
Abstract
Abstract
Introduction:
Few animal studies have evaluated the pharmacological effects of Δ9-tetrahydrocannabinol (THC) in relation to its pharmacokinetic properties. Understanding this relationship is essential, however, if comparisons are to be drawn across conditions—such as sex, age, and route of administration—which are associated with variations in the absorption, metabolism, and distribution of THC. As a first step toward addressing this gap, in this report, we describe a rapid, sensitive, and accurate method for the quantification of THC and its main oxidative metabolites, and apply it to representative rodent tissues.
Materials and Methods:
The sample workup procedure consisted of two steps: bulk protein precipitation with cold acetonitrile (ACN) followed by phospholipid removal by elution through Captiva-Enhanced Matrix Removal cartridges. The liquid chromatography/tandem mass spectrometry (LC/MS-MS) protocol utilized a commercially available C18 reversed-phase column and a simple methanol/water gradient system. The new method was validated following Food and Drug Administration (FDA) guidelines, and was applied to the quantification of THC and its main oxidative metabolites—11-hydroxy-Δ9-tetrahydrocannabinol (11-OH-THC) and 11-nor-9-carboxy-Δ9-tetrahydrocannabinol (11-COOH-THC)—in plasma and brain of mice treated with a single intraperitoneal dose of THC (10 mg/kg).
Results:
ACN precipitation and column elution effectively depleted matrix constituents—most notably choline-containing phospholipids—which are known to interfere with THC analysis, with average recovery values of >85% for plasma and >80% for brain. The LC conditions yielded baseline separation of all analytes in a total run time of 7 min (including re-equilibration). The 10–point calibration curves showed excellent linearity (R2>0.99) over a wide range of concentrations (1–1000 pmol/100 μL). Lowest limit of quantification was 2 pmol/100 μL for all analytes, and lowest limits of detection were 0.5 pmol/100 μL for THC and 11-OH-THC, and 1 pmol/100 μL for 11-COOH-THC. Intraday and interday accuracy and precision values were within the FDA-recommended range (±15% of nominal concentration). An application of the method to adult male mice is presented.
Conclusions:
We present a fast and sensitive method for the analysis of THC, which should facilitate studies aimed at linking the pharmacokinetics and pharmacodynamics of this compound in animal models.
Introduction
The pharmacological properties of Δ 9 -tetrahydrocannabinol (THC), the psychotropic constituent in cannabis, continue to be at the center of substantial research efforts.1–5 Most studies on the health impact of THC utilize adult male rodents, but the expanding legal use of cannabis products by women and men of all ages has highlighted the need to understand the impact of this drug in animal models that better capture variations in sex, age, and route of exposure. The validity of these comparative studies depends, however, on the ability to relate the pharmacodynamics of THC with its unique pharmacokinetic profile and metabolic transformations. Indeed, it is known that several key determinants of THC distribution and metabolism—including plasma lipid-binding proteins, cytochrome P450 (CYP) enzymes, and body fat stores—markedly change with sex and age.6–8
The first step in addressing this issue is to develop analytical methods that allow the rapid and accurate quantification of THC and its primary oxidative metabolites not only in blood and other accessible biological fluids but also in organ systems where these lipophilic compounds act and/or tend to accumulate (e.g., brain and white adipose tissue). Several useful protocols intended for toxicological or forensic measures of THC in accessible body fluids are available.9–12 However, such protocols cannot be directly applied to internal organs without consideration for tissue-specific matrix effects, which include unpredictable shifts in chromatographic behavior and ion suppression. 13 This study describes a fast, accurate, and sensitive method that applies recent developments in matrix removal technology and liquid chromatography/tandem mass spectrometry (LC/MS-MS) to the quantification of THC and its two main oxidative metabolites—11-hydroxy-Δ 9 -tetrahydrocannabinol (11-OH-THC) and 11-nor-9-carboxy-Δ 9 -tetrahydrocannabinol (11-COOH-THC)—in rodent tissues.
Materials and Methods
Solvent and chemicals
Authentic THC, 11-OH-THC, and 11-COOH-THC standards and their corresponding 2 H-containing derivatives were obtained from Cerilliant (Round Rock, TX). THC for animal administration was purchased from Sigma-Aldrich (St. Louis, MO). LC/MS-grade water and methanol were from Honeywell (Muskegon, MI). LC/MS-grade acetonitrile (ACN), isopropanol, and acetone were from Sigma-Aldrich. Formic acid (FA) was from Thermo Fisher (Houston, TX); 1-stearoyl-2-hydroxy-sn-glycero-3-phosphocholine (18:0 lysophosphatidylcholine, 18:0 LPC) and 1-stearoyl-2-arachidonoyl-sn-glycero-3-phosphocholine (18:0–20:4 phosphatidylcholine, 18:0–20:4 PC) were from Avanti Polar Lipids (Alabaster, Alabama).
Standard preparation
Stock solutions (100 μM) of authentic THC, 11-OH-THC, and 11-COOH-THC and their corresponding 2 H-containing internal standards (ISTDs) were prepared in methanol. External standards were prepared to yield stock solutions of the following concentrations (in μM): 20, 10, 2, 1, 0.1, and 0.01, which were kept at −80°C until use. ISTD stock solutions were prepared to yield 10 μM final concentration for each standard.
Equipment
Chromatographic separations were carried out using a 1200 series LC system (Agilent Technologies, Santa Clara, CA), consisting of a binary pump, degasser, thermostated autosampler, and thermostated column compartment coupled to a 6410B triple quadrupole mass spectrometric detector (MSD; Agilent Technologies).
Animals
Male C57BL6/J mice (20–25 g) and Sprague-Dawley rats (225–250 g) were purchased from Charles River Laboratories (Wilmington, MA). The animals were housed in groups of four in rooms maintained on a 12-h light/12-h dark cycle (lights on at 6:30 AM and off at 6:30 PM) under constant temperature conditions (22±2°C) and relative humidity (55–60%). Food and water were available ad libitum. Rats were used as source of plasma and brain tissue. Mice were used for in vivo experiments. All procedures were approved by the Institutional Animal Care and Use Committee at the University of California, Irvine, and carried out in strict accordance with the National Institutes of Health guidelines for care and use of experimental animals.
Drug and treatments
For in vivo experiments, THC was dissolved in a vehicle consisting of Tween 80/saline (5:95 v/v), 14 and was administered by intraperitoneal (IP) injection. Mice were sacrificed at set time points (15, 30, 60, and 120 min) after THC or vehicle administration.
