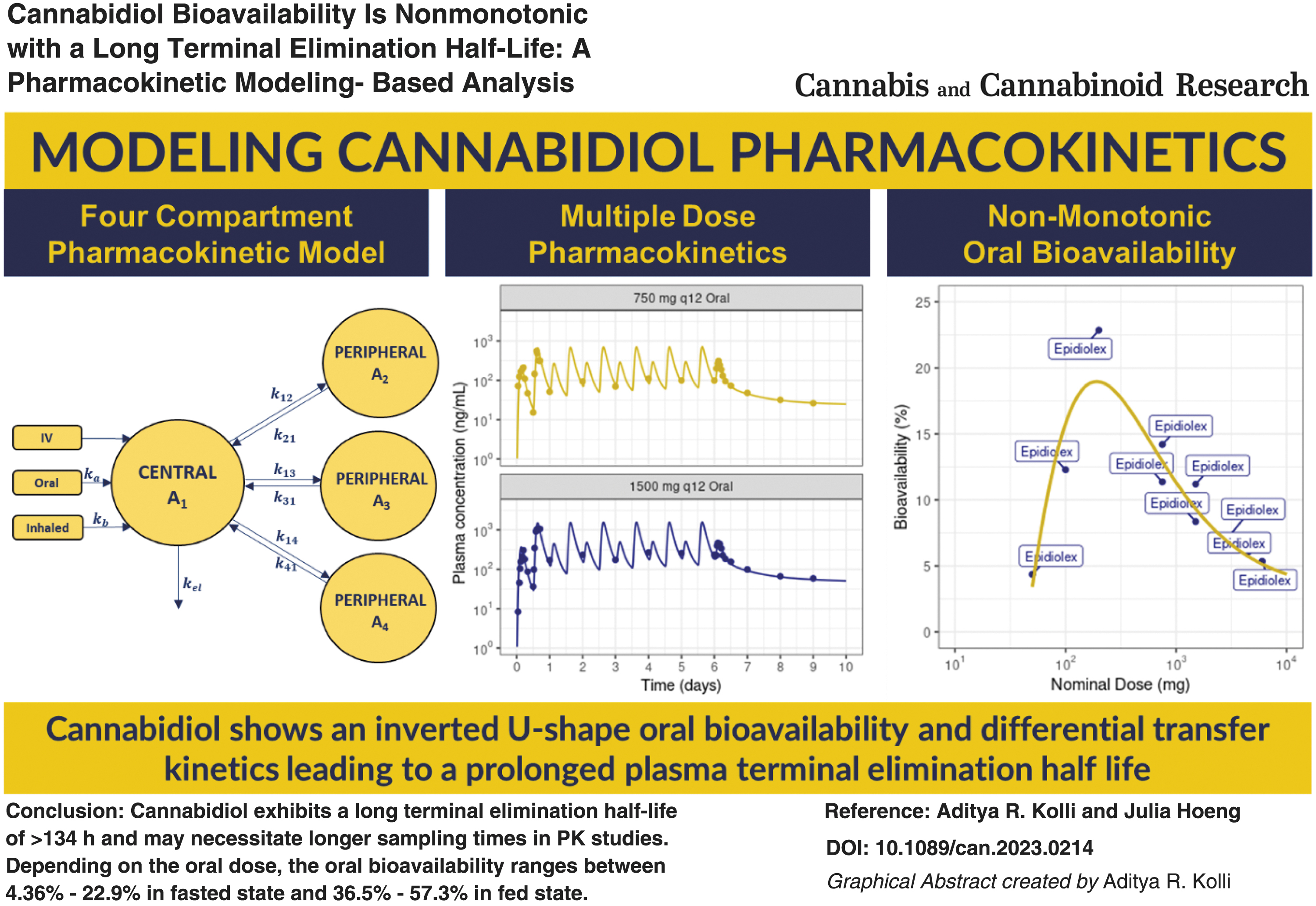
A four-compartmental PK model (Fig. 1) comprising one central compartment and three peripheral compartments was employed to describe the plasma concentration–time data of CBD. The oral absorption of CBD over time (t) was modeled using a Weibull function (WB).10,12 The model Equations (1)–(8) are given below,
Schematic of a four compartmental PK model for CBD. CBD, cannabidiol; PK, pharmacokinetic.
where oraldepot and inhaleddepot represent dosing depots for their respective routes of administration; A1 represents the amount of CBD in central compartment, V1 stands for the volume of central compartment; A2, A3, and A4 denote the amounts of CBD in the three peripheral compartments; k12, k21, k13, k31, k14, and k41 are the rate constants governing distribution between central and peripheral compartments; kel is the elimination rate constant; ka is the apparent absorption rate, γ represents the Weibull absorption shape factor for oral delivery; kb is the absorption rate for inhaled CBD; and Cplasma is the concentration of CBD in plasma.
