
Abstract
Introduction:
Recognition of fungal surface β-glucan by pattern recognition receptor Dectin-1 is a critical process for fungal clearance in the lung. In humans, persistent fungal infection is observed in individuals with particular Dectin-1 polymorphism. We have identified that nitric oxide (NO) modifies critical cysteines in pattern recognition molecules to disassemble and alter protein function. There is a hydrophobic S-nitrosylation motif present in surfactant protein-D (SP-D) that is also present in Dectin-1. We hypothesized that Dectin-1 can be modified by nitrosative stress potentially leading to impairment of fungal clearance.
Materials and Methods:
Recombinant Dectin-1 was incubated with
Results:
Dectin-1 was S-nitrosylated by
Conclusions:
These data provide mechanistic data to support the role of Dectin-1 nitrosylation as a mediator of reduced fungal clearance in the face of increased NO exposure.
Introduction
The innate immune system plays a critical role in host defense in the lung. Alveolar macrophages are the predominant cells in pattern recognition and clearance of pathogens through pattern recognition receptors (PRRs). Among these receptors, a C-type lectin-like receptor Dectin-1 (dendritic cell associated lectin-1) was identified as the β-glucan receptor.1–5 It specifically binds to β-glucan on the fungal surface and induces a series of signaling cascades to initiate clearance.2,4,6–11 Upon engagement, Dectin-1 triggers different protective responses through intracellular signaling, including activation of the Syk kinase/CARD9 pathway, which appears to be critically important for Dectin-1.12–16 These responses include fungal uptake and killing as well as inducing expression of cytokines and chemokines.17–26 It has been reported that Dectin-1 deficiency is related to persistent fungal infection.26–29 Furthermore, genetic knock-out Dectin-1 gene30–33 results in exacerbation of fungal infection and reduces Th1 and Th17 regulatory T cell function, including deficiency of the cytokines IL-17 and IL-22.34–41 It is likely that there is cooperative signaling between Dectin-1 and other PPRs, and such interactions have been observed for TLR-2 and TLR-4.17–19,23,42–50
It has been shown that NO is capable of nitrosylating thiol residues to form S-nitrosothiol (SNO) and that this modification can have signaling consequences within a number of systems.51–54 In another scavenger molecule, surfactant protein-D (SP-D), critical thiol residues are involved in the assembly of the native multimer.55–58 Nitrosylation of these thiols results in multimer disassembly and a switch in molecular function that allows SP-D to bind calreticulin and promote acute macrophage activation.57,58 The cysteines involved in the regulation of SP-D function are located within a hydrophobic pocket in the tail domain of the molecule.55–58 Examination of the sequence of Dectin-1 reveals the presence of a cysteine residue, Cys54, within a hydrophobic pocket (Fig. 1A). We reasoned that this cysteine may also be a target for NO modification that would alter its assembly in the plasma membrane and alter its role as a PRR. In this article, we demonstrate that Dectin-1 is indeed a target for NO modification and that the formation of SNO on Dectin-1 leads to its shedding from the cell surface. Concomitant with the loss of Dectin-1 is a reduced ability of macrophages to phagocytose fungal material. We propose that NO-mediated modification of Dectin-1 may be a significant mechanism of regulating innate immune function and fungal clearance capacity.