Tissue collection
Mice and rats were anesthetized with isoflurane and killed by cervical dislocation or decapitation, respectively. Brains were removed and dissected on an ice-cold glass plate and immediately flash frozen on dry ice. Blood was collected by cardiac puncture into an ethylenediaminetetraacetic acid (EDTA)-rinsed syringe and transferred into 4-mL tubes containing spray-coated K2EDTA. Plasma was separated by centrifugation at 1450×g at 4°C for 15 min and transferred into polypropylene tubes. All tissue samples were stored at −80°C until analyses.
LC conditions
Analytes were separated on an Eclipse XDB C18 column (1.8 μm, 3.0×50.0 mm; Agilent Technologies). The mobile phase consisted of water containing 0.1% FA as solvent A and methanol containing 0.1% FA as solvent B. The flow rate was kept at 1.0 mL/min. The gradient conditions were as follows: starting 75% B to 89% B in 3.0 min, changed to 95% B at 3.01 min, and maintained till 4.5 min to remove any strongly retained materials from the column. Equilibration time was 2.5 min. The column temperature was maintained at 40°C and the autosampler at 9°C. The total analysis time, including re-equilibrium, was 7 min. The injection volume was 5 μL. To prevent carryover, the needle was washed in the autosampler port for 30 s before each injection using a wash solution consisting of 10% acetone in water/methanol/isopropanol/ACN (1:1:1:1, by volume).
MS conditions
The mass spectrometer was operated in the positive electrospray ionization (ESI) mode, and analytes were quantified by multiple reaction monitoring (MRM) of the transitions reported in Table 1. The capillary voltage was set at 3500 V for all transitions. The source temperature was 300°C and gas flow was set at 12.0 L/min. Nebulizer pressure was set at 40 psi. Collision energy and fragmentation voltage were set for each analyte as reported in Table 1. The MassHunter software (Agilent Technologies) was used for instrument control, data acquisition, and data analysis.
Mass Spectrometry Parameters for Analytes Under Study
Sample preparation
As summarized in Figure 1, plasma (0.1 mL) was transferred into 8-mL glass vials (Thermo Fisher, catalog no.: B7999-3) and proteins were precipitated with 0.5 mL of an ice-cold crashing solution consisting of ACN plus 1% FA and 50 pmol ISTDs. Brains were homogenized in ice-cold ACN with 1% FA and spiked with ISTD (50 pmol). Both plasma and brain samples were mixed for 30 s and centrifuged at 2800×g at 4°C for 15 min. After centrifugation, the supernatants were loaded onto Captiva-Enhanced Matrix Removal (EMR)-Lipid cartridges (1.0 mL, 40 mg), a kind gift from Agilent Technologies, and eluted by applying vacuum (3–5 mmHg). For brain fractionation, cartridges were prewashed with water/ACN 1:4 to activate the EMR sorbent. For plasma fractionation, no pretreatment was necessary. Tissue pellets were washed with water/ACN (1:4, 0.2 mL), mixed for 30 s, and centrifuged as described above. The supernatants were collected, transferred onto EMR cartridges, eluted, and pooled with the first eluate. The cartridges were washed with water/ACN (1:4, 0.2 mL). After the final elution, the vacuum was increased gradually to 10 mmHg to ensure full analyte recovery. Eluates were dried under N2.
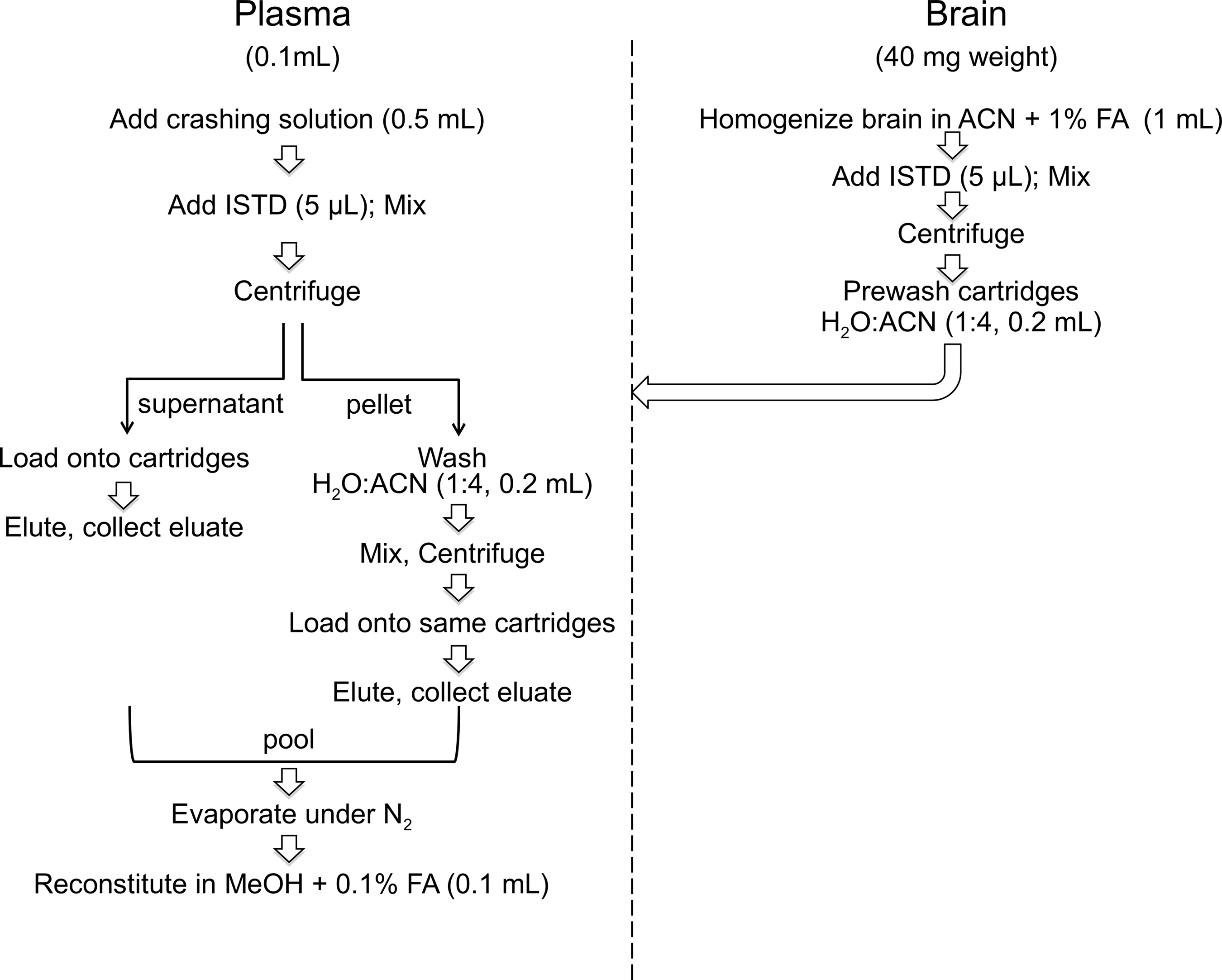
Sample preparation workflow. ACN, acetonitrile; FA, formic acid; ISTD, internal standard.