The four-compartment model was simultaneously fitted to both single intravenous data 13 and long-term multiple oral dosing PKs 1 to estimate model parameters (Table 1). The coefficient of variation was calculated for the long-term multiple oral dosing PKs. Model predictions within a factor of two of the experimental data were considered acceptable. To assess goodness-of-fit (GOF) and model performance, the mean absolute percentage error (MAPE) and the coefficient of determination (R 2 ) for measured and predicted values were determined using a linear regression model.
Cannabidiol Four Compartment Pharmacokinetic Model Parameter Estimates
Fixed.
%CV, coefficient of variation.
GOF was assessed, and a visual check was performed to select the four-compartmental model. The compartmental distributional rate constants (k12, k21, k13, k31, k14, and k41), V1, and kel were held constant, whereas oral bioavailability or inhaled systemic percentage delivered dose (F) and the rate of absorption (ka and kb) were estimated to predict PKs for different oral and inhaled doses. Next, the Cedergreen–Ritz–Streibig model
14
was applied to evaluate the oral bioavailability at various doses using Equation (9).
where α represents the rate of hormetic effect, b is the slope around 50% maximal bioavailability, c is the bioavailability at infinite dose, d has no interpretation, e is the dose at which bioavailability is 50% of maximal bioavailability, and f governs the rate of change at doses close to 0. A more comprehensive explanation of Equation (9) is provided by Cedergreen et al. 14 The terminal elimination half-life of CBD is calculated based on the slope of the last 15% of the simulated PK curve. CBD oral bioavailability is known to largely vary based on the oral formulation.
Hence to evaluate the influence of oral dose on clinical PKs, data selection was limited to “Epidiolex.” Furthermore, all inhalation-based CBD PKs were selected. Human CBD plasma concentration–time profiles from one study evaluating intravenous dosing, 13 six studies evaluating the oral administration of Epidiolex,1,2,8,15–17 and three studies evaluating inhalation-based delivery8,13,18 were obtained from the literature.
Experimental data from the literature were digitized using WebPlotDigitizer (https://apps.automeris.io/wpd/) and no proprietary PK data were generated in this study. The model was developed in R (v4.1.2) 19 using “mrgsolve” (v0.11.1), 20 parameter estimation was performed using “GenSA” (v1.1.7), 21 and plots were generated using “ggplot2” (v3.3.5). 22 The model parameters and model code are provided in Supplementary Data.
