
Abstract
Abstract
Three-Dimensional (3D) liver microtissues, specifically prepared from primary human hepatocytes (PHH) in coculture with nonparenchymal cells (NPCs), have been shown to be a valuable tool for in vitro toxicology. However, a lack of thorough characterization on a functional, transcriptomic, and proteomic level of such models during long-term cultivation is evident. By integrating multiple omics technologies, we provide in this study an in-depth long-term characterization of 3D microtissues composed of PHH from three different donors cocultured with primary NPCs. The 3D human liver microtissues (hLiMTs) exhibited stable adenosine triphosphate (ATP) content and albumin secretion over 5 weeks. Histological analysis indicated a healthy liver tissue with polarized expression of bile salt export pump (BSEP) and multidrug resistance protein 2 (MRP2) in a structure reminiscent of bile canaliculi. The 3D microtissues exhibited stable basal and inducible cytochrome P450 activities up to 5 weeks in culture. Analysis of 40,716 transcripts using RNA arrays revealed distinct similarities to native human liver gene expression. Long-term culture showed a stable phenotype up to 5 weeks, with differences in liver gene expression primarily attributed to individual donors. Proteomic profiling of 2200 unique proteins by label-free LC-MS/MS revealed a relatively stable protein expression where only 7.3% were up- or downregulated more than twofold from day 7 to 35 in culture. Taken together, these results suggest that hLiMTs represent a responsive and physiologically relevant in vitro liver model that maintains stable function over 5 weeks and is therefore well suited for repeated-dose toxicity testing.
Introduction
I
A number of different techniques are used to mimic more physiologically relevant conditions for liver cells in vitro by culture in hydrogels, scaffolds, microcarriers, or micropatterned surfaces.5,8–14
An alternate method for 3D culture is cellular self-assembly, for which no scaffolds or hydrogels are needed. This approach allows for good size reproducibility since only one microtissue unit per well is formed using a defined cell number. 15 Moreover, the production, maintenance, and endpoint analysis of 3D microtissues can be automated by liquid handling robotic devices. 16 This automation capability is essential for increasing throughput of assays and reducing costs per data point, both crucial requirements in a high-throughput setting. Furthermore, the technology utilizes very little input material, because each 3D microtissue consists of only a couple of thousand cells, and the small size of the microtissue circumvents limitations in oxygen and nutrient supply.17,18 The reduced input material requirements allow for production of a large number of 3D microtissues from unique primary material, such as hepatocytes and nonparenchymal cells (NPCs). Aside from the choice of the culture format, the selection of the appropriate cell source is equally important for the prediction of human hepatotoxic drugs. Even though well characterized, immortalized liver-derived cell lines are available (i.e., HepRG, HepG2, and Huh7); their suitability is hampered by either their low metabolic activity 19 or their relatively low sensitivity level compared to primary human hepatocytes when tested against known hepatotoxic drugs. 20 Primary human hepatocytes are known to best preserve the hepatocellular phenotype, maintaining xenobiotic metabolism, such as cytochrome P450 (CYP) activity, and reflect individual genetic polymorphisms in CYP activity.21,22 Moreover, with the availability of cryopreserved primary liver cells, repeated reproducible production of human liver microtissues (hLiMTs) with consistent properties is achieved.
We have previously shown that scaffold-free cellular self-assembly of primary human hepatocytes into hLiMTs, cocultured with NPCs, preserves viability and basic liver functionality for more than 4 weeks in culture. 15 In a more recent study, 3D spheroid hepatic cultures were evaluated for their value for hepatotoxicity risk assessment in drug discovery. The study, which involved a panel of 110 drugs with and without clinical concern for drug-induced liver injury (DILI), demonstrated twofold higher sensitivity and similar specificity for predicting hepatotoxic liabilities compared to 2D primary human hepatocyte cultures. 23 In this study, we provide a further in-depth characterization of this 3D liver microtissue model on a morphological, functional, transcriptional, and proteomic level. This study also provides insights on how donor–donor differences are reflected in hLiMTs and the basic biological characterization needed for further toxicological assessments.
Materials and Methods
3D human liver microtissues
Cryopreserved primary human hepatocytes (IPHH)_01, IPHH_02, IPHH_03 and cryopreserved primary human NPCs (IPHN_03) were obtained from BioreclamationIVT (Brussels, Belgium). For specific cell donor information refer to Supplementary Table S1 (Supplementary Data are available at www.liebertpub.com/aivt). 3D InSight™ Human Liver Microtissues (MT-02-002-04; InSphero AG, Schlieren, Switzerland) were produced as described previously, 15 each hepatocyte lot (IPHH_01, IPHH_02, and IPHH_03) combined with the same NPC lot (IPHN_03). After tissue formation, the 3D microtissues were maintained in 3D InSight Human Liver Maintenance Medium–AF (CS-07-001a-01; InSphero AG) at 37°C in a humidified 5% CO2 cell culture incubator. The culture medium was replenished every 3 to 4 days during the whole cultivation period of 35 days.
Spheroid size analysis
Images of spheroids were generated using a Zeiss Axiovert 25 microscope (5 × objective; Carl Zeiss GmbH, Oberkochen, Germany) in combination with a Canon Powershot camera (zoom factor: 8.9 × ; Canon, Inc., Tokyo, Japan). Spheroid size was determined by ImageJ (set scale: 2430 pixel/1000 μm, elliptic selection), using the diameter of the assay plate (1 mm) as calibrator.
Intracellular adenosine triphosphate content
Intracellular adenosine triphosphate (ATP) was determined using CellTiter-Glo® Luminescent Cell Viability Assay 2.0 (G9242-3; Promega, Madison, WI), following a slightly modified protocol. In brief, PBS (w/o magnesium and calcium) was mixed with the CellTiter-Glo Reagent (1:1). Before addition of 40 μL of assay buffer mix to each well containing hLiMTs, media supernatants were removed. After vigorous mixing in the assay plate, the whole assay buffer mix was transferred to white assay plates (Cat. No. 675075; Greiner Bio-one, Kremsmuenster, Austria) and incubated for 20 minutes in the dark while shaking on a horizontal plate shaker. Luminescence was measured by a Tecan M200 Pro Infinite plate reader.
CYP analysis
Spheroids were incubated with CYP substrates (phenacetin, midazolam, diclofenac, bupropion, bufuralol, coumarin, and S-mephenytoin) for 24 hours, and metabolites (acetaminophen, 1-OH-midazolam, 4-OH-diclofenac, OH-bupropion, 1-OH-bufuralol, 7-OH-coumarin, and 4-OH-mephenytoin, respectively) from single microtissue were quantified in supernatants by liquid chromatography-mass spectrometry (LC-MS) technology, performed by Pharmacelsus GmbH (Germany).
For CYP induction studies, microtissues were exposed to prototypic CYP inducers (500 μM phenobarbital, 40 μM omeprazole, and 10 μM rifampicin) for 72 hours with daily medium change. The above-mentioned CYP substrate cocktail was added for 24 hours to the microtissues before analysis of the corresponding metabolites in the supernatants.
Human albumin analysis
The human albumin concentration of the harvested supernatants from single microtissues (24 hours after medium replenishment) was determined using the human Albumin ELISA Quantitation Set (E80-129/E101; Bethyl Laboratories, Inc., Montgomery, TX). The assay was performed according to the manufacturer's instructions.
Immunohistochemistry of hLiMTs
Microtissues for immunohistochemistry analysis were harvested 7 days after seeding, washed once with PBS (with magnesium/calcium), and incubated with Carnoy solution (Morphisto, Frankfurt, Germany) overnight at 4°C. Fixed spheroids were embedded in Eppendorf tubes with 2% Agarose and further processed for paraffin embedding and sectioning of 4–5 μm thick sections. Standard hematoxylin and eosin (H&E), periodic acid-Schiff (PAS), and the following immunohistochemical stainings were performed: bile salt export pump (BSEP) (sc-74500; Santa Cruz Biotechnology, Inc., Dallas, TX), CD68 (NCL-L-CD68; Novocastra Laboratories Ltd., New Castle, United Kingdom), multidrug resistance protein 2 (MRP2) (ALX-01-016; Enzo Diagnostics, Inc., Lausen, Switzerland), and CK19 (NCL-CK19; Novocastra Laboratories Ltd.).
Immunofluorescence of 3D hLiMTs
Microtissues (coculture of IPHH_01/IPHN_03) for immunofluorescence analysis were harvested after 20 days in culture, fixed with 4% PFA for 20 minutes at room temperature, washed with PBS, equilibrated overnight in 30% sucrose solution (w/v), and frozen in OCT embedding matrix (CellPath Ltd., Newtown, United Kingdom). Spheroid cryosections (8 μm) were stained for CYP3A4 (PAP011, 1: 5000; Cypex Limited, United Kingdom), albumin (sc51515, 1:200, Santa Cruz), E-20 cadherin (13-1700, 1:300; Thermo Scientific), and MRP2 (ab3373, 1:100; Abcam, United Kingdom). Confocal images were acquired using a Zeiss LSM 510 microscope (Carl Zeiss AB, Stockholm, Sweden).
Gene expression analysis
The RNA of 72 pooled microtissues from different culture time points (day 7, 14, 21, 28, and 35) and donors (IPHH_01, IPHH_02, IPHH_03, all cocultured with IPHN_03) was isolated using Maxwell® RSC simplyRNA Tissue Kit (Promega). RNA from human livers was obtained from the previously described livers 24 and identified as HL43, HL49, and HL51. RNA integrity and quality were assessed using the Agilent TapeStation RNA ScreenTape (Agilent Technologies, Palo Alto, CA). The Affymetrix Human Gene 2.1 ST Array (40,716 transcripts) was used for microarray analysis and performed at the Karolinska Institutet core facility for Bioinformatics and Expression Analysis (BEA, Stockholm, Sweden).
The Affymetrix Expression Console software was used to normalize the Affymetrix CEL file data using the Robust Multichip Average method. Only transcripts identifying exons (one per gene) with expression >4.9 (log2) were considered for further analysis. Significant differentially expressed genes (p < 0.05) in the indicated two-group comparisons were identified with a two-tailed heteroscedastic t-test. Classification of the selected genes according to their biological functions was performed using the Ingenuity Pathway Analysis software (Ingenuity Systems, Redwood, CA). Heatmap representations and hierarchical clustering were performed using the Qlucore software (Qlucore AB, Lund, Sweden). Principal component analyses (PCAs) were performed on the basis of the complete transcriptomic or proteomic data set using Qlucore Omics Explorer 3.1 (Qlucore AB).
Materials and sample preparation for proteomics analysis
hLiMTs were cultivated in InSphero assay plates (InSphero AG). Twenty single microtissues from each condition were pooled in an Eppendorf tube. Samples were prepared as described previously. 25 Peptide concentrations in final samples were measured with a SPECTROstar nano spectrophotometer from BMG Labtech using absorbance at 280 nm.
Mass spectrometric acquisition
Two microgram peptides per sample were analyzed on a self-packed analytical column (75 μm ×30 cm) with 3 μm Magic C18AQ (Bruker) at 50°C, using an EASY-nLC nano liquid chromatography system connected to a Q Exactive mass spectrometer (Thermo Scientific). The peptides were separated by a 2-hour nonlinear gradient from 0 to 44% B at 300 nL/min followed by a linear increase to 100% B in 2 minutes and 100% A for 8 minutes. Solvent A was 1% acetonitril (ACN) in aqueous 0.1% formic acid, and solvent B was 97% ACN in aqueous 0.1% formic acid. For data-dependent acquisition (DDA; shotgun LC-MS/MS) and data-independent acquisition (DIA) mass spectrometry the similar methods as published in Bruderer et al. were used. 25 For DDA analyses (protein inventory) on the Q Exactive a modified Top12 method from Kelstrup et al. 26 was used. The full scan was performed between 400 and 1200 m/z. The automatic gain control target for the MS/MS scan was set to 1e6. Normalized stepped collision energy was 22.5%, 25%, and 27.5%. The DIA method consisted of a survey scan at 70,000 resolution from 400 to 1220 m/z (automatic gain control target of 5e6 and 109 ms max injection time). Then, 19 DIA windows were acquired at 35,000 resolution (automatic gain control target 3e6 and 327 ms max injection time). Normalized stepped collision energy was 22.5%, 25%, and 27.5%. The spectra were recorded in profile type.
Mass spectrometric raw data analysis
The DDA spectra were analyzed with the MaxQuant analysis software (Version 1.5.1.2 27 ) using the default settings with the following modifications: The minimal peptide length was set to 6; search criteria included carbamidomethylation of cysteine as a fixed modification, oxidation of methionine and acetyl (protein N-terminus) as variable modifications; and the mass tolerance for the precursor was 4.5 and 20 ppm for the fragment ions. The DDA files were searched against the human UniProt fasta database (release December 11, 2014, 20,187 entries) and the Biognosys' iRT peptide sequences (11 entries). The identifications were filtered to obtain a false discovery rate (FDR) of 1% on peptide and protein level.
For the generation of the spectral libraries, nine DDA measurements of the microtissues from three culture time points and three donors were performed. DDA spectra were analyzed as described above, and a spectral library was generated using the built-in workflow in Spectronaut, similar to SpectraST. 28 The obtained library contained annotation of precursors and fragment ions, as well as normalized retention times (iRT 29 ).
The DIA data were analyzed with Spectronaut 6, a mass spectrometer vendor independent software from Biognosys. 25 The default settings using the iRT alignment feature were used for the Spectronaut search. Interference correction on MS2 level was enabled. The FDR was set to 1% at peptide level. Local normalization was performed on the peptide quantities to compensate for loading differences and spray bias. 30
Only peptides identified in all samples were considered for further statistical and pathway analysis, which was performed as described above for the gene expression analysis.
Results
Morphological characterization of hLiMTs
Fixated, paraffin embedded 3D microtissues composed of PHH from different hepatocyte donors, cocultured with NPCs, were sectioned and stained with different methods (Fig. 1A). H&E staining of 7-day-old microtissues showed dense, tissue-like cellular contacts with presence of hepatocytes with cuboidal shape and absence of a necrotic core. PAS stain shows extensive incorporation of glycogen in hLiMTs indicating active glycogen metabolism. Immunohistochemistry for Kupffer cell marker CD68 revealed presence of Kupffer cells throughout the 3D microtissue. Positive staining for endothelial cells (CD31) could not be observed with this specific coculture combination (data not shown). However, integration of endothelial cells was evident when using a different hepatocyte lot (Supplementary Fig. S1), suggesting that incorporation efficiency can depend on the coculture combination. The present NPC lot did not lead to a significant incorporation of other NPCs. Stimulation of Kupffer cells within these hLiMTs with lipopolysaccharide resulted in interleukin (IL)-10, tumor necrosis factor α (TNFα), and IL-1β secretion (Supplementary Fig. S2A). Inducibility of cytokine secretion could be preserved over 5 weeks of culture as demonstrated by IL-6 measurements upon 48 hours of lipopolysaccharide (LPS) stimulation (Supplementary Fig. S2B), suggesting stable integration of Kupffer cells, which was also recently demonstrated by positive CD68 staining for more than 4 weeks in culture. 31 Hepatobiliary transporter proteins BSEP (ABCB11) and MRP2 (ABCC2) showed polarized expression, indicating the presence of an apical surface with presence of bile canaliculi. Expression of basolateral marker protein SR-BI was reported as well, 32 substantiating the distinct polarization of hepatocytes. In addition, immunofluorescence analysis of cryosections of hLiMTs showed presence of hepatocyte functionality markers albumin, CYP3A4, and CYP2E1. High expression of E-Cadherin suggests tight intercellular interactions. Moreover, co-staining for MRP2 and BSEP adds to the observations from paraffin sections that hepatocytes are polarized, also after prolonged culture time (20 days) (Fig. 1B). Of note, while centrally located cells show expected canalicular colocalization of MRP2 with BSEP, in peripheral cells MRP2 seems to be redistributed to the basolateral membrane exposed to the outside of the microtissue. Staining for the biliary epithelial marker CK19 lets us suggest that MRP2 expression may coincide with the presence of cholangiocytes, which have been described to express MRP2, 33 and which are present as residual cells in the hepatocyte fraction, primarily located at the periphery of hLiMTs (Supplementary Fig. S3). Interestingly, peripheral cells do also stain partially positive for the mesenchymal marker vimentin (Supplementary Fig. S4).
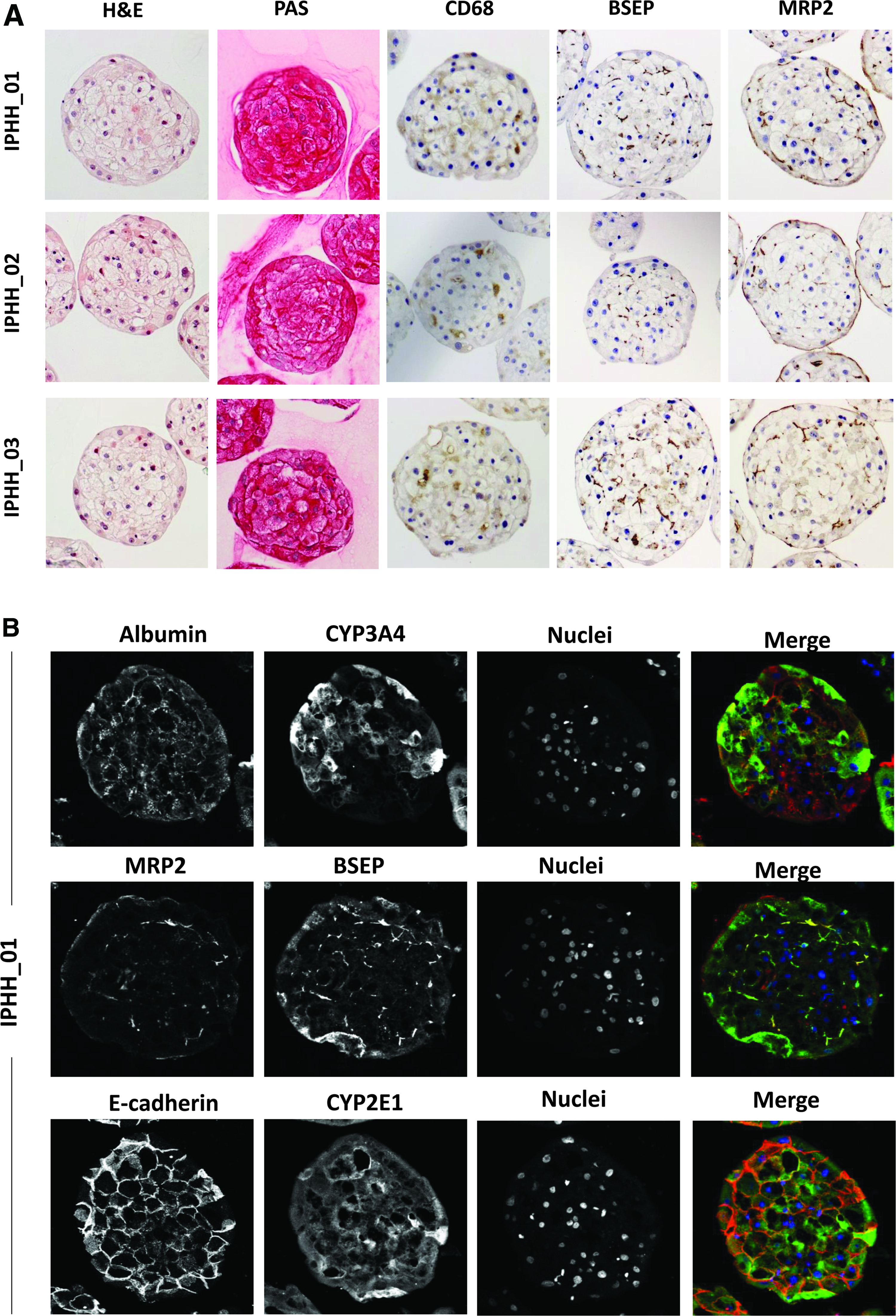
Morphological characterization of 7-day old hLiMTs.
Assessment of stability, viability, and functionality over time
hLiMTs were monitored over 5 weeks of culture time for changes in microtissue diameter, ATP content, and albumin secretion (Fig. 2A–C). Microtissues from three different hepatocyte donors in coculture with NPCs showed high stability, with no significant loss of microtissue size. Microtissue viability, as determined by ATP content, was also stable over time. The microtissues from hepatocyte lot IPHH_02, however, displayed less ATP content per microtissue, which is in line with the smaller size of these spheroids (Fig. 2A). Supernatants of 24-hour exposures of the different microtissues were analyzed for albumin secretion and normalized to the initial number of hepatocytes at seeding. The albumin secretion was very stable over 28 days of culture time, but dropped from day 28 to 35 in culture in all three microtissue cultures. This suggests that the intrinsic functional lifetime of hepatocytes in this culture system is limited to 4–5 weeks in culture.
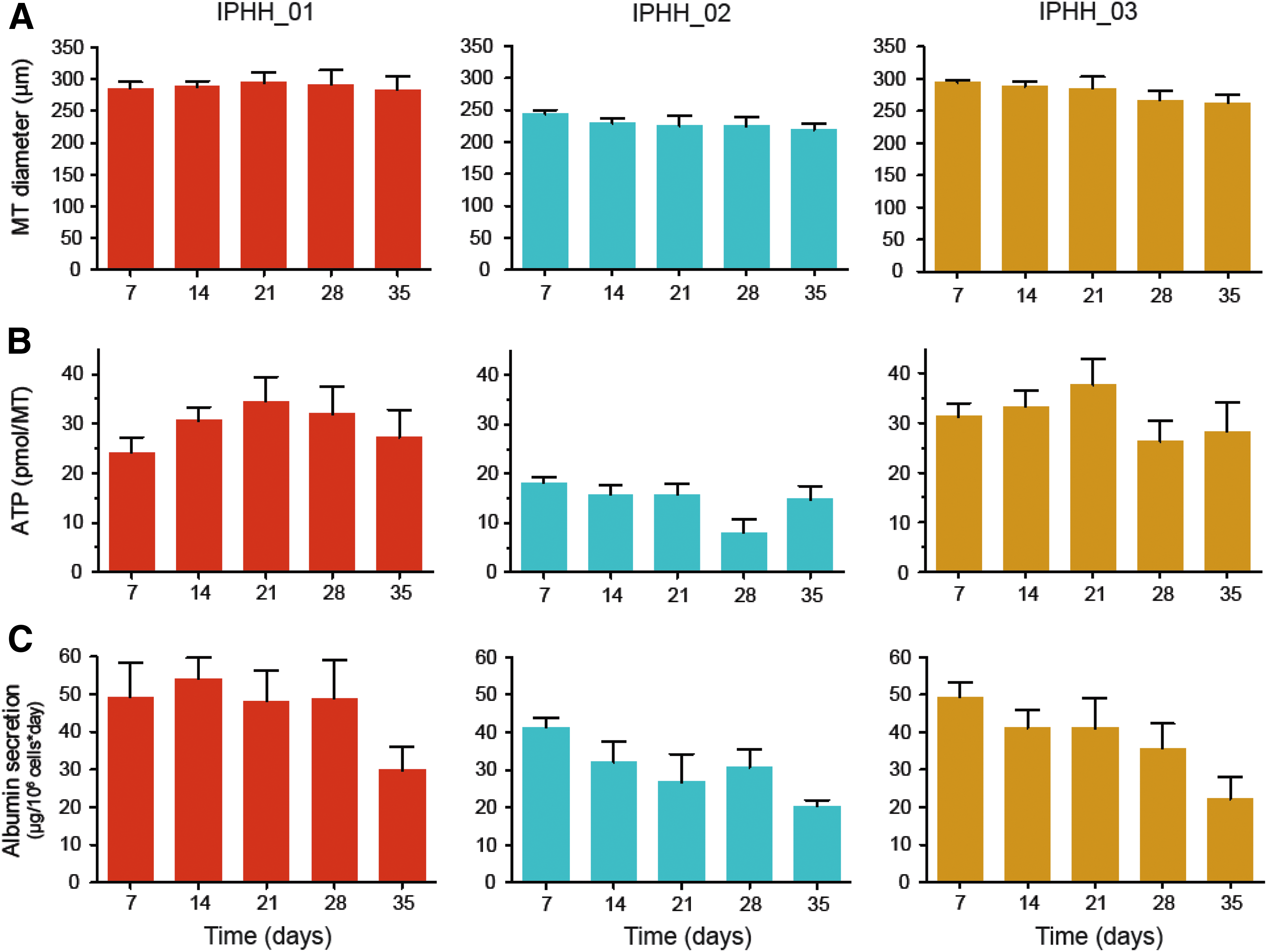
Analysis of hLiMTs derived from three individual hepatocyte donors (IPHH_01, IPHH_02, and IPHH_03) in coculture with NPCs.
Gene expression profiling of hLiMTs
To investigate the gene expression profile of the hLiMTs over time, RNA was isolated from freshly thawed mixtures of hepatocytes and NPCs (day 0 samples) and derived microtissues Affymetrix Human Gene 2.1 ST Arrays. For comparisons RNA from liver biopsies from three unrelated donors were included. PCA revealed a clustering of the microtissue samples from all three donors collected at days 7–35, distinct from freshly thawed, mixed cell suspension (hepatocytes and NPCs) and liver samples of unrelated donors (Fig. 3A). The highest difference was observed in PC1, whereas less difference was observed for PC2. Consistency of gene expression over time was demonstrated by the fact that the number of differently expressed genes from day 7 to 14 was only 26 (0.4%) out of 6531 analyzed genes (Fig. 3B). This number increased to a maximum of 162 genes when day 35 samples were compared to day 7 samples. This reflects a gene expression change of 2.5% during a 4-week culture period, suggesting relative transcriptional stability of hLiMTs over several weeks in culture. The long-term stability of the gene expression pattern was also reflected in a subset of genes responsible for the detoxification of xenobiotics. Genes relevant for administration, distribution, metabolism, and excretion (ADME)/Tox applications remained largely stable within each liver microtissue type over the 35-day culture time (Fig. 3C) except for CYP2C8, which displayed gradually declining expression levels. Analyzing the individual gene expression of the donors by unsupervised hierarchical clustering identified significant differences between donors, illustrated by the fact that samples cluster by donor rather than by culture time (Supplementary Fig. S5A).
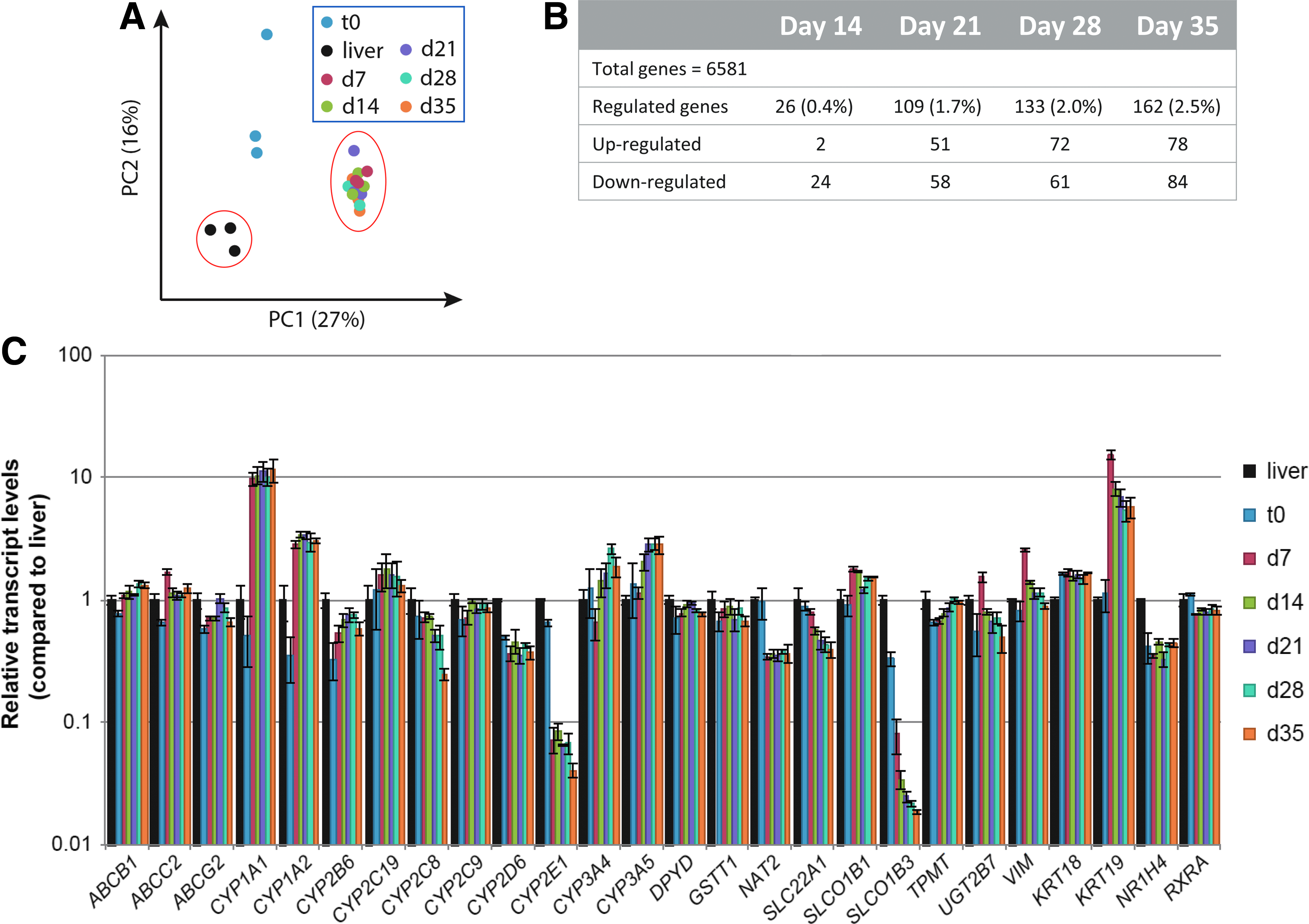
Transcriptomic profiling of clinical liver specimens, as well as freshly thawed cell suspension and derived hLiMTs from three individual hepatocyte donors (IPHH_01, IPHH_02, and IPHH_03) in coculture with NPCs (IPHN_03).
Although expression of most ADME/Tox relevant liver genes was comparable to native liver, a few genes did seem to be differentially regulated upon transfer to 3D cell culture conditions compared to either fresh (unrelated) liver samples or day 0 cell suspension used for the generation of hLiMTs. Simultaneous upregulation (CYP1A1, CYP1A2, CYP3A4, and CYP3A5) and downregulation (CYP2C8, CYP2E1, and OATP1B3 (SLCO1B3)) of some ADME genes are probably slight adaptations to new in vitro conditions rather than the result of a gradual epithelial-to-mesenchymal transition (EMT) dedifferentiation process. This assumption is supported by an in-depth transcript expression analysis of representative epithelial and mesenchymal markers over the course of the entire culture period. It can be observed that mesenchymal markers only transiently increase (fibronectin [FN1], vimentin [VIM]) and return toward baseline levels over the course of 35 days or remain unaffected (alpha-SMA (ACTA2), N-cadherin (CDH2)) (Supplementary Fig. S6). Epithelial markers on the other hand, such as CK18 (KRT18), ZO-1 (TJP1), E-cadherin (CDH1), or hepatocyte-specific genes like albumin (ALB) or BSEP (ABCB11), remain stably expressed or even stabilize at higher expression levels.
Protein profiling of 3D hLiMTs
To establish the phenotypic drift of hLiMTs over time on the protein level, proteome analysis was performed from available microtissue samples after 7 to 35 days of culture. A total of 2273 proteins expressed in all samples and unique (identified with at least one proteotypic peptide) were included in the analysis. As in the gene expression data, phenotypic stability of hLiMTs was further observed by the low number of proteins that appeared significantly differentially abundant (p ≤ 0.05) over time in comparison to day 7 (Fig. 4A), starting from 43 differentially abundant proteins (day 14) to 165 proteins (day 35). Thus, the maximal observed change in protein abundance was only 7.3% of all proteins tested. PCA of the different donor samples identified t0 samples (cell suspension), which clustered separately from microtissues (Fig. 4B) similar to what has been observed in transcriptomics analysis. The microtissues over time revealed a subtle but continuous shift in proteome expression over time with day 7 samples being most distinct from all other time points. This is also reflected in a heatmap visualization of the entire proteome over time, demonstrating that day 7 samples are distinct from all other time points (Supplementary Fig. S5B). From day 14 on, changes in the proteome seemed to be quite moderate as the samples clustered according to donor, then time, in unsupervised hierarchical clustering. A closer examination of ADME/Tox relevant proteins demonstrates a stable expression from day 7 on, with a few exceptions of declining expression over time, such as for CYP2C8, CYP2E1, OCT1 (SLC22A1), and OATP1B1 (SLCO1B1). Again, as observed in the transcriptome, such changes of protein expression level do not seem to follow a general behavior, as could be expected through a continuous dedifferentiation process or during EMT. Similar to the transcript analysis, protein expression of mesenchymal markers was stable (N-cadherin (CDH2)) or only transiently increased (vimentin (VIM)), while epithelial markers were either stable (E-cadherin (CDH1)) or even increased (CK18 (KRT18), albumin [ALB]) over the 35-day incubation period. These observations indicate that changes in the proteome expression reflect a transient adaptive process, giving rise to a stable phenotype rather than a continual dedifferentiation or transdifferentiation process as commonly observed in 2D hepatocyte cultures.
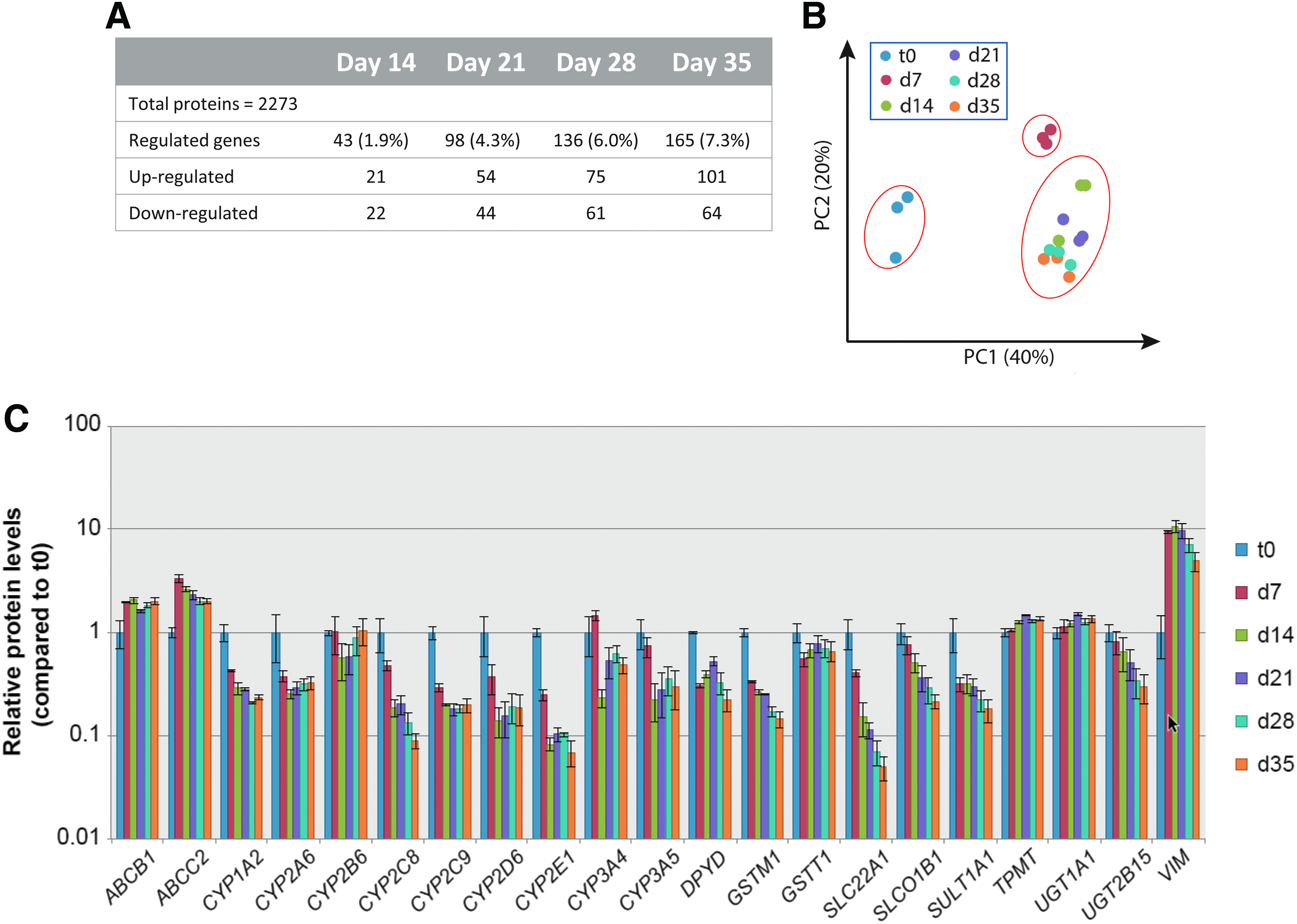
Proteomic profiling of freshly thawed cell suspension and derived hLiMTs from three individual hepatocyte donors (IPHH_01, IPHH_02, and IPHH_03) in coculture with NPCs (IPHN_03).
Assessment of cytochrome activity and inducibility of hLiMTs
To profile basal CYP activity of hLiMTs derived from PHH from different hepatocyte donors, microtissues were cultured under uninduced low-dexamethasone conditions. A cocktail of different CYP substrates was added to hLiMTs to monitor substrate conversion into metabolites as indicator of P450 activity. A linear relationship between 1, 2, 3, 6, 24, and 30 hours of exposed microtissues for substrate conversion was found (data not shown). Therefore subsequent experiments were performed for only one incubation time (24 hours). Supernatants of single hLiMTs were collected after exposure to CYP substrates for 24 hours after each week of a 5-week consecutive culture period, and metabolites were quantified by LC-MS (Fig. 5A).
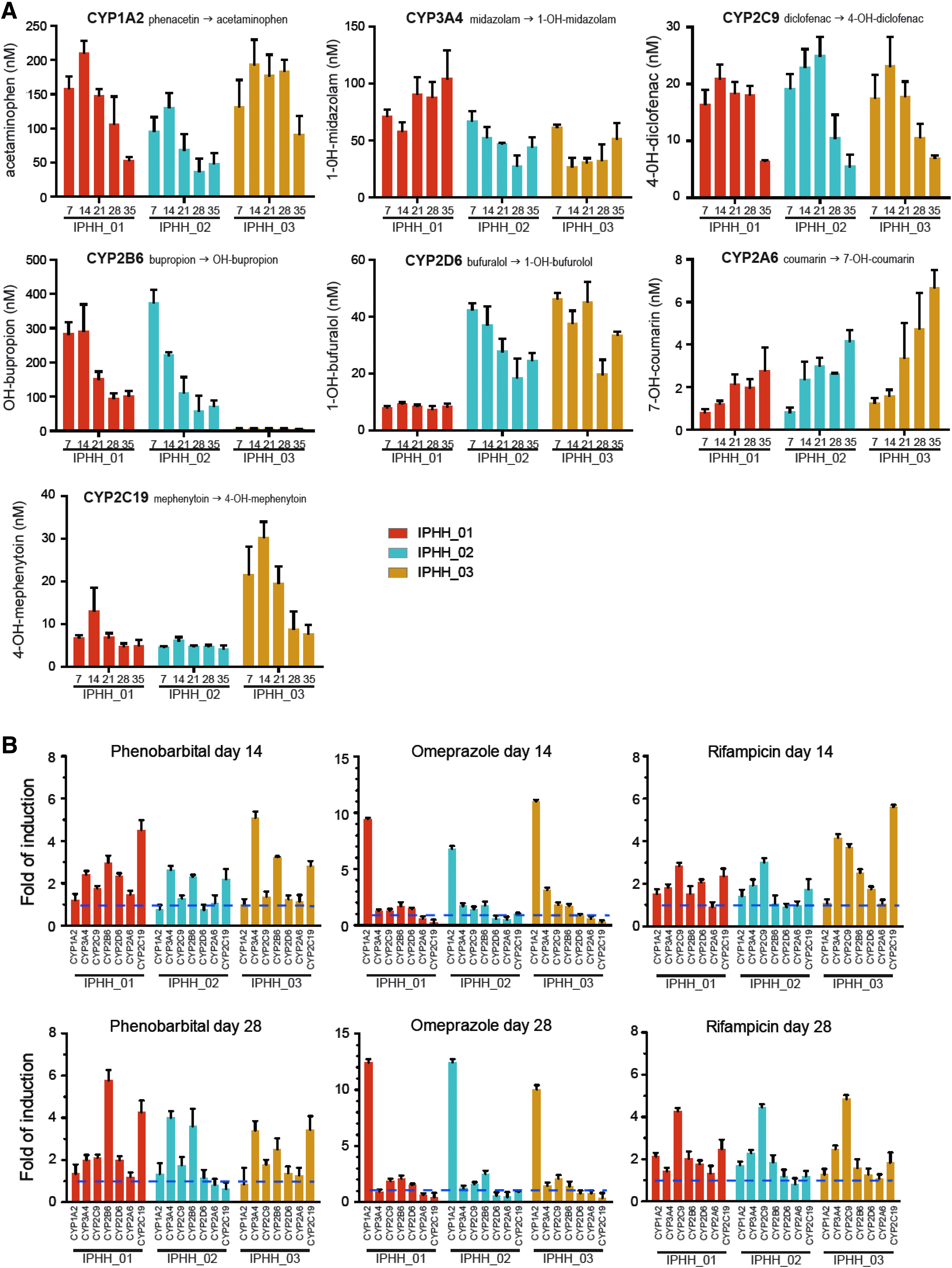
Assessment of basal CYP activity and inducibility of CYP activity of hLiMTs derived from three individual hepatocyte donors (IPHH_01, IPHH_02, and IPHH_03) in coculture with NPCs (IPHN_03).
The basal activity of the tested CYPs appeared to be preserved for several weeks in culture. However, a tendency for decline of CYP activity was seen for CYP1A2, 2C9, 2B6, and 2C19, which is most prominent after 4 weeks in culture, consistent with the loss of albumin secretion after this time in culture. Substantial differences were observed for the basal expression of certain CYPs between hepatocyte donors. For example, IPHH_01 displayed very little CYP2D6 activity; IPHH_03 showed very little CYP2B6 activity but had the highest CYP2C19 activity of the tested donors. The relative CYP activities among the different donor cells were mirrored by the corresponding protein expression levels (Supplementary Fig. S7), as well as in the case of IPHH_01 by the respective homozygous 2D6*4 genotype, which is known to represent a poor metabolizer. 34
We next tested the inducibility of cytochromes with prototypic inducers phenobarbital, omeprazole, and rifampicin. The inducers were added during 72 hours to the microtissues with daily re-dosing, followed by addition of CYP substrates for 24 hours. To test for the long-term functionality of this response, the induction was performed in the second culture week (day 14) and the fourth culture week (day 28). The fold induction of substrate turnover was calculated to the corresponding solvent control. Phenobarbital induced the enzyme activities of CYP3A4, 2B6, and 2C19 more than twofold (Fig. 5B). Omeprazole was a very specific inducer for CYP1A2 activity; Rifampicin induced CYP3A4, 2C9, and to a lesser extent the 2C19 activity. Overall, comparable inducibility between day 14 and 28 was observed, indicating that the microtissues are still susceptible to xenobiotic stimuli even after 4 weeks in culture.
Discussion
The use of 3D hepatocyte spheroid cultures is rapidly increasing in academic research and in the pharmaceutical industry. Increased knowledge of the biological capabilities and limitations of these liver model systems is critical for understanding the outcome of different experiments designed to evaluate different liver-specific functions. However, often the used models are insufficiently characterized for their biological functionality, which hinders interpretation of experimental data. This study provides a comprehensive biological baseline characterization of a commercially available 3D InSight human liver spheroid model for long-term culture.
Previous publications on the biological functionality of 3D liver spheroids focused mainly on rat liver models in different coculture configurations.35–39 In addition, 3D mouse liver spheroid models,40–42 as well as 3D cultures of human hepatocyte cell line spheroids, produced with HepG2 43 or HepaRG 44 were reported. Recently two articles were published, one showing the comparison of 1-week-old 3D liver spheroids with intact liver on a proteomic level 45 and another describing the metabolic signature of liver spheroids over the course of 3 weeks. 46 However, very little data are available on gene and protein expression changes over long-term culture of primary human liver spheroids.
As a follow-up to our previous studies,15,23 we extended the characterization of this model system by gene expression and proteomic profiling, as well as functional and morphological analysis. The morphological data show expression of typical liver marker protein, such as the periportally located albumin and perivenous CYP3A4 protein. Interestingly, the CYP3A4 protein was detected only in a subset of hepatocytes, indicating that the phenotype of the cell originating from the periportal region is preserved, similar to recently reported data. 45 The presence of Kupffer cells was detected over the whole culture period by active cytokine secretion when stimulated with lipopolysaccharide (Supplementary Fig. S2). Although the Kupffer cells originated from a different donor, basal activity was low while tissue viability and function remained stable over several weeks of culture, suggesting that Kupffer cells are compatible with hepatocytes from different donors. In addition, the presence of BSEP (ABCB11) and MRP2 (ABCC2) supports the conclusion that extensive bile canaliculi are established within hLiMTs due to the presence and polarized expression of these hepatobiliary transporters (Fig. 1A, B). This suggests that excretion of toxic waste products such as bile acids into bile canaliculi is active. The functionality of these transporters was previously tested with fluorescent bile acid analogs cholyl-lysyl fluorescein (CLF) and chloromethylfluorescein diacetate (CMFDA), which were actively transported to the bile canaliculi. 47 The observed atypical redistribution of the MRP2 (ABCC2) transporter to the basolateral membrane of peripheral cells (Fig. 1B), as also observed in Bell et al., 45 might represent biliary cells as a minor constituent in hepatocyte fractions, as suggested by positive CK19 (KRT19) staining (Supplementary Fig. S3).
Phenotypically, both gene expression and proteomic profiling indicate that only small changes in transcript and protein expression occur, once microtissues have been formed and gained their full functionality ∼7 days after seeding, with only 2.5% of genes and 7.3% of proteins significantly regulated over the entire culture period up to 35 days. This further supports the observation of the extended viability and hepatic functionality of this 3D cell culture format and implies phenotypic stability during long-term culture. However, PCA revealed phenotypic differences of microtissues compared to both native unrelated liver samples and hepatocyte/NPC suspensions before seeding, whereas liver microtissue samples collected over a 4-week period were tightly clustered. This indicates that the microtissue cultures are somewhat distinct from the “raw material” (freshly thawed liver biopsies or liver cell suspensions), yet maintain a stable phenotype during long-term culture. However, differences between native liver, cell suspension, and microtissues seem to be less prominent when analyzing liver enriched genes and proteins relevant to ADME/Tox such as transporters and phase I and phase II enzymes. We observed with a few exceptions (i.e., CYP1A1, CYP1A2, CYP2E1, and OATP1B3 (SLCO1B3)) surprisingly similar expression levels throughout the different samples and time points (Fig. 3C).
Similar to the gene expression PCA, the protein expression profile from cell suspensions (hepatocytes/NPCs) was clearly distinct from microtissue cultures (Fig. 4B). This was also reflected in some of the ADME genes with day 0 cell suspension demonstrating higher protein expression levels than in corresponding microtissues (Fig. 4C), which contrasts with the observation on the transcript levels (Fig. 3B). This discrepancy might be explained, in part, by the different half-lives of transcripts and corresponding proteins (9 hours vs. 46 hours in average 48 ) during the adaptational process from the time of organ harvest to in vitro culture.
Comparative analysis of transcriptomics and proteomics in other studies only showed moderate correlations, 49 explained due to multiple factors such as posttranslational modifications and cellular location of proteins. Likewise, Liu et al. recently reviewed the relationships between transcripts and proteins under various scenarios. They concluded that transcript levels alone are not sufficient to predict protein levels and that correlations may depend on many factors such as applied techniques used for quantification of transcriptome and proteome, cell state, and posttranscriptional and posttranslational processes. 50 These factors might also explain differential observations between transcriptomics and proteomics in this dataset.
Functional analysis of CYPs over a 5-week cultivation period showed that activities of most cytochromes are stably maintained. However, clear differences between the used hepatocyte lots were observed, which reflect native cytochrome polymorphism in individual human donors. Depending on the research question, a strategy for avoiding large donor-related variation in CYP activities is the use of pooled primary human hepatocytes from different donors, which more closely reflect the average response of a population but might miss the interindividual variability in response.
A hallmark of biological responsiveness of native liver is the ability to upregulate detoxification or activation of xenobiotics by increased activity of cytochromes. The prototypic inducers of CYPs, phenobarbital (CYP3A4, 2B6, and 2C9), omeprazole (CYP1A2), and rifampicin (CYP3A4, 2C9, 2B6, and 2C19) showed the induction of appropriate cytochrome activity. The expected inhibition of CYP2C19 by omeprazole was also observed in two out of three donors. The fold inductions varied between 2- and 15-fold, which is in the expected range for a functional substrate-turnover assay since higher fold inductions are typically only observed with gene-expression endpoints; however, it is recommended to use both enzyme activity and RNA expression endpoints due to interplay between inhibition and induction of P450 enzymes. 51 The low dexamethasone content of the culture medium thus allowed for good basal cytochrome activity, while maintaining CYP inducibility even after 4 weeks in culture.
While the transcriptome and proteome of fresh liver samples and/or cell suspension are distinct from microtissue cultures, the CYP450 activity profile highlights the functionality and stability of 3D liver microtissue cultures over time. This suggests that the 3D configuration of hLiMTs preserves typical liver gene expression and function for up to 5 weeks in culture. Given the regenerative potential of liver tissue, the initial alterations of transcriptome and proteome in the early phase of microtissue formation may represent a repair and recovery process rather than a terminal dedifferentiation or epithelial-to-mesenchymal transdifferentiation commonly observed in 2D PHH cultures.52,53 This theory is supported by the observation that, at the protein level, classical EMT markers (fibronectin, vimentin, alpha-SMA, and N-Cadherin) are only slightly or transiently induced, peaking around 7–14 days of culture, and revert toward baseline levels, while classical epithelial markers (CK18, E-Cadherin, and ZO-1) or liver specific markers (albumin, BSEP) remain stably expressed or even increase over time (Supplementary Fig. S6). This is in alignment with the proposed concept of EMT/mesenchymal-to-epithelial transition (MET) during liver regeneration, in which epithelial cells undergo partial and transient EMT to revert later to hepatocytes or ductal cells through MET. 54 This hypothesis is supported by the observed positive vimentin staining at the periphery of 3D microtissues (Supplementary Fig. S4), suggesting that hepatocytes directly exposed to cell culture media might undergo partial EMT. Alternatively, temporary elevation of fibronectin, vimentin, and alpha-SMA could also be indicative of residual stellate cell activation as a consequence of organ procurement, the cell isolation, and freeze/thaw process, which eventually resolve in the course of hLiMT culture where stellate cells reacquire a more quiescent state.
Because a 3D spheroid model can only be as good as its starting material, careful selection of hepatocyte lots and Kupffer/NPC sources is important. For this model, several hepatocyte lots were tested and suitable lots selected. This selection for suitable donor lots is only feasible by having access to a repository of cryopreserved human hepatocytes and NPCs from commercial suppliers and which are available prequalified and quality control tested. Cryopreserved cells have also the advantage to be ordered in multiple vials per lot, which assures reproducible quality and results. The achieved homogeneity in spheroid size (Fig. 2A) allowed for low standard deviations between wells and assays. Thus, with the described hLiMTs, a highly biologically functional model is available, which is suited for various applications in liver research, such as the study of drug metabolism and pharmacokinetics, drug–drug interactions, hepatobiliary uptake 32 and efflux, 47 as well as applications in nanotoxicology. 31 In addition, the 3D liver spheroids proved suitable for reflecting liver pathologies, such as cholestasis, steatosis, and viral hepatitis,45,55 as well as for prediction of DILI.23,45
Conclusion
We have shown the stability of the hLiMTs from 7 to 35 days of culture, with preserved morphology and important liver functions. Transcriptomic and proteomic signatures assessed over 5 weeks confirm the stability of the model system during extended culture times, but also reveal the dynamics of primary liver cells, reflecting the adaptive process the cells undergo to cope with the stressful isolation procedure, freeze/thaw cycle, and exposure to new in vitro culture conditions. Importantly, after an initial adjustment to such new in vitro conditions (7–14 days), hLiMTs remain relatively stable on both a transcriptomic and proteomic level over time. This implies that hLiMT culture can be subjected to experimental procedures around 7 days after tissue formation after which the model remains rather stable with respect to phenotype and function and which would allow an experimental time window of up to 25 days. In long-term cultures, the difference observed between different samples was better assigned to variation among individual donors than time in culture. The described 3D liver spheroid model therefore proves to be suitable for many applications in in vitro liver research and has the potential to become a standard model in liver toxicology.
Footnotes
Acknowledgments
The authors thank Franziska Stolz and Richard Prazak for expert technical assistance. This work was supported by the Swedish Research Council (grant number 2760) and the Swiss State Secretariat for Education, Research and Innovation and the European Union [Eurostars Project grant number E!8511 “HTS-DILI”].
Author Disclosure Statement
S.M., K.R., J.M.K., and W.M. are employees of InSphero AG. V.M.L. and M.I.-S. are cofounders and owners of HepaPredict AB.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.