In some experiments, samples were first spiked with standards and then subjected to EMR fractionation. In this case, the dried samples were reconstituted in methanol containing 0.1% FA (0.1 mL) and transferred to deactivated glass inserts (0.2 mL) placed inside amber glass vials (2 mL; Agilent Technologies, catalog no.: 5182-0716). In other experiments, samples were subjected to EMR fractionation and standards were added onto the dry residues, which were then reconstituted in methanol with FA 0.1% (0.1 mL), and transferred to deactivated glass inserts placed in amber glass vials (2 mL).
Selectivity
Selectivity was assessed in blank plasma and brain matrices (no THC). Each blank sample was visually inspected for interference of matrix components at the retention times of analytes under study.
Calibration curves
Ten-point calibration curves (1 pmol/100 μL to 1000 pmol/100 μL) were prepared in methanol containing 0.1% FA. Matrix calibration curves were prepared in plasma and brain by adding standards either before (pre-spiked EMR) or after EMR fractionation (post-spiked EMR). The final concentration of the ISTDs was 50 pmol/100 μL.
Quality controls
Quality control (QC) samples were prepared at three different concentrations using the same procedure described above for the standard curve: 5 pmol/100 μL for low quality control (LQC), 50 pmol/100 μL for middle quality control (MQC), and 500 pmol/100 μL for high quality control (HQC).
Limit of detection and quantification
Lower limit of detection (LLOD) and lower limit of quantification (LLOQ) were determined using a signal-to-noise (S/N) ratio of ≥3.0 and ≥10.0, respectively. For the determination of LLOD and LLOQ, standards were prepared by serial dilution at concentrations of 0.02, 0.05, 0.1, 0.2, and 0.5 pmol/100 μL and were run in triplicate.
Linearity, precision, and accuracy
Linearity was assessed using the 10-point pre-spiked EMR and post-spiked EMR calibration curves prepared in plasma and brain, evaluating the slope and the intercept of the regression line. Linear regression analysis was carried out using the 1/x 2 weighting factor. Acceptable correlation coefficient (R 2 ) was set to ≥0.95.
Five replicates of the three QC concentrations, LQC, MQC, and HQC, were prepared as pre-spiked and post-spiked EMR matrices and were used to determine accuracy and precision. Each QC concentration was run in triplicate on three separate days along with calibration curves in the corresponding matrices, to determine interday accuracy and precision. Precision was evaluated by calculating percent relative standard deviation (%RSD) of sample replicates within each day. Accuracy was determined as relative percent error from nominal concentration and calculated as follows: [(measured concentration)/(nominal concentration)]×100. Following the Food and Drug Administration (FDA) Bioanalytical Method Validation Guidelines, 15 acceptable mean values for precision and accuracy should be ±15% of the actual value and ±20% for LOQ.
Stability
Analyte stability was determined in plasma or brain homogenates at three different reference standard concentrations: 5, 50, and 500 pmol/100 μL. Samples were subjected to various temperature and time of storage conditions. For short-term stability studies, samples were kept at 9°C for 12 h. For freeze-thaw stability studies, samples were subjected to three freeze-thaw cycles with 12 h of freezing at −20°C. Long-term stability was performed storing samples at −20°C and analyzing them after 10 days. Bench-top stability was determined at 25°C for 6 h.
Matrix effect and recovery
Matrix effect and recovery were calculated using the method of Matuszewski et al. 16 Three sets of samples were prepared by spiking ISTDs and reference standards at three different QC concentrations (5, 50, and 500 pmol/100 μL). Set A consisted of neat solvent spiked with ISTDs and reference standards; set B consisted of post-spiked EMR samples; and set C consisted of pre-spiked EMR samples. The matrix effect was calculated as (setB/setA – 1)×100. Recovery was calculated as (setC/setB)×100.
The matrix effect was also studied using a post-column infusion method. 17 THC, 11-OH-THC, and 11-COOH-THC were diluted with methanol containing 0.1% FA (10 μM) and directly infused into the MSD. Infusion was performed with a single syringe pump, through a T-connector that combined the post-column flow with the LC column flow into the MSD. THC, 11-OH-THC, and 11-COOH-THC were individually infused at 0.3 mL/h and baseline responses were monitored using selected reaction monitoring transitions for each analyte. After steady state was reached, blank methanol (used as control) and pre-EMR (only protein precipitation, without cleanup) and post-EMR plasma or brain matrices were injected into the column and LC data were collected for each matrix.
Phospholipid removal
Tissue phospholipids were fractionated by LC using an Eclipse XDB C18 column (3.5 μm, 2.1×100 mm; Agilent Technologies). The mobile phase consisted of water containing 5.0 mM ammonium formate as solvent A and methanol containing 5.0 mM ammonium formate as solvent B. A gradient was run from 65.0% B to 95.0% B over 40 min, at 1.0 mL/min. Re-equilibration was performed for 2.5 min. For MSD, gas flow was set at 12.0 L/min with a temperature of 120°C, nebulizer pressure was set at 50 psi, and capillary voltage at 3500 V. Fragmentation and collision energies were set at 150 V and 30 V, respectively. All transitions were followed in ESI-positive mode using fragmentation and collision energies of 150 V and 30 V, respectively. Dwell times were set at 200 ms. Several PC species were monitored, which all yielded a product ion with m/z=184.4 (choline phosphate). The following transitions18,19 were used: (m/z=808.4>184.4); (m/z=806.4>184.4); (m/z=786.4>184.4); (m/z=784.4>184.4); (m/z=760.4>184.4); (m/z=758.4>184.4); (m/z=704.4>184.4); (m/z=524.4>184.4); (m/z=524.4>184.4); (m/z=520.4>184.4); and (m/z=496.4>184.4). To confirm phospholipid removal, total lipid extracts from brain before sample cleanup (pre-EMR) and extracts subjected to sample cleanup with EMR cartridges (post-EMR) were analyzed by thin-layer chromatography (TLC) 20 (20×10 cm 60-Å silica gel plates, layer thickness 0.2 mm; Sigma-Aldrich). TLC plates were washed twice by elution with 1:1 methanol/CHCl3 and activated at 180°C, 2 h, before use. Polar lipids were eluted with a mixture of chloroform/methanol/acetic acid/water (85:15:10:3.5, by volume) as solvent system. Phospholipids were visualized by using the molybdenum blue reagent (Sigma-Aldrich).