Results
Intravenous PKs of CBD
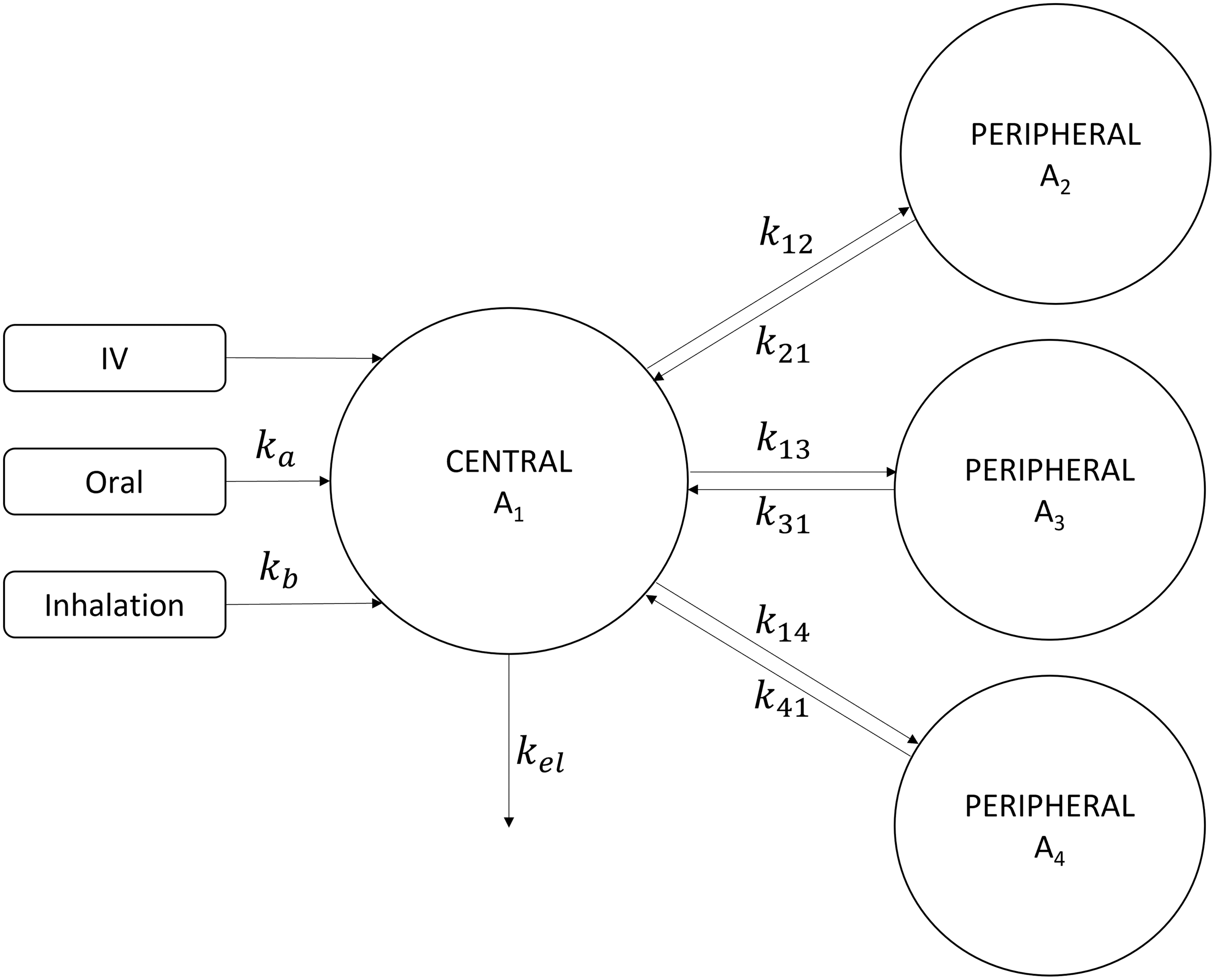
The three-compartmental PK model developed previously, 10 and the four-compartmental PK model (Fig. 1) effectively described the plasma concentration–time (0–72 h) profile for a single intravenous infusion of 20 mg CBD (Fig. 2A). The simulated PKs for both models showed good agreement to experimental PK data (Fig. 2A) with R2 of 0.99 and MAPE values<14.8%. The three- and four-compartmental PK models described plasma CBD concentrations within a twofold deviation of experimental measurements (Fig. 2B).
Three- and four-compartmental PK model-predicted CBD plasma concentration–time profiles
The plasma terminal elimination half-life of CBD for a three- and four-compartmental PK model is ∼15 h for a single dose 3-day PKs and is in agreement to literature reported values for a single dose. 1 An extrapolation of the three-compartmental PK model simulated PKs beyond 3 days resulted in a CBD plasma terminal elimination half-life of 20.6 h, indicating the differential tissue distribution of CBD. However, the three-compartmental PK model was inadequate to describe long-term multiple dosing PKs (presented in later section), and a four-compartment PK model was selected for further analysis.
The four-compartment PK model estimated that plasma terminal elimination half-life is >134 h due to nonlinear PKs of CBD. Given the multiphasic elimination of CBD, the terminal elimination half-life and choice of PK model can vary based on the terminal end-points measured and dosing regimens evaluated.
Oral bioavailability and inhaled systemic dose of CBD
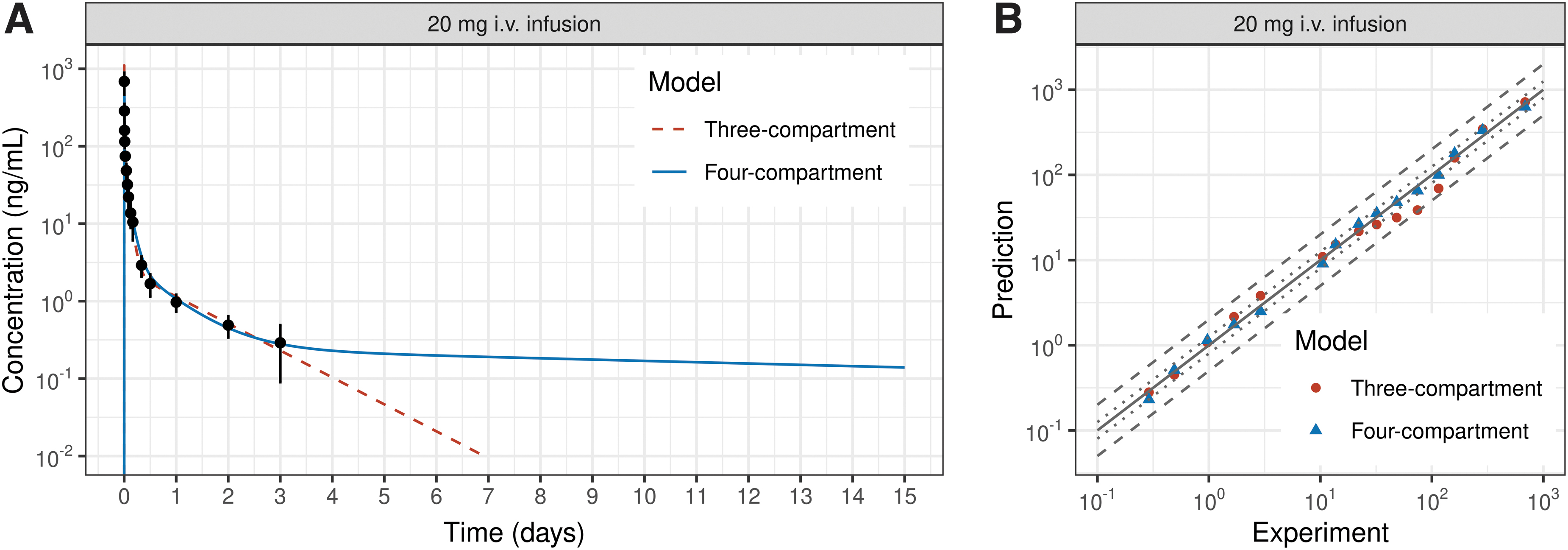
The four-compartmental PK model-predicted PKs for smoking 20 mg CBD-containing cannabis placebo cigarettes (Fig. 3) is estimated to deliver 31% of the nominal dose 13 (Fig. 4). Studies have evaluated inhalation-based CBD delivery using specialized devices such as the Volcano Medic® vaporizer. 18 Inhalation of 100 mg of vaporized CBD using Volcano Medic vaporizer resulted in a model-predicted Cmax of 188 ng/mL with 9.79% of the nominal dose delivered to systemic circulation (Figs. 3 and 4). In contrast, 100 mg of CBD-dominant cannabis vaporized using Volcano Medic vaporizer resulted in a higher model-predicted Cmax of 497 ng/mL, with 25.9% of the nominal dose delivered.
