
Abstract
Abstract
In the process of a paradigm shift in toxicity testing, many efforts have focused on how to integrate and interpret information for biological events occurring at molecular and cellular level to be predictive of adverse effects at organism or population level to be useful, for example, for regulatory decision-making. The adverse outcome pathway (AOP) concept provides such a framework of knowledge-based safety assessment of chemicals that links mechanistic information with an apical endpoint. Here we outline an AOP that links the activation of peroxisome proliferator-activated receptor α (PPARα) to reproductive tract malformations and impaired fertility in males. The development of this AOP relies on evidence collected from rodent models and incorporates human mechanistic and epidemiological data. Interest in PPARα action as a mechanistic basis for effects on the reproductive system arises from the relationships between activation of this receptor and impairment of steroidogenesis leading to reproductive toxicity. The PPARα-initiated AOP is a first step for structuring current knowledge about mode of action (MoA), which is neither androgen receptor-mediated nor via direct aromatase inhibition. In the current form, the pathway lays a strong basis for linking an endocrine MoA with an adverse effect, a prerequisite requirement for the identification of endocrine disrupting chemicals.
Introduction
T
Endocrine activity involves a range of biological pathways, including (but not limited to) estrogen, androgen, thyroid, peroxisome proliferator-activated receptors (PPARs), retinoids, and actions through other nuclear receptors. A variety of man-made chemical classes such as synthetic drugs and pesticides and naturally occurring substances (e.g., genistein, daidzein) are active toward endocrine systems in biological organisms. With hormones orchestrating virtually all physiological processes, the identification of relevant (in vivo) endpoints that indicate specific perturbation of endocrine processes is challenging. This makes it difficult to identify and hence regulate substances that can (potentially) cause adverse effects by disrupting the endocrine system. The OECD has had a program of test guideline development for identifying substances that may be EASs or EDs since the late 1990s. Many new tests have been validated and older tests updated (e.g., OECD TG 443, 407). The “toolbox” of tests can be found in the OECD Conceptual Framework for Testing and Assessment of EDs. 3
In addition, new approaches to understand the mechanisms of toxicity, including endocrine disruption, are being explored; one of them being adverse outcome pathways (AOPs). 4 An AOP is an analytical construct that describes a sequential chain of causally linked events at different levels of biological organization that lead to an adverse health or ecotoxicological effect. AOPs are the central element of a toxicological knowledge framework being built to support chemical risk assessment based on mechanistic reasoning. Organization of existing knowledge into AOP descriptions provides a systematic and transparent assembly of the evidence that supports extrapolation from initial or intermediate measures of biological perturbation to useful predictions of potential hazard. With this in mind, an AOP describes key building blocks, key events (KEs), and key event relationships (KERs) linking a molecular initiating event (MIE) and adverse outcome (AO), in sufficient detail to support the application of a wide range of mechanistically based data in risk assessment and regulatory decision-making.
The OECD has undertaken a major activity of supporting the development of AOPs. 5 The AOPs are captured in the AOP-Wiki (www.aopwiki.org), which is part of a broader AOP knowledge base (AOP-KB www.aopkb.org) whose elements are currently under development. The goal is to assemble a comprehensive collection of accessible AOPs that provide a base to develop new and more accurate safety assessment approaches for substances. The utility of AOPs for regulatory application is defined to a large extent by the confidence and precision with which they facilitate extrapolation of data measured at low levels of biological organization (often in vitro) to predict the higher level outcomes. In parallel, they promote the use of alternative methods increasing our understanding of biological processes and contributing to decreasing the use of animals in toxicological research.
This review describes an AOP (PPARα activation in utero leading to reproductive tract malformations and impaired fertility in males) that was developed within the OECD AOP process. 6 Although the involvement of PPARα in the pathway has been investigated for almost 20 years, its role is still under scientific discussion.7,8 The uncertainty and lack of adequate experiments confirming or refuting its role was recently expressed during the OECD organized review of this AOP in September 2015 (https://aopwiki.org/wiki/index.php/Review:OECD_External_Review_September,_2015_-_Aop:18). The reviewers supported the evidence calls as moderate to strong for all other subsequent KEs and KERs leading to the AO. This AOP, together with 12 other pioneer AOPs, was reviewed twice in a process managed by the OECD.