Results
Sample preparation
The sample workup protocol involved two main steps (Fig. 1): bulk protein precipitation with ice-cold ACN followed by centrifugation and elution of the supernatant on solid-phase extraction columns (Captiva-EMR cartridges, Agilent, see Materials and Methods). The column fractionation step was included to eliminate matrix-associated choline-containing phospholipids, which are a primary source of interference in the LC/MS-MS analysis of THC. 16 The successful removal of these contaminants was verified using two independent methods. First, we monitored 11 abundant species of PC and LPC in extracts of rat plasma and brain using an LC/MS-MS protocol described under Materials and Methods. Figure 2 illustrates representative LC chromatograms obtained with extracts of plasma (Fig. 2A) and brain (Fig. 2B) analyzed before (red trace) or after (blue trace) column fractionation. With either matrix, only small amounts of PC or LPC (<1%) were detectable in post-column analyses. In a second set of experiments, brain extracts were subjected to TLC analysis before and after passage through the column (Fig. 2C). Visualization with molybdenum blue showed that most phospholipids were effectively removed by the cleanup procedure.
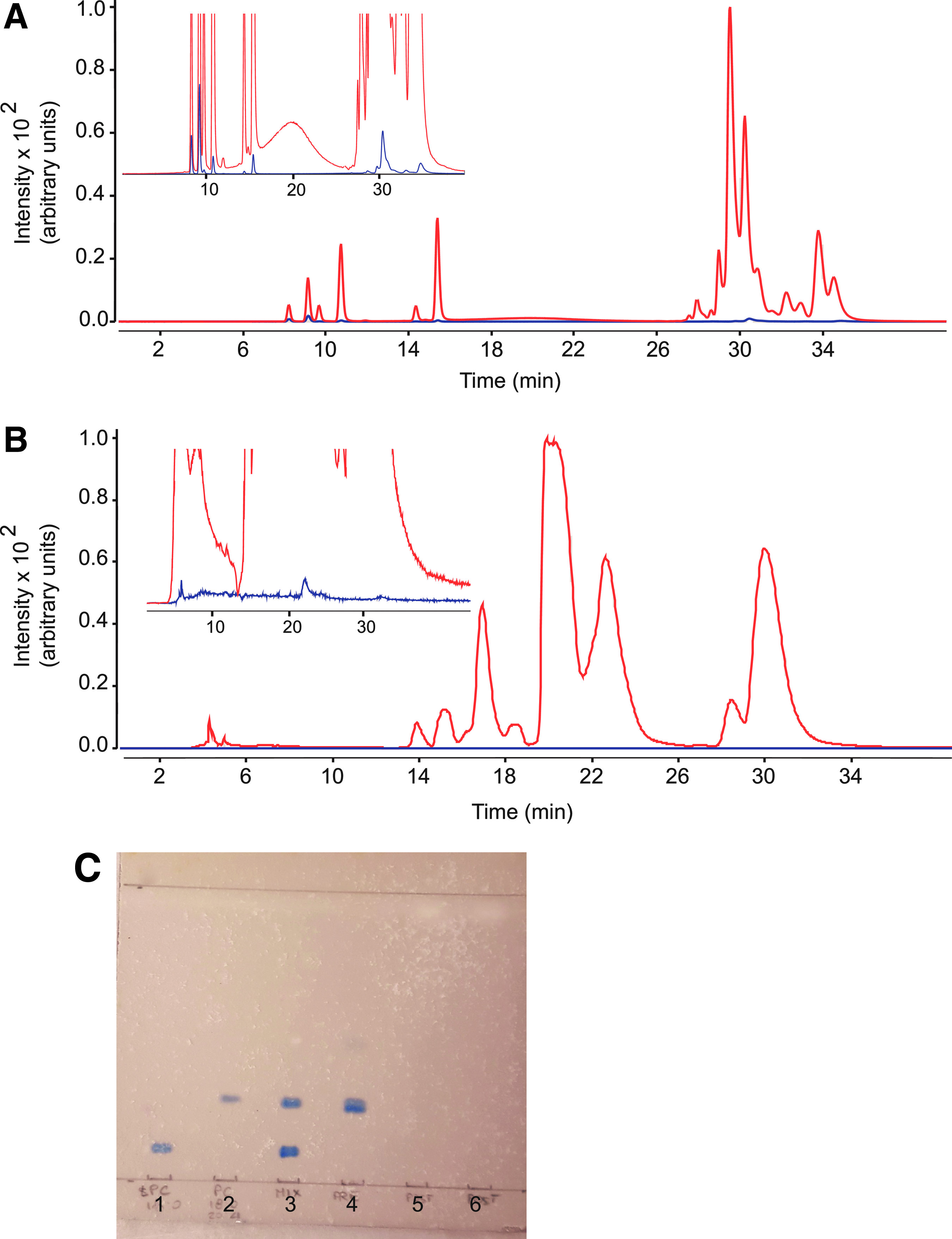
Verification of phospholipid removal by elution through EMR cartridges.
LC/MS-MS analysis
Figure 3 depicts a representative LC separation of THC, 11-OH-THC, and 11-COOH-THC, using the column and gradient conditions described under Materials and Methods. The retention times were 1.3 min for 11-OH-THC/ 2 H-11-OH-THC, 1.5 min for 11-COOH-THC/ 2 H-11-COOH-THC, and 2.5 min for THC/ 2 H-THC, and the total run time (including re-equilibration) was 7 min. The optimal MRM transitions, identified for all analytes using the MassHunter (Agilent) software, are reported in Table 1. Analyte carryover between runs was minimal (<0.3%) and no interfering material was observed in extracts of rat plasma or brain at the retention times of target analytes.