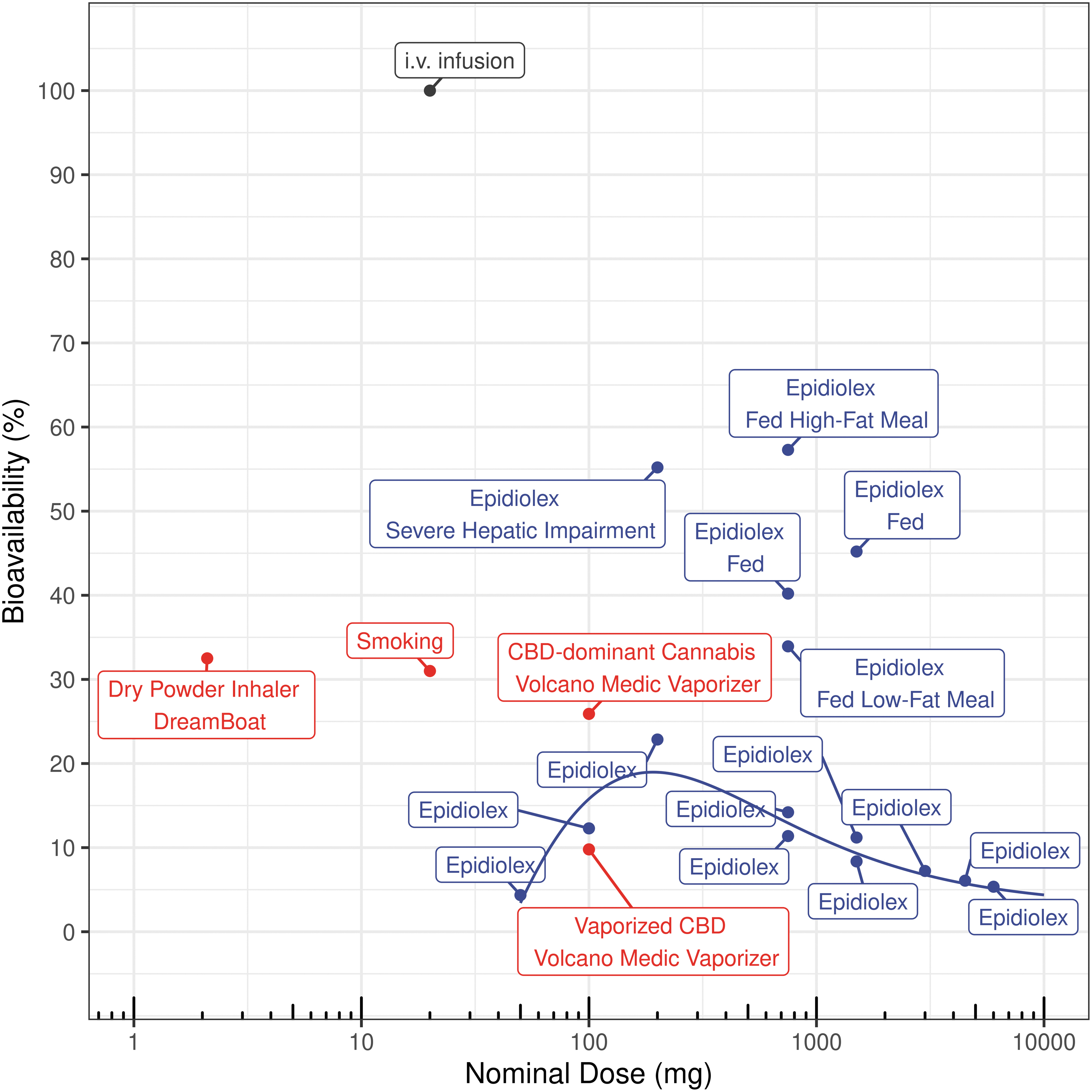
Oral bioavailability and inhaled bioavailability (i.e., systemic delivered dose [%]) for different nominal doses of CBD (dots). The line represents the Cedergreen–Ritz–Streibig model-fitted oral bioavailability of various doses of CBD administered in fasting state. The values are also available in Supplementary Table S1. “Epidiolex” is the bioavailability in fasting state condition.