The first (internal) review took place in March 2015 and an external review followed in September 2015. In the internal review process, 3 experts nominated by the Extended Advisory Group on Molecular Screening and Toxicogenomics (EAGMST) checked compliance of the AOP structure and content with guidance documents. 9 In the external part of the review process, scientific/technical content of the AOP was assessed by six independent experts in the field. The external review was conducted by experts not involved in the development of the specific AOP, who had scientific expertise in the hazard area/endpoint covered by the AOP.
Due to the uncertainties connected to experimental data defining the role of PPARα in the pathway, the external reviewers proposed two options: designate MIE as unknown or change to the next KE. Both of the options are not optimal as an AOP per definition should contain an MIE and designation of subsequent events as MIE would be more generic (e.g., decrease of steroidogenic enzymes). Currently, the final decision has not been made by the EAGMST. The final outcome of the review was that there was some uncertainty over the MIE, but the AOP as a whole provides a useful framework for regulatory chemical assessment, development of targeted testing strategies, and identifying further research needs.
The Core Elements of the AOP
The AOP was developed linking PPARα activation with male reproductive/developmental-related adverse outcomes (found at AOP-Wiki: www.aopwiki.org/aops/18). The development of this AOP relies on the evidence collected from rodent models and incorporates human mechanistic and epidemiological data.
The KEs in the pathway comprise the activation of PPARα (MIE), leading to decreases in translocator protein (TSPO; KE1a) and/or steroidogenic acute regulatory (StAR) protein (KE1b), the disruption of cholesterol transport in mitochondria (KE2), reduction in testosterone synthesis (KE3) and, hence, circulating hormones (KE4). This indirectly leads to the AOs of malformation of the reproductive tract in males (AO1) with impaired fertility (AO2) as a possible consequence (Fig. 1).
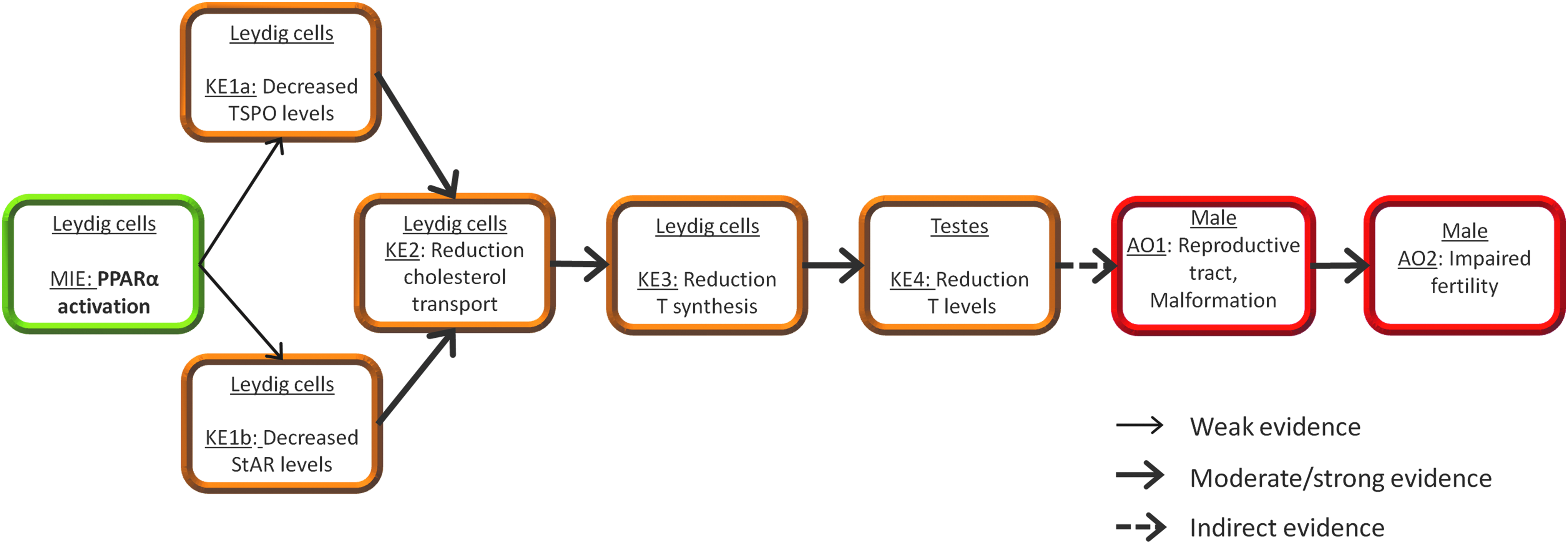
The schematic representation of PPARα activation in utero leading to impaired fertility in males. The narrow lines indicate weak key event relationships, whereas dashed lines indicate an indirect key event relationship. Green box represents the MIE; orange key, events that might lead to disturbance of homeostasis; in red, adverse outcomes. MIE, molecular initiating event; PPARα, peroxisome proliferator-activated receptor α.