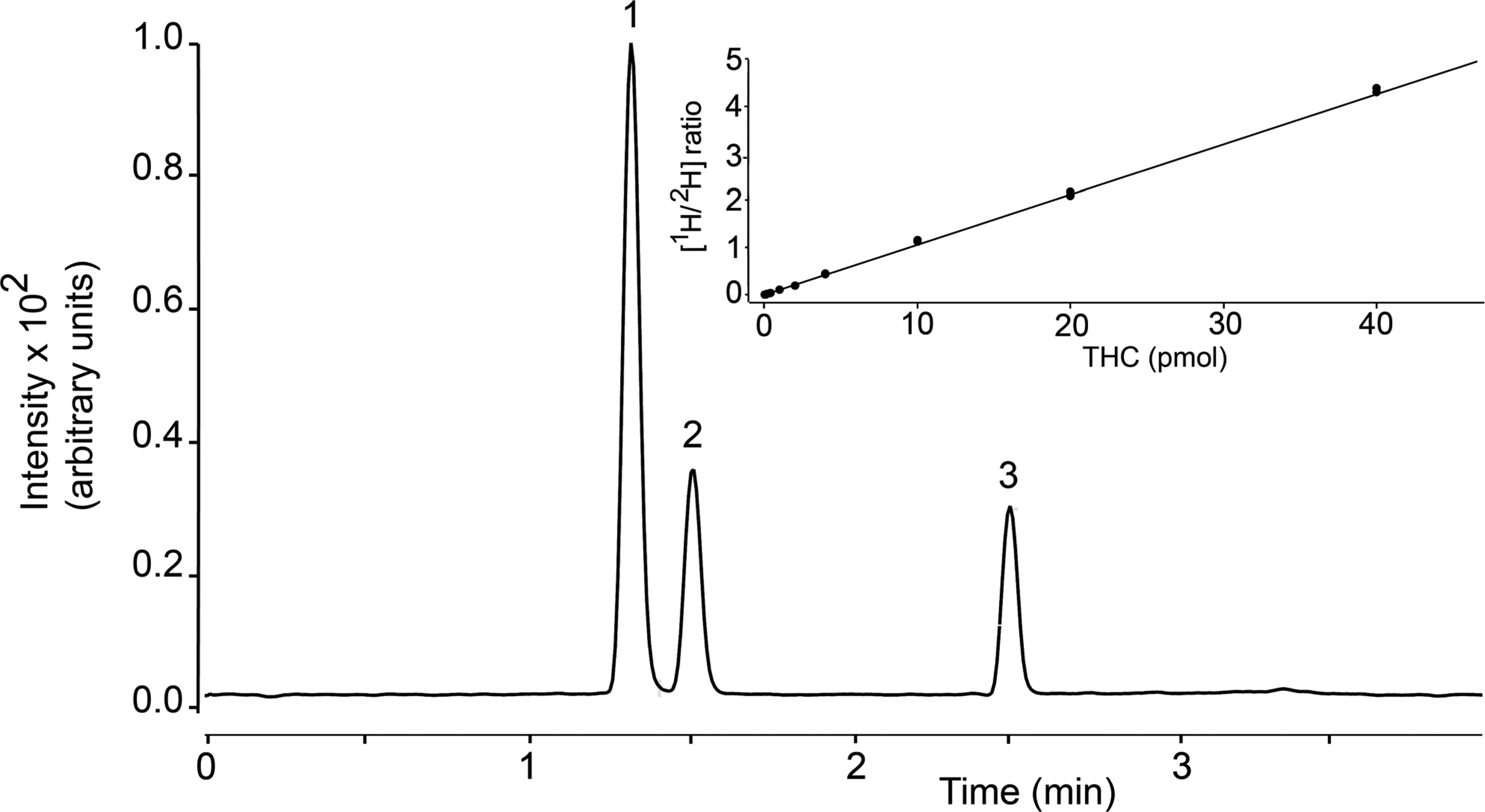
Representative chromatogram showing LC separation of THC and metabolites. (1) 11-OH-THC, (2) 11-COOH-THC, (3) THC. Inset: Isotope dilution calibration curve for THC. 11-COOH-THC, 11-nor-9-carboxy-Δ 9 -tetrahydrocannabinol; 11-OH-THC, 11-hydroxy-Δ 9 -tetrahydrocannabinol; LC, liquid chromatography; THC, Δ 9 -tetrahydrocannabinol.
Quantification
A representative 10-point calibration curve for THC is reported in Figure 3 (inset). Curve linearity for all analytes was determined in the absence or presence of matrix (plasma or brain) using a 1/x 2 weighting factor. Without matrix, we obtained R2 values of 0.99 for THC, 0.997 for 11-OH-THC, and 0.995 for 11-COOH-THC. In spiked plasma, R2 values were 0.992 for THC, 0.997 for 11-OH-THC, and 0.995 for 11-COOH-THC; in spiked brain, R2 values were 0.995 for THC, 0.994 for 11-OH-THC, and 0.995 for 11-COOH-THC. An S/N ratio ≥3 was used to define the LOD, which was found to be 0.5 pmol/100 μL for THC and 11-OH-THC, and 1 pmol/100 μL for 11-COOH-THC. An S/N ratio ≥10 was used for the LOQ, which was 2 pmol/100 μL for all analytes.
Precision and accuracy
Average intraday and interday precision and accuracy data were collected for all analytes in both plasma and brain matrix, before and after column fractionation. Five separate QCs at three different standard analyte concentrations (5, 50, and 500 pmol/100 μL) were prepared, and run in triplicate for three consecutive days. Precision was determined by calculating the %RSD. Accuracy was determined as relative percent error on nominal quantity. Accuracy and precision values for pre-column and post-column plasma containing QCs are reported in Table 2. Corresponding values for brain are shown in Table 3. In all cases, accuracy and precision were within FDA recommendations (±15% of nominal concentration).
Intraday and Interday Accuracy and Precision of Analyte Quantification in Plasma
Standards were added (A) before or (B) after EMR fractionation. Analyses were run on three consecutive days.
%RSD, percent relative standard deviation; EMR, Enhanced Matrix Removal; SD, standard deviation.
Intraday and Interday Accuracy and Precision of Analyte Quantification in Brain
Standards were added (A) before or (B) after EMR fractionation. Analyses were run on three consecutive days.