Although the Cmax for the dry powder inhaler is lower due to a low nominal dose of 2.1 g, the percentage delivered dose (32.5%) is higher than that of the Volcano Medic vaporizer (Figs. 3 and 4). The decline in systemic delivered dose with increased nominal dose can be attributed to inhalation device and formulation-driven efficiency (Fig. 4).
As formulation influences oral bioavailability, studies evaluating oral solution of CBD in sesame oil (Epidiolex) were selected. CBD PKs for oral doses ranging between 50 and 6000 mg CBD (Fig. 3) were fitted to estimate oral bioavailability and the rate of absorption (Fig. 4 and Supplementary Table S1). In the fasted state, the bioavailability of CBD exhibited a nonmonotonic pattern with an inverted U-shaped profile, which could be described using a Cedergreen–Ritz–Streibig model (Supplementary Table S2).
The Cedergreen–Ritz–Streibig model estimated that a dose of 191 mg could potentially yield maximum bioavailability of 19% for fasted-state oral administration. At a lower dose of 50 mg CBD, the oral bioavailability was estimated to be ∼4.36%, as most of the CBD may undergo first-pass metabolism in the gut wall and liver, resulting in a lower overall bioavailability. For oral doses of 750 mg and more, the bioavailability potentially reached the solubility limit in the gut fluid, resulting in a decline in oral bioavailability 12 (Fig. 4). For example, a 6000 mg of oral dose of CBD yielded an oral bioavailability of 5.35%. The bioavailability of a 750 mg oral dose with low- and high-fat meal-fed states increased to 36.5–57.3%, accompanied by up to a fivefold rise in the plasma Cmax and AUC (Figs. 3 and 4).
This increase in Cmax and AUC for a 750 mg oral dose of CBD administered in fed state (high-fat meal) exceeded the systemic exposure achieved with a 6000 mg oral dose of CBD administered in fasted state (Supplementary Table S1). These results underscore the importance of the absolute dose and fast/fed state conditions on oral bioavailability of CBD.
In subjects with no hepatic impairment, the administration of a 200 mg of oral dose resulted in a 22.9% oral bioavailability. In contrast, in subjects with severe hepatic impairment, 16 oral bioavailability increased to 58.9%, as a greater amount of CBD was able to bypass hepatic first-pass metabolism (Fig. 4). Furthermore, the clearance (kel) of CBD in severe hepatic impairment was lowered by 40.5% leading to a 5.15-fold increase in resulting exposure (AUC) (Fig. 3 and Supplementary Table S1).