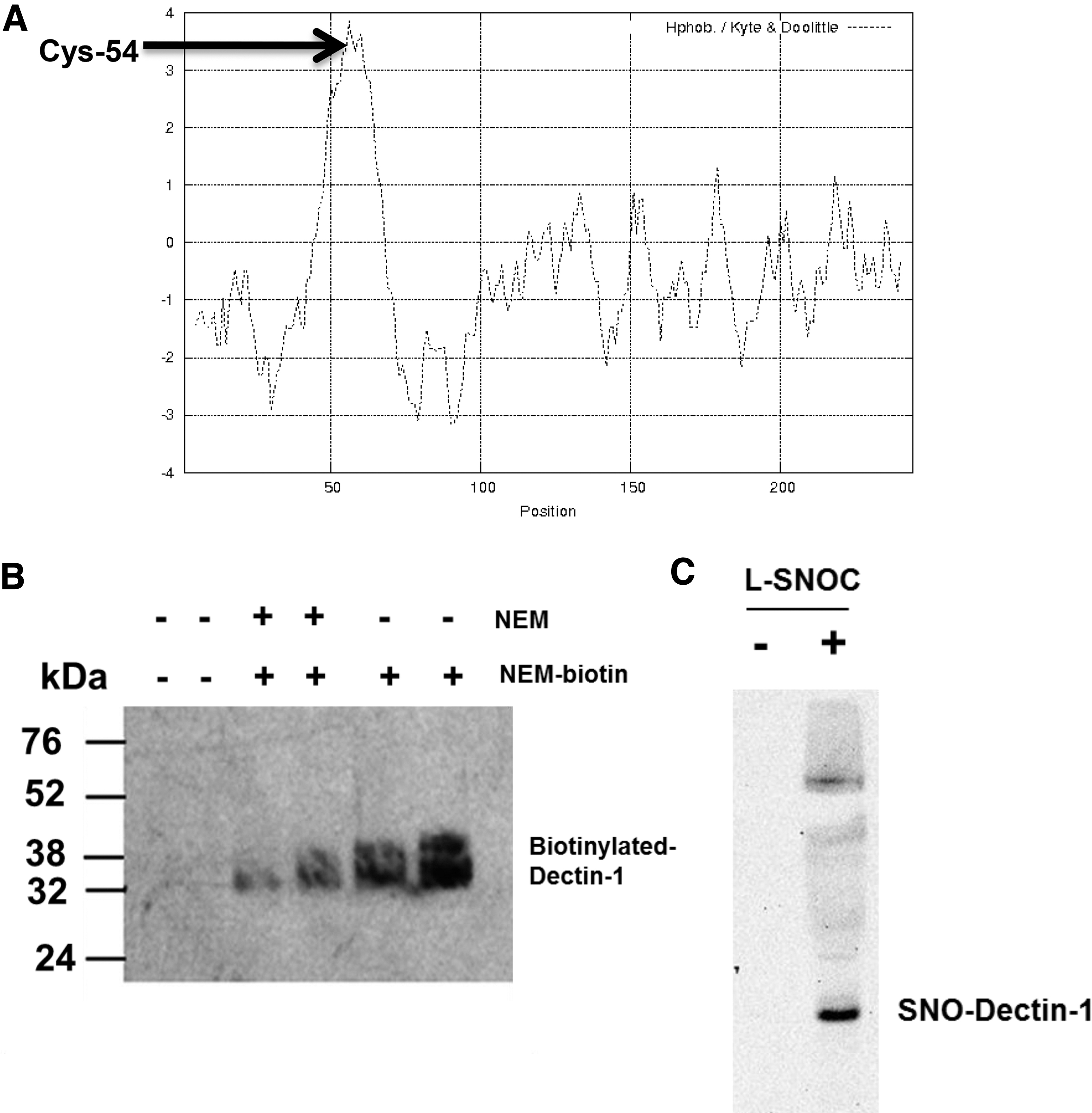
In vitro alkylation of reduced cysteine residues and S-nitrosylation of Dectin-1.
Materials and Methods
Cell culture
A murine macrophage cell line Raw 264.7 was obtained from ATCC (Catalog No. TIB-71). Cells were cultured in 100 mm plastic dishes containing Dulbecco's modified Eagle's medium (DMEM) (Gibco, St. Louis, MO) supplemented with 10% fetal bovine serum (Gibco) in a CO2 incubator (5% CO2 in air) at 37°C and subcultured every 3 days.
Dectin-1 alkylation and in vitro S-nitrosylation
Recombinant Dectin-1 (R&D systems) in 10 mM phosphate buffer was incubated with N-ethylmaleimide (NEM)-biotin at 50°C for 10 minutes and denatured by lithium dodecyl sulfate sample (LDS) buffer at 95°C for 10 minutes. The resulted protein was isolated by 4%–12% Tris glycine gel electrophoresis at 200 V for 40 minutes and transferred to a PVDF membrane. After blocking with 5% nonfat milk, the membrane was incubated with horseradish peroxidase (HRP) conjugated anti-biotin antibody (Sigma, St. Louis, MO) for 1 hour. Signal was detected using the enhanced chemiluminescence kit (Amersham), and blots were exposed to Kodak Biomax MS film.