Recovery and matrix effect
Recovery, which was determined as (Response pre-column/Response post-column)×100, ranged from 86.8% to 99.8% for plasma and from 81.9% to 102.8% for brain (Table 4). The matrix effect was investigated using the post-column infusion 21 and the post-extraction spike methods. 16 Post-column analyte infusion into the ion source combined with injection of matrix into the column allows one to identify regions of a chromatographic profile that are sensitive to the matrix effect. Figure 4 illustrates the results obtained injecting pre-column plasma extract (red trace), while monitoring the MRM transition for THC (m/z 315.2>193.1). We observed substantial ion suppression in a region of the chromatogram that encompassed the retention time of THC (overlaid black trace). No such suppression was observed when either post-column plasma extracts (blue trace) or methanol (green trace) was injected. Moreover, no suppression was noted when monitoring target ions for 11-OH-THC (m/z 331.1>313.2) or 11-COOH-THC (m/z 345.2>299.2; not shown). Figure 5 illustrates the results of studies with brain extracts, which produced varying degrees of ion suppression at the retention times of THC (Fig. 5A), 11-OH-THC (Fig. 5B), and 11-COOH-THC (Fig. 5C).
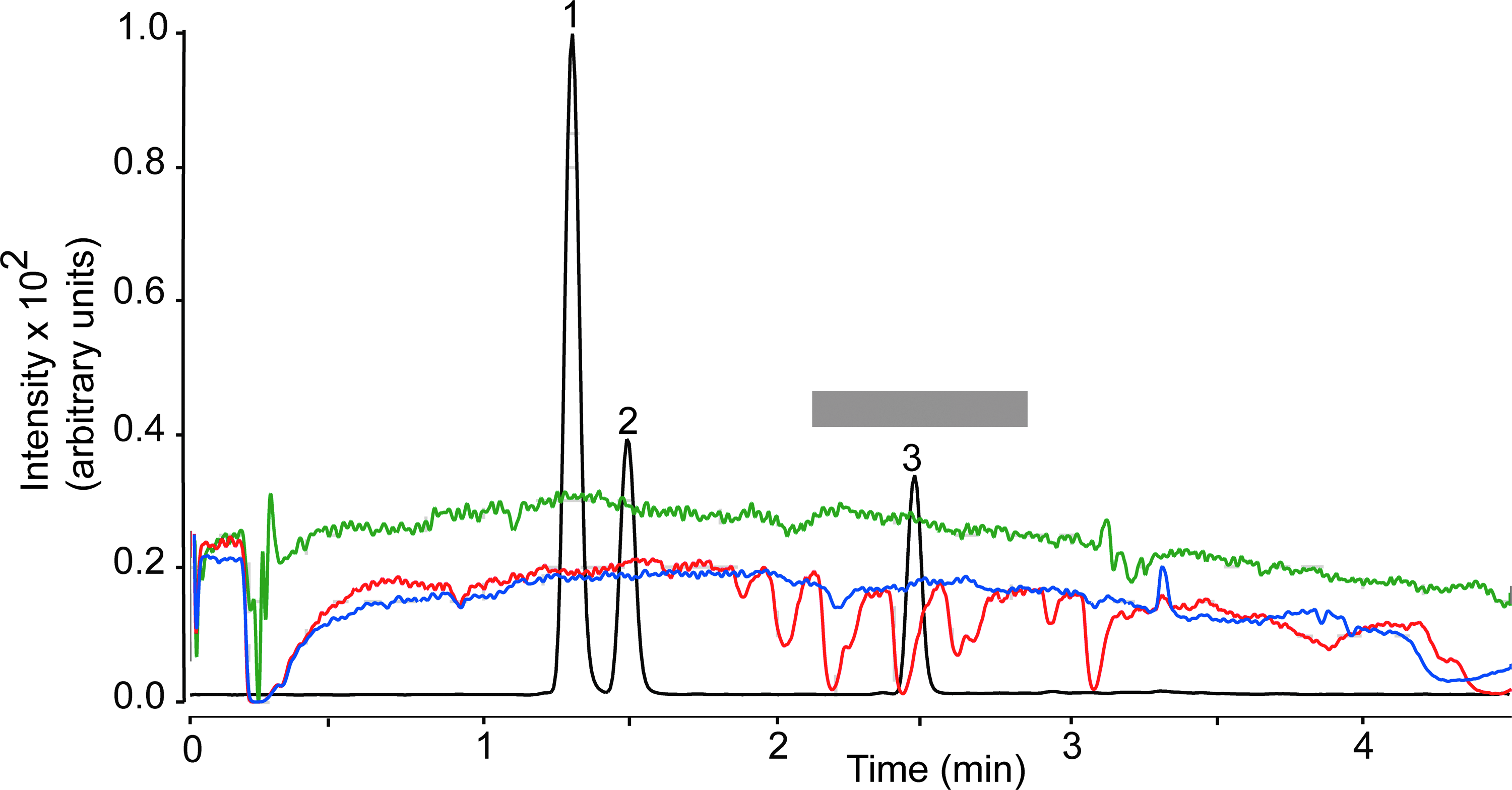
Plasma matrix effect for THC (MRM transition m/z 315.2>193.1). Injection of plasma extracts before EMR (red) or after EMR (blue). Methanol injection (green). The gray bar highlights a region of signal suppression corresponding to the retention time of THC. Black trace, overlaid LC tracing shows the elution of 11-OH-THC (1), 11-COOH-THC (2), and THC (3). MRM, multiple reaction monitoring.