CBD PKs for multiple doses
Multiple dosing regimens and long-term PKs of CBD evaluated previously 1 were simulated using the three- and four-compartment PK models. The model-predicted bioavailability of morning dose of 750 and 1500 mg CBD, administered in fasting state, was estimated to be 14.1% and 11.2%, respectively. However, the model-predicted bioavailability for evening CBD dose, administered in fed state, was ∼45%. Although the three-compartmental PK model predicted short-term PK in both fasting and fed states, it inadequately described the terminal PK measurements (Fig. 5).
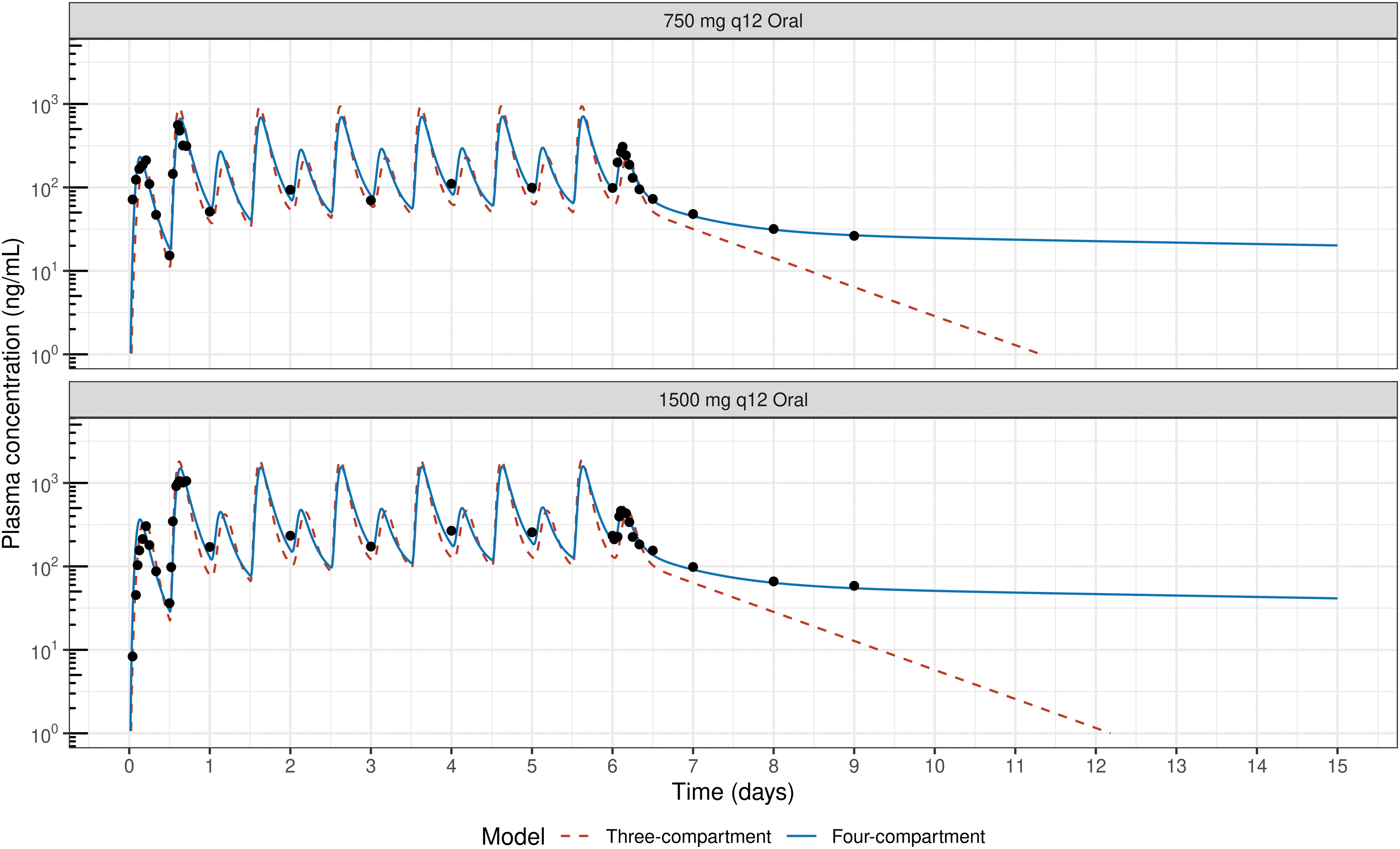
The four-compartmental PK model was able to better predict the multiple dosing PK including the terminal PK measurements with a prolonged terminal elimination half-life (Fig. 5). The four-compartmental PK model estimated that CBD trough concentration (Ctrough) and Cmax for the 750 mg two times a day (bis in die, BID) dosing regimen 1 to be 93.9 and 712 ng/mL respectively; for the 1500 mg BID dosing regimen, 1 these values were 202 and 1587 ng/mL (Fig. 5).
The four-compartmental PK model, with differential intercompartmental rate constants (k12, k21, k13, k31, k14, and k41) and long terminal elimination half-life, indicates delayed distributional equilibrium. Hence, the Ctrough over time for different doses of CBD was derived (Fig. 6). Ctrough reached steady state after 150 days of daily dosing for the simulated dosing regimens. However, 95% of the maximal Ctrough was estimated to be reached on day 70 for once-a-day (QD) dosing regimen. Similarly, plasma Cmax reached 95% of the maximal concentration on day 36.
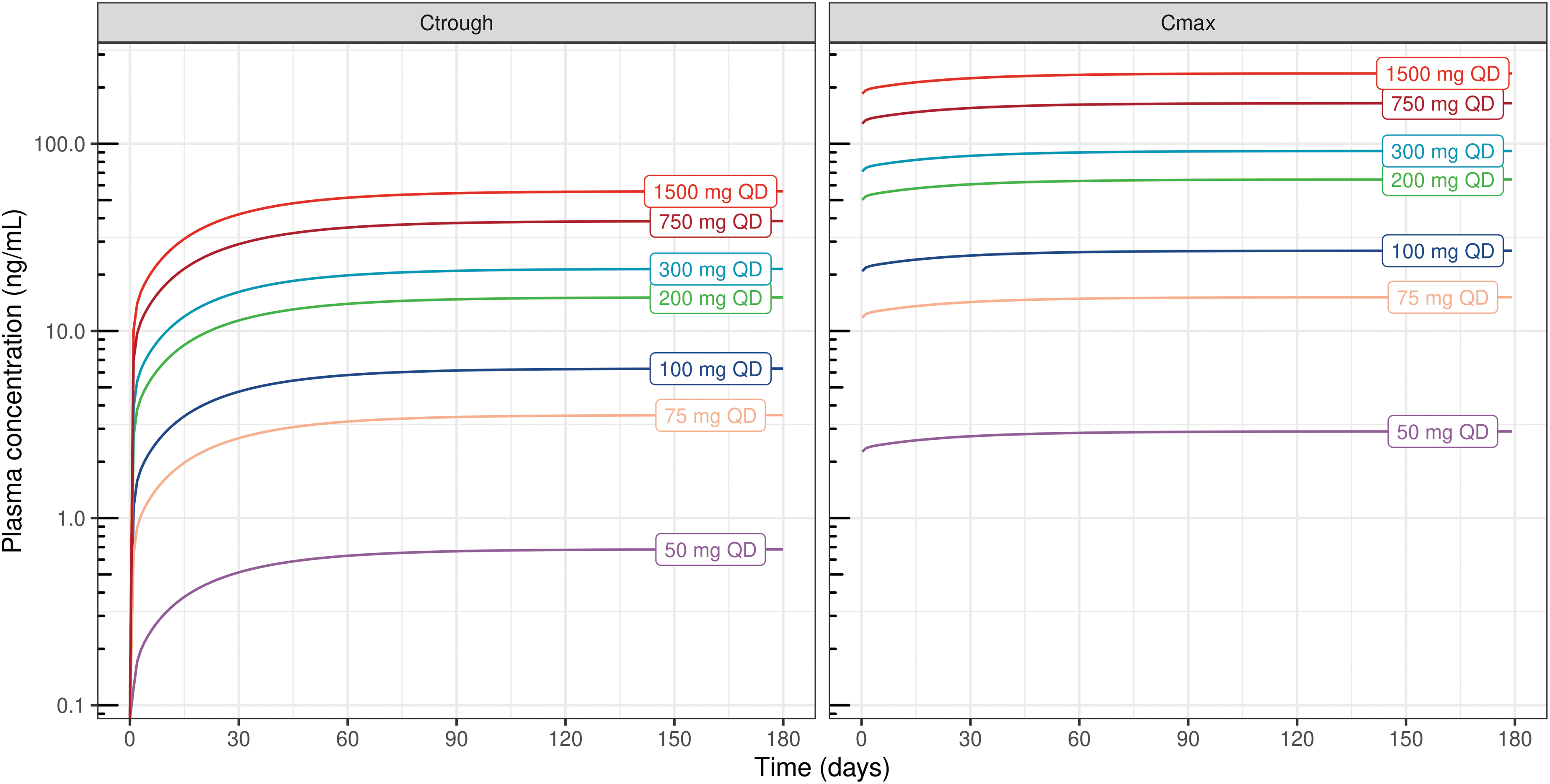
CBD plasma trough (Ctrough) and maximum (Cmax) concentrations for different oral doses of CBD for daily administration of 180 days by assuming fasting state. QD, once-a-day dosing.
Discussion
The four-compartmental PK model describes the human plasma concentrations–time profiles for intravenous infusion, oral and inhalation-based delivery of CBD, and estimates a long terminal elimination half-life of >134 h due to delayed distribution equilibrium. Such prolonged blood plasma terminal elimination half-life has been reported for other cannabinoids such as Δ 1 -tetrahydrocannabinol (98.4 h). 23 Since the three-compartment PK model for single intravenous infusion describes the CBD PKs well (Fig. 2), the four-compartment PK model was not previously explored. The three-compartment PK model describes CBD PKs for single or multiple oral dosing regimens over a short period but is inadequate to capture long-term PK profiles (Figs. 3 and 5).