To generate S-nitrosylated Dectin-1, recombinant Dectin-1 (10 μg/mL) was exposed to
Biotin-switch assay for detection of SNO-Dectin-1
Detection of SNO-Dectin-1 was performed through an adaptation of the biotin switch method.55–58 In vitro S-nitrosylated Dectin-1 samples were suspended in HEN buffer (25 mM Hepes, pH 7.7/0.1 mM EDTA/0.01 mM neocuproine), and 20 μM N-ethylmaleimide (NEM) at 37°C for 30 minutes to block free thiols. Excess NEM was removed by protein precipitation using cold acetone. Protein pellets were resuspended in HENS buffer (HEN 1% sodium dodecyl sulfate [SDS]), SNO bonds were decomposed by adding 20 mM sodium ascorbate. The newly formed thiols were then linked with the sulfhydryl-specific biotinylating reagent N-[6-biotinamido)-hexyl]-1-(2-pyridyldithio) propionamide (Pierce). Biotinylated proteins were precipitated with Streptavidin-agarose beads and Western blot analysis was performed to detect the amount of Dectin-1 remaining in the samples.
Western blots
Immunoblotting for Dectin-1 was performed using an equal protein 30 μg of cell lysate or recombinant protein per lane. Samples were loaded on a 4%–12% precast NuPAGE Bis-Tris polyacrylamide gel (Invitrogen).55–58 Proteins were electrophoresed using MES-SDS buffer at 200 V for 35–45 minutes per the manufacturer's instructions (Invitrogen). Native gel electrophoresis was performed using a Novex 4% Tris-glycine gel (Invitrogen). Samples were mixed with a cold native gel sample buffer (Invitrogen) before loading. Electrophoresis was run at room temperature at a constant voltage of 80 V for 5 hours. Proteins were then transferred from the gel to a nitrocellulose membrane using the XCell II Mini-Cell and sandwich blot module (Invitrogen) in Tricine-10% methanol-0.01% SDS transfer buffer (Invitrogen) at 30 V for 1 hour. Blots were blocked for 1 hour at room temperature with 10% nonfat milk and then incubated with primary Dectin-1 antibody (R&D system) (1:500 in 1% nonfat milk) overnight at 4°C. Blots were then incubated with 1:5000 goat anti-rabbit IgG-horseradish peroxidase for 2 hours at room temperature. Signal was detected using the enhanced chemiluminescence kit (Amersham), and blots were exposed to Kodak Biomax MS film.
RNA extraction
Total cellular RNA was isolated from Raw 264.7 cells using Tri-Reagent (Molecular Research Center, Cincinnati, OH).55–58 In brief, the total RNA was extracted by a single step, guanidium thiocyanate–phenol–chloroform extraction. After centrifugation at 13,000 × g for 15 minutes at 4°C, the RNA-containing aqueous phase was precipitated in isopropanol. RNA precipitates were washed once in 75% ethanol and resuspended in 30 μL of ribonuclease-free water.
Reverses transcription
Total RNA (1 μg) was subjected to reverse transcription using the reverse transcription system (Promega, Madison, WI) for 1 hour at 42°C. The reaction was terminated by incubating the reaction mixture at 99°C for 5 minutes and then kept at 4°C. The resulting complementary DNA (cDNA) was used as a template for PCR amplification.
Real-time quantitative polymerase chain reaction
Real-time quantitative polymerase chain reaction (qPCR) was performed with Applied Biosystems (ABI) Prism 7300 Sequence Detection System (Perkin Elmer). The target genes of IL-6 and β-actin were amplified by ABI gene assays. The cycle conditions were set as follows: 95°C for 10 minutes followed by 40 cycles of 95°C for 15 seconds and 60°C for 1 minutes. All controls and samples were run in triplicate in the same plate. β-Actin messenger RNA (mRNA) levels of the samples in the same plate were analyzed by real-time PCR to normalize the mRNA contents among the samples tested.
NF-κB-dependent secretory embryonic alkaline phosphatase assay
Raw-Blue cells, an NF-κB reporter cell line, (InvivoGen) (1 × 105 cells in 100 μL) were grown in 96-well Falcon flat-bottom culture plates (Becton Dickinson). The cells were incubated with S-nitrosoglutathione (GSNO) for 24 hours and medium was changed and incubated with curdlan for additional 24 hours. A QUANTI-Blue™ (InvivoGen) solution, which is a detection medium developed to determine the activity of any alkaline phosphatase present in a biological sample, was prepared following the manufacturer's instructions. 50 μL of medium and 150 μL of resuspended QUANTI-Blue solution were added to each well of a flat-bottom 96-well plate. The plate was incubated for 60 minutes at 37°C and the secretory embryonic alkaline phosphatase (SEAP) levels were determined using a spectrophotometer at 620–655 nm. Each experiment was performed in triplicates.