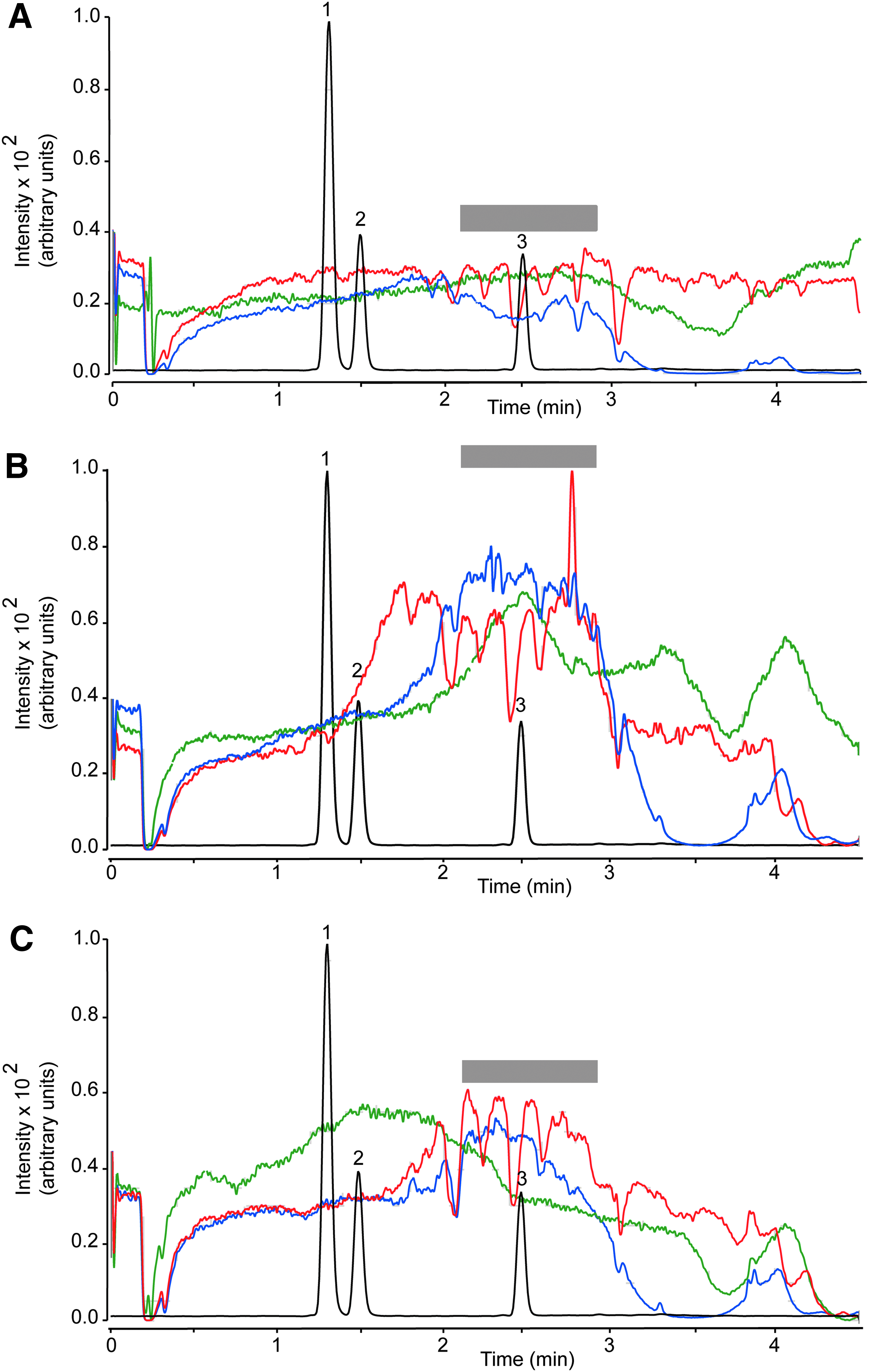
Brain matrix effect for
Analyte Recovery Following Fractionation from Plasma and Brain
Recovery (%) was calculated as (Response pre-EMR/Response post-EMR)×100, where response pre-EMR is the average area for the analytes, which have gone through the fractionation process. Response post-EMR is the average area for the same quantity of analyte spiked into the extracted matrix after the fractionation procedure (n=2/condition, run in triplicate).
To obtain a quantitative assessment of ion suppression, we next used the post-extraction spike method, 16 in which authentic standards were added to the samples after workup. Replicates of neat solvent and post-column plasma and brain spiked with 5, 50, or 500 pmol of each analyte were prepared and analyzed, and the matrix effect was calculated as described in Materials and Methods. The data are reported in Table 5.
Matrix effect (%) was calculated as (Response post-EMR/Response neat solvent – 1)×100, where response post-EMR is the average area for the analytes, which have gone through the fractionation process. Response from neat solvent is the average area for the same quantity of analyte spiked into neat solvent (n=2/condition, run in triplicate).
Stability
We evaluated analyte stability in plasma and brain extracts under four conditions: short-term storage (12 h at 9°C), long-term storage (10 days at −20°C), bench-top residence (6 h at 25°C), and resistance to three freeze-thawing cycles. We set acceptance criteria at ±15% of nominal concentration. The results, reported in Tables 6 and 7, showed that all analytes were stable under most circumstances. Notably, however, THC underwent minor losses at low and middle concentrations in plasma, and after two cycles of freezing and thawing.
Plasma Stability of Analytes Under Study
Brain Stability of Analytes Under Study
Method application
The method was applied to the quantification of THC and its metabolites in plasma and brain extracts of mice treated with a single dose of THC (10 mg/kg, IP) and killed after 15, 30, 60, or 120 min. As shown in Figure 6A, THC levels rose quickly in plasma, reaching a maximal concentration of 23.4 nmol/mL within 30 min of injection. The levels of 11-OH-THC and 11-COOH-THC also peaked at 30 min, but were substantially lower than those of THC. In brain (Fig. 6B), where THC is independently metabolized by enzymes of the CYP system, 22 THC and THC-OH levels rose in parallel and reached maximal concentrations (THC: 1.9 pmol/mg; THC-OH: 1.7 pmol/mg tissue) 60 min after THC injection. THC-COOH levels remained very low, yet clearly detectable, throughout the experiment.
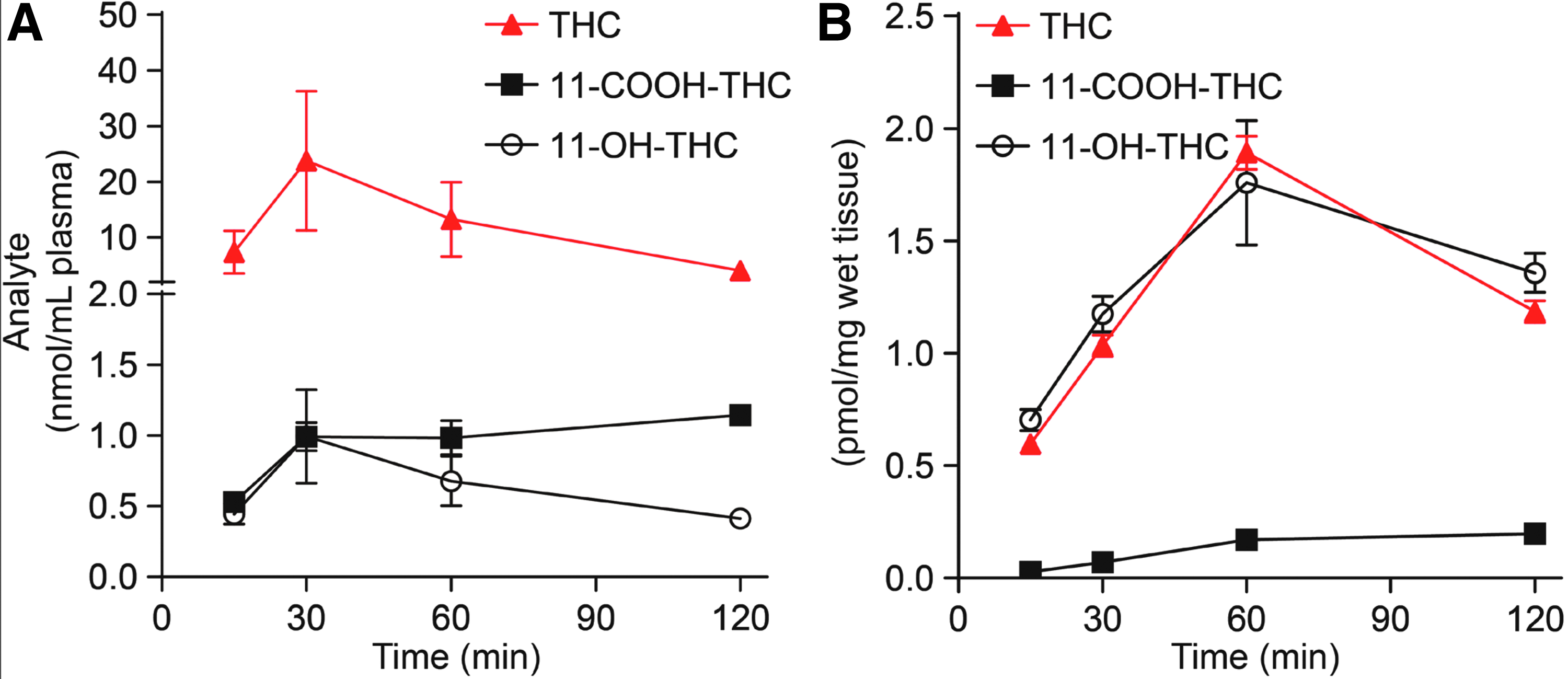
Time course of THC, 11-OH-THC, and 11-COOH-THC levels in
Discussion
This study presents a rapid, sensitive, and accurate method for the quantification of THC and its main oxidative metabolites. The method was specifically designed for application in rodent tissues, and was validated following FDA guidelines. 15 Excellent protocols for THC analysis are already available,9,19,23–25 but this method offers several technical advantages, including almost complete elimination of interfering choline-containing phospholipids and improved control on ion suppression—two major obstacles in the analysis of THC by LC/MS-MS.18,26 Possible applications for the new method include, among others, studies aimed at examining the impact of age, sex, and route of administration on the pharmacokinetic properties of THC in different rodent species.
Choline-containing phospholipids interfere in the LC/MS-MS analysis of THC18,26 and pose a substantial challenge to the analyst. The column fractionation procedure presented in this study allowed us to remove ≥99% of these interfering lipids from plasma and brain extracts, without significant losses in analyte recovery. The LC conditions afforded optimal peak selectivity and baseline separation for the three target analytes (THC, 11-OH-THC, and 11-COOH-THC) in a short run time (7 min, including re-equilibration). Moreover, calibration curve linearity was excellent for all analytes (R 2 >0.99) over a wide range of concentrations (1–1000 pmol/100 μL). Several studies have utilized LC-MS/MS to measure THC in animal tissues,27–31 but to the best of our knowledge, this method is the first to incorporate important variables such as phospholipid removal and matrix effects, while retaining rapidity and ease of application.
THC is metabolized by the CYP system present in liver and other tissues. 22 The CYP isoenzymes CYP2C9 and, to a minor extent, CYP2C19 and CYP3A4, 32 generate first the hydroxylated product, 11-OH-THC, and then the carboxyl derivative, 11-COOH-THC.22,33,34 Both 11-OH-THC and 11-COOH-THC undergo phase II metabolism, the former through the action of uridine diphosphate-glucuronosyltransferase (UGT)1A9 and UGT1A10, and the latter through UGT1A1 and UGT1A3. 35 The polar glucuronide metabolites produced in these reactions are cleared by the kidney. 35 Each of these steps may be influenced by sex and age. 36 For example, in elderly people, age-dependent changes in hepatic and renal clearance and body fat mass can enhance the bioavailability of THC and prolong its plasma half-life. 37 Sex can also affect THC distribution and metabolism. For example, female rodents express greater amounts of hepatic CYP isozymes and aldehyde oxygenase activity, which may facilitate the conversion of THC into the bioactive metabolite, 11-OH-THC. 38 Sex- and age-dependent variability of THC pharmacokinetics and metabolism39,40 underscore the need to integrate pharmacodynamics investigations on this agent with parallel studies on its disposition. The availability of a sensitive and reliable method for THC quantification in rodent tissues should facilitate such studies.
Our results underscore several problems that must be taken into consideration in the analysis of THC and its first-pass metabolites. First, as it often happens with hydrophobic molecules, the accuracy and precision of THC measurements are reduced at the low end of the calibration curve: even though the accuracy and precision data reported in this study are within FDA recommendations (±15% of nominal concentration), care should be exerted to keep this potential confounder within acceptable limits. Second, interference by ion suppression is a concrete issue in THC quantification, which should be diligently monitored when switching from one matrix to another (e.g., from plasma to brain). Finally, and importantly, when stored in the presence of tissue matrix, THC can undergo time-dependent degradation, even at low temperatures. Our stability study showed that low and mid concentrations of THC suffered from minor, but statistically detectable losses after extraction from plasma and brain. It is advisable, therefore, to limit tissue storage time when analyte levels are expected to be low.
In conclusion, as studies on the health impact of THC expand to include both male and female animals across their lifespan, it is essential that the pharmacodynamics properties of this agent are evaluated in relationship with its varying biological disposition. The development of robust analytical methods to quantify THC and its metabolites, such as the one presented in this study, is a necessary step in this direction.
Footnotes
Acknowledgment
The study was supported by NIDA Center of Excellence grant P50DA044118.
Author Disclosure Statement
No competing financial interests exist.