The four-compartmental PK model, with its varied intercompartmental kinetics, describes the long-term CBD PK for both single and multiple oral dosing regimens (Figs. 2, 3, and 5). Such complex and differential distribution kinetics are observed for other compounds, such as anesthetics, requiring a four compartmental PK model to describe long-term PKs. 24 These multiple compartments potentially represent various groups, including the vessel-rich group, muscle group, slow-to-moderately perfused tissue group, and adipose tissue group. In rodents, CBD accumulates in different tissues, with adipose tissue showing levels 100-fold higher than muscle and liver due to its high lipophilicity. 25
CBD has also been reported to accumulate in rat lymphatics, with lymph CBD concentrations 250-fold higher than in plasma. 26 Upon repeated dosing, CBD levels have been detected in various joint tissues 27 and synovial fluid 28 in rodents. Such varied tissue distribution in rodents could also be present in humans. The human PK model estimates varying rates of CBD distribution to multiple compartments (Fig. 1) resulting in multiphasic elimination with vastly different PK profiles. However, the changes in Cmax and Ctrough beyond 70 days of QD oral dosing are predicted to be minimal (Fig. 6).
Furthermore, identifying the rise in Ctrough beyond 70 days in humans could be experimentally challenging considering the large variability in bioavailability. Different values for the terminal elimination half-life of CBD are reported in the literature because CBD PK studies are limited by design and analytical limits of detection, making the estimation of “true” terminal elimination half-life challenging. One potential approach to overcome this is to administer a loading dose followed by a maintenance dose over several days and then abstaining to determine the terminal elimination half-life. Importantly, due to the challenges in accurately capturing terminal elimination PK profiles, PK models based on short-term sampling times may be inadequate for describing CBD PKs.