Flowcytometric analysis
To determine Dectin-1 expression by direct immunofluorescence, 3 × 105 Raw 264.7 cells were suspended in 100 μL of 1 × PBS and were incubated with phycoerythrin (PE)-conjugated-Dectin-1 antibody (2D11) (R&D system) for 30 minutes at 4°C. Fluorescein-conjugated control antibody was isotype-matched IgG2a (R&D system). The cells were then washed twice with PBS and were fixed with 2% paraformaldehyde in PBS. Fluorescence was analyzed on an EPICS-Elite flow cytometer (Beckman Coulter Electronics).
Phagocytosis assay
The ability of Raw 264.7 cells to phagocytose zymosan was assessed. 3 × 105 Raw 264.7 cells were seeded in replicates of 5–6 in 96-well tissue culture plates with opaque sides and optically clear bottoms (Costar; Corning Life Sciences). Raw 264.7 cells were cultured to 60% confluence and treated with GSNO, lipopolysaccharides (LPS) or IL-4 for 24 hours and incubated with DMEM medium containing 0.1% bovine serum albumin (BSA) for at least 2 hours to abolish any activation signals. Raw 264.7 cells were challenged with fluorescein isothiocyanate (FITC)-labeled zymosan particles using a multiplicity of infection of 10:1 for 2 hours to allow phagocytosis to occur. Trypan blue (250 μg/mL; Molecular Probes) was added for 10 minutes to quench the fluorescence of extracellular yeast, and fluorescence was determined using a Spectramax Gemini EM fluorometer at settings of 485 excitation/535 emission (Molecular Devices). The phagocytic index was calculated as previously described in relative fluorescence units.
Statistical analysis
All experiments were replicated independently at least three times. Means of triplicate experiments were compared using one-tailed unpaired t tests or with one- or two-way analyses of variance (GraphPad Prism version 4.03 program).
Results
Quaternary structure of Dectin-1
A Kyte-Doolittle hydropathy plot for the Dectin-1 sequence was constructed using a window size of nine residues. A positive value indicates a region of hydrophobicity. Cysteine 54 is located within the tail domain, which is the most hydrophobic portion of the molecule (Fig. 1A). The positioning of a reduced cysteine within such a hydrophobic region has been proposed as a motif for the formation of an SNO.54,55,58 In multimeric proteins, the presence of a cysteine within a hydrophobic region can also be indicative of a structural disulfide bond. Therefore, we chose to examine whether cysteines within Dectin-1 were available for alkylation as that is a prerequisite for being able to form SNO. Figure 1B shows that Dectin-1 is capable of being alkylated by NEM as there is a significant increase in biotin-linked protein upon incubation that can be blocked by prior alkylation. This demonstrates that Dectin-1 contains at least one cysteine residue that is in a reduced state.
The presence of a reduced cysteine, potentially within a hydrophobic pocket, is consistent with Dectin-1 being a target for S-nitrosylation. Therefore, we examined whether it was possible to form nitrosylated Dectin-1 by treating recombinant protein with the SNO donor
S-nitrosylation of Dectin-1 results in Dectin-1 shedding from macrophage surface
Dectin-1 forms a C-type lectin structure on the surface of cells that can bind β-glucan and in which cysteines play a critical structural role.2,4,59 SP-D also has a C-type lectin structure with critical cysteines that is destabilized by nitrosylation.55,57,58 Furthermore, we have shown that high efflux of NO, from inducible nitric oxide synthase (iNOS) induction, results in S-nitrosylation of SP-D disrupts its quaternary structure and disassembles the protein. 55 We hypothesized that SNO-Dectin-1 formation may result in Dectin-1 shedding from the cell surface. To test this hypothesis, we examined the effects of treating macrophages (Raw 264.7 cells) with the SNO donor GSNO. When Raw cells were incubated with GSNO, Dectin-1 was shed to the medium, as determined by Western blot, in a dose-dependent manner (Fig. 2A upper panel). This is indicative of SNO treatment destabilizing the Dectin-1 multimeric complex and releasing monomeric units to the medium. To confirm that the appearance of Dectin-1 in the medium was associated with loss of Dectin-1 from the cell surface of Raw cells, we used a flow cytometry approach. After SNO treatment cells were stained with PE-labeled anti-Dectin-1 and the fluorescence profile assessed (Fig. 2A lower panel). GSNO treatment reduces the quantity of Dectin-1 on the cell surface as seen by the leftward shift in the histogram. Notably, there is a population of cells that displays less fluorescence than the IgG controls and this becomes the predominant population after treatment with 500 μM GSNO.
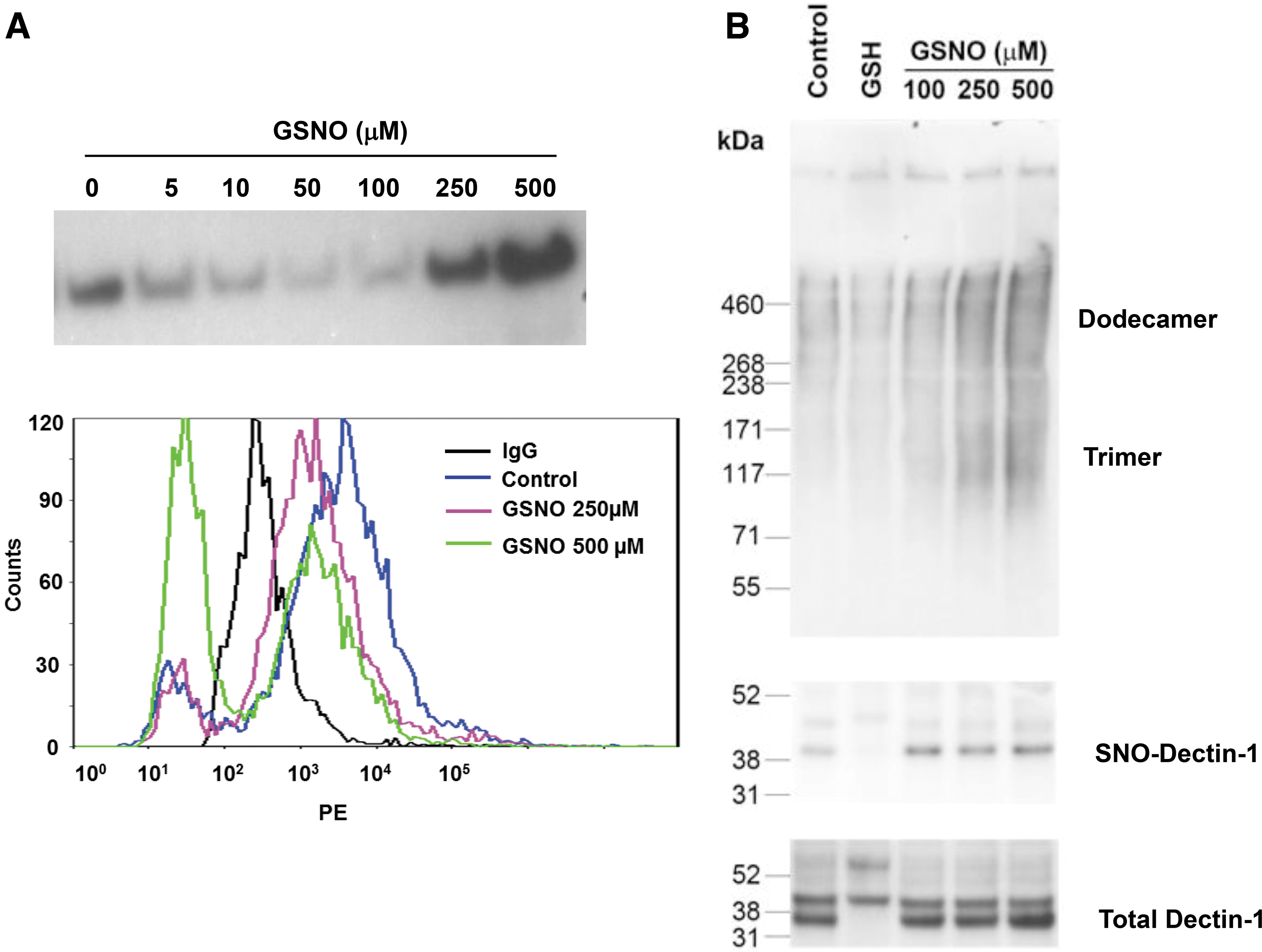
S-nitrosylation induces Dectin-1 shedding from cell surface and alters its quaternary structure.
By comparison with SP-D we proposed that the ability of GSNO treatment to cause Dectin-1 to be shed from the cell surface resulted from destabilization of its multimeric structure. Similar to other C-type lectins the multimeric structure of Dectin-1 is critical to its function and its presence on the cell surface. Therefore, we chose to examine whether GSNO treatment destabilized Dectin-1 multimerization in Raw cells. After Raw cell treatment with GSNO, Dectin-1 structure was analyzed by native gel electrophoresis and nitrosylation confirmed by biotin switch (Fig. 2B). Gels were performed with equal loading of total Dectin-1 and, thus, the quantities seen on the native gel are reflective of the relative amounts of the individual multimers. The fully multimeric form of Dectin-1 is too large to enter the native gel and can be seen at the loading site. However, both dodecameric and trimeric Dectin-1 can both be clearly observed. There is a GSNO-mediated increase in both dodecameric and trimeric Dectin-1 that is not observed by simple treatment with glutathione. This appearance of lower molecular weight multimers is indicative of structural disruption and is accompanied by the appearance of SNO-Dectin-1 as shown by biotin switch.
S-nitrosylation of Dectin-1 decreases curdlan and zymason induced NF-κB activity and IL-6 expression
Having identified that Dectin-1 was S-nitrosylated and SNO-Dectin-1 formation resulted in shedding from the macrophage surface, we next determined how SNO formation altered Dectin-1 function. Dectin-1 upon binding one of its agonists, such as curdlan or zymosan, mediates induction of NF-κB activation. To assess this function, we used the Raw-Blue reporter system in which Raw cells have been transformed to express SEAP upon NF-κB. Challenge of Raw-Blue cells with curdlan resulted in NF-κB activation as seen by increased SEAP activity in the medium (Fig. 3A). Pre-treatment with either 250 or 500 μM GSNO reduced SEAP release significantly by ∼50%. This is consistent with GSNO mediating Dectin-1 shedding and thus reduced NF-κB function in response to curdlan.
S-nitrosylation of Dectin-1 reduces NF-κB activation and IL-6 mRNA expression by treatment with zymosan or curdlan.
One of the downstream targets of NF-κB within macrophages is IL-6. Therefore, we also assessed the ability of both curdlan and zymosan to induce IL-6 expression and the effects of SNO pretreatment upon that induction (Fig. 3B). IL-4 and LPS were used as both a positive (IL-4) and a negative (LPS) control for expression. Both curdlan and zymosan induced IL-6 expression and this induction was enhanced by pre-treatment with IL-4, while it was inhibited by LPS, which induces Dectin-1 shedding. Pre-treatment with GSNO also inhibited both zymosan and curdlan induction of IL-6 expression. These data are consistent with the effects of nitrosylation on Dectin-1 shedding (Fig. 2A).
S-nitrosylation reduces phagocytic capacity of macrophages
One of the principle effects of β-glucan recognition by Dectin-1 is the initiation of a phagocytic response. Therefore, to examine whether SNO modification of Dectin-1 had functional consequences on macrophage function, we examined whether pre-treatment with GSNO could reduce macrophage phagocytosis. Incubation of Raw cells with zymosan beads led to significant incorporation (Fig. 4). Pretreatment with IL-4 did increase this incorporation but not significantly. In contrast, LPS pre-treatment - as predicated due to its effects on Dectin-1 shedding - significantly reduced bead incorporation. Pre-treatment with either 250 or 500 μM GSNO reduced bead incorporation to the same level as seen with LPS, indicating that nitrosation of Raw cells was equally capable of reducing phagocytic function. These results are in accordance with nitrosylation acting as a mediator of Dectin-1 shedding and thus regulating macrophage function.
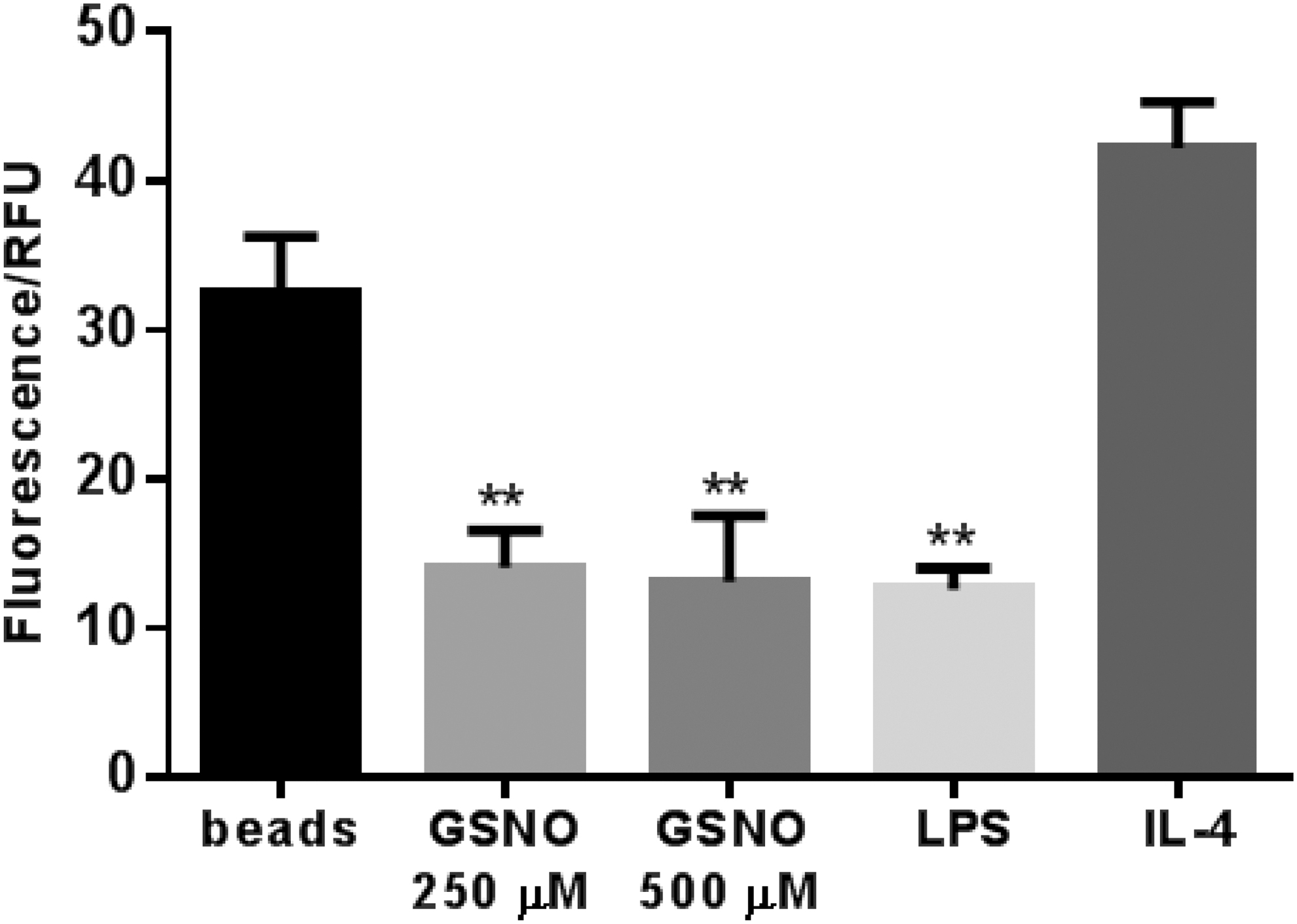
S-nitrosylation of Dectin-1 decreases phagocytosis of zymosan particles. Raw 264.7 cells were plated onto 96-well plates (1 × 105 cells/well) and incubated with or without GSNO (250 and 500 μM), LPS (100 ng/mL), or IL-4 (10 ng/mL) for 24 hours. The media was removed and Texas red conjugated Zymosan A (Saccharomyces cerevisiae) BioParticles™ (5 × 105 particles/well) were then added to macrophages for 2 hours. The wells were subsequently aspirated, and extracellular particles were quenched by briefly adding trypan blue. Cells were then washed, and intracellular fluorescence was measured (Ex 595 nm/Em 615 nm) by a fluorescent plate reader (Molecule DevicesMe3). Data presented as means ± SD of triplicate cultures, a representative of three independent experiments. **p < 0.01 versus Beads' group.
Discussion
In this study, we demonstrate that there is a cysteine residue in Dectin-1 in a hydrophobic region (Fig. 1A) and this cysteine is in a reduced state (Fig. 1B), as it is able to be alkylated by NEM. This positioning of a cysteine within a hydrophobic domain has been shown to make pattern recognition molecules such as Dectin-1 targets for NO modification. Here, we demonstrate that NO S-nitrosylates Dectin-1 to form SNO-Dectin-1 (Fig. 1C), which results in Dectin-1 shedding from the macrophage surface and disassembly of its oligomeric structure in vitro (Fig. 2). Dectin-1 is a major β-glucan receptor on leukocytes. Surface expression of macrophage Dectin-1 is regulated by cytokines and growth factors. Here we demonstrate that nitrosylation of macrophages biases function away from the Dectin-1 mediated response, as it results in increased NF-κB function and reduced IL-6 expression. Engagement of Dectin-1 on myeloid cells by β-glucan triggers a variety of cellular responses, including dendritic cell maturation, ligand uptake by endocytosis and phagocytosis, respiratory burst and production of arachidonic acid metabolites, certain cytokines and chemokines. Dectin-1 is the principal β-glucan receptor for the glucans present on millions of species and, thus, Dectin-1 has a clear role in anti-fungal activity. Our data shows that nitrosylation of Dectin-1 impairs phagocytosis of zymosan particles (Fig. 4), suggesting that nitrosative stress affects macrophage fungicidal function.
The impetus for this study came from our previous study that NO modified molecules have a broad pulmonary signaling consequence in regulation of innate immunity in the lung.55–58,60–63 Lung inflammation induces expression of iNOS and accelerates NO production. NO can react with a wide range of reactive targets, including metal centers, thiols, DNA, lipids, and reactive oxygen species to produce a range of NO-modified biomolecules. NO has long presented a curious dichotomy within biological systems, namely that it is both an important physiological regulator and the mediator of many pathologies. It is clear in the lung that NO is required for the control of lung vessel and airway dilation, immune defense, and the maintenance of barrier function. NO is also a key mediator of acute lung injury, bronchopulmonary dysplasia, respiratory distress syndrome, and asthma. The contradictory behavior of NO is highlighted by the successes and failures of inhaled NO, which has revolutionized the treatment of persistent pulmonary hypertension of the newborn and, potentially, bronchopulmonary dysplasia, but has failed to help—and may even harm—patients with acute respiratory distress syndrome.
The principal NO signaling pathway is through activation of guanylyl cyclase and the cyclic-GMP (cGMP) pathway. However, pathways other than cGMP have become of increasing interest in recent years. In particular, the capability of NO to induce signaling functions through the post-translational modification of proteins. Among these modifications, S-nitrosylation is an important regulator of protein function. S-nitrosylation is a covalent modification of thiol groups by formation of a thionitrite, SNO group, and can occur either through direct reaction, metal catalysis, or through the formation of higher nitrogen oxides. Over the past decade, hundreds of proteins have been shown to become S-nitrosylated and, in many cases, this modification is accompanied by altered function. The potential for SNO as a post-translational regulator of protein function has been highlighted by recent proteomic and targeted studies.
We have shown that NO modifies the lectin SP-D to form SNO-SP-D - resulting in disassembly - which switches this lectin from an anti-inflammatory function to an inflammatory activator.55,58 We have identified that certain cysteine residues are critical for protein quaternary structure and are the targets for S-nitrosylation.55,58 In SP-D two cysteine residues in the most hydrophobic region of the protein are S-nitrosylated by NO in a bleomycin injury model. When we analyzed the hydrophobicity of Dectin-1, we found that Cysteine 54 is located in the most hydrophobic region of the protein (Fig. 1A) and here we demonstrate that Dectin-1 is a target for nitrosylation. Furthermore, the signaling consequences of SP-D nitrosylation appear to be related to disassembly of its quaternary structure. In this context, it is interesting to note that nitrosylation of Dectin-1 also leads to structural disruption. It seems possible that this NO-mediated regulation of structure through modification of hydrophobic cysteines may be a fundamental signaling mechanism.
S-nitrosylation of Dectin-1 has direct functional consequences as it impairs the fungicidal activity of macrophages. These provide a link between nitrosative stress and innate immune regulation. It has long been known that chronic inflammation can increase susceptibility to fungal infection. For instance, fungal infection is a major factor in HIV-related mortality and in our study; we have seen chronic inflammation in Sftpd−/− (SP-D knock-out) mice results in reduced pneumocystis clearance. 62 Mice lacking Dectin-1 have exacerbated infection with pneumocystis and lower survival rates.8,64,65 Nitrosative stress induced shedding of Dectin-1 and inhibited phagocytosis may provide the pathological link between chronic inflammation and fungal infection. A recent study showed that Dectin-1 polymorphism is associated with persistent infection of fungi. 66 Thus, it may be interesting to consider whether these polymorphisms affect susceptibility to NO and structural disruption. Understanding the importance of NO downregulation of pattern receptor provides a potential strategy for targeting NO therapeutic approaches to increase fungal clearance.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by NIH Grant HL086621.