In its current form, the pathway lays a strong basis for linking an endocrine mode of action (MoA) with an adverse effect, a prerequisite requirement for the identification of EDs. Identifying the mechanism of action is important because some MoAs are known to operate in model species but not humans (or vice versa), meaning that the related adverse effect could be discounted (or accounted for) when considering relevance to man. The PPARα is a ligand-activated transcription factor that belongs to the nuclear receptor family, which also includes the steroid and thyroid hormone receptors. Interest in PPARα action as a mechanistic basis for effects on the reproductive system arises from the relationships between activation of this receptor and steroidogenesis, impairment of which may impact on fertility. 10
PPARs play important roles in the metabolic regulation of lipids, of which cholesterol, in particular, being a precursor of steroid hormones, makes the link between effects on lipid metabolism and effects on reproduction.
The AOP draws heavily on data generated on data-rich chemicals (mainly phthalates) and aims to provide a means of systematic collection of relevant information, which could be used to assess reproductive toxicity potential. The phthalates are among the most widespread environmental contaminants worldwide. The wide exposure of the general population to phthalates11–13 has raised concern about possible detrimental health effects.14–17 Some phthalates are developmental and reproductive toxicants in rats, largely affecting the male reproductive system.18–24 The adverse endocrine effects of antiandrogens, including the phthalates, have recently been shown to act in a dose-additive manner.7,25,26 Moreover, recent reviews of epidemiological and experimental data have concluded that phthalate antiandrogenicity is plausible in adult men.27,28 This underscores the importance of delineating this AOP, gaining further knowledge on its events, identifying chemicals acting via this MoA and understanding relevance of the pathway to humans.
The following sections describe the events of the AOP in detail, outlining the strength of evidence for the KEs and the relationships between them.
MIE: PPARα Activation in Leydig Cells
The PPARα belongs to the steroid/thyroid/retinoid receptor superfamily of transcription factors (NR1C). PPARα is expressed at high levels in tissues that perform significant catabolism of fatty acids (FAs). 29 The receptor is also present in skeletal muscle, intestine, pancreas, lung, placenta, and testes.30,31 PPARs are activated by FAs and their derivatives. They are sensors of dietary lipids and are involved in lipid and carbohydrate metabolism, immune response, and peroxisome proliferation.32,33
PPARα is also a target of hypothalamic hormone signaling and was found to play a role in embryonic development. 34 According to the reviews of David, 8 Latini et al., 10 and Corton and Lapinskas, 35 PPARα activators have been shown to alter steroidogenesis and in consequence impair reproduction. In addition, a predictive model of rat reproductive toxicity based on more than 500 assays mapped to hundreds of genes and spanning many MoAs showed an association of PPARα activity with reproductive toxicity. 36 According to this model, 10 PPARα agonists, including bis(2-ethylhexyl) phthalate (DEHP), mono(2-ethylhexyl) phthalate (MEHP), and perfluorooctanoic acid (PFOA), shared a relatively common reproductive toxicity profile of a decrease in reproductive performance (i.e., decreased fertility).
The PPAR family is composed of three isotypes (PPARα, PPARβ, and PPARγ). They form heterodimers with the retinoid X receptor (RXR) and adopt an active conformation in the presence of an agonist. The characteristic feature of the PPAR ligand binding cavity is its size, which is three to four times larger than that of the other nuclear receptors, which increases the number and type of chemical substances that may act as ligands. 32 The capacity to fit and bind these diverse chemical structures results in distinctive gene expression profiles, which ultimately lead to different clinical outcomes.37,38 As an example, rosiglitazone is a full agonist for PPARγ exerting strong activation of adipogenic genes. In contrast, MEHP is only a partial agonist and showed only mild stimulation. 38 Perturbation of PPAR signaling can also alter other nuclear receptor signaling pathways, such as the retinoic acid and thyroid hormone signaling pathways, by