Numerous formulations containing varying amounts of CBD and administered through different routes result in diverse systemic exposures. 10 A comprehensive understanding of CBD oral bioavailability is crucial for therapeutic development. CBD bioavailability assessment across different doses exhibits a nonmonotonic pattern with an inverted U-shaped profile (Fig. 4). As CBD undergoes extensive first-pass metabolism, low doses may be almost entirely metabolized, resulting in an extremely low bioavailability, whereas the decline in bioavailability at higher doses may be due to potential saturation of CBD solubility in the gastrointestinal fluid. 12
The PK model suggests that doses between 150 and 300 mg may offer maximum oral bioavailability in fasted state and requires to be evaluated in clinic. In contrast, CBD administered in a fed state shows a significantly higher bioavailability of 36.5–57.3% depending on the type of meal consumed, resulting in up to a fivefold increase in systemic exposure (AUC). 2 Moreover, the oral bioavailability and overall systemic exposure are elevated in hepatic impaired patients. This increased and differential bioavailability underscores the significance of hepatic first-pass metabolism of CBD in normal subjects.
Although various devices are available to vaporizing different materials, such as cannabis whole plant, flower tops, and extracts, the emitted dose from Volcano Medic Vaporizer is estimated to be at ∼57.8%, which is then captured in balloons for inhalation. 29 However, ∼23.6% of the dose is lost due to condensation on to the walls of the balloon and other parts of the vaporizer, resulting in significant device loses. 29 Although the dose is consumed over 6–10 breaths in ∼10 min, the physicochemical properties of the emitted aerosol are not fully characterized to estimate airway-deposited (or absorbed) doses. 30
Nevertheless, the PK model estimates that inhaled CBD is rapidly absorbed into systemic circulation, with 9.79% of vaporized CBD and 25.9% CBD-dominant cannabis from nominal doses being delivered to systemic circulation. Most vaporizers require multiple inhalations, can deliver high doses through thermal aerosolization, and may be associated with generation of byproducts. These aspects remain to be further evaluated. In contrast, low inhaled doses can be delivered using a dry powder inhaler, which has similar levels of device losses but results in a 32.5% nominal dose being delivered. 8
Although the percentage of oral bioavailable dose (fasted and fed states) and inhaled systemic dose may be comparable (Fig. 4), the generation and circulation of CBD metabolites are expected to be significantly lower in inhalation-based delivery, making it the preferred route of administration.
The experimental PK data obtained from different studies are limited and no specific experimental data have been generated in this study to enable such PK modeling-based analysis, and hence, limitations exist. The volume of the central compartment estimated based on a single intravenous infusion study 13 is applied to describe PKs in other studies. It remains unclear whether the estimated oral bioavailability of 200 mg in the fasted state represents absolute bioavailability or is influenced by variations in the volume of central compartment.
However, the relative bioavailability of 200 mg CBD in fed state is higher than that of the fasted state (Supplementary Table S1). The Cedergreen–Ritz–Streibig model estimates a maximum oral bioavailability of 19% in the fasted state (Fig. 4). Furthermore, it remains uncertain whether oral bioavailability in the fed state follows a nonmonotonic pattern and requires additional studies evaluating dose–exposure relationship. In addition, the mucociliary clearance (MCC) and the oral absorption of inhaled CBD were not incorporated in the PK model, as the MCC-cleared dose would either undergo first-pass metabolism or is poorly absorbed, resulting in minimal contribution to systemic PKs.
CBD is an inhibitor of CYP enzymes and long-term exposure could potentially reduce CYP-mediated clearance, thereby extending the terminal elimination half-life. However, a previous study 31 reported minimal changes in CYP-mediated clearance during clinically relevant repeated dosing of up to 1500 mg/day. Although CBD has been shown to undergo enterohepatic circulation in rats, 32 moderate or higher oral doses of CBD in humans did not exhibit a characteristic second peak and were not considered. 1
In conclusion, CBD exhibits a long terminal elimination half-life of >134 h in humans, necessitating longer sampling times in PK studies. CBD also has varied distribution equilibrium, and it may take >70 days of daily dosing to reach steady-state Ctrough. Although the oral bioavailability in a fasted state is estimated to range between 4.36% and 22.9%, depending on the administered dose, the bioavailability in a fed state is estimated to be between 36.5% and 57.3%. Notably, the human oral bioavailability of CBD administered in a fasted state follows a nonmonotonic pattern with an inverted U-shaped profile.
Inhaled CBD is estimated to deliver 33.9% of nominal dose to the systemic circulation; due to device-related losses, the overall exposure to generated metabolites will be lower than that achieved through oral dosing, making inhalation-based CBD delivery an efficient option. Although this preliminary model framework requires further validation at the population level, it provides critical insights for designing long-term CBD dosing regimens for PK studies aimed at treating various therapeutic indications.
Footnotes
Acknowledgments
We thank Philip Morris International for providing the computational resources and Vectura Fertin Pharma for paying the article open access fee. The authors also thank Renata Murgasova for the discussions.
Authors' Contributions
A.R.K. contributed to conceptualization, methodology, software, formal analysis, data curation, validation, visualization, and writing—original draft. J.H. was involved in conceptualization and writing—review and editing.
Author Disclosure Statement
A.R.K. is a paid employee of Philip Morris International. J.H. is an employee of Vectura Fertin Pharma.
Funding Information
No funding was received for the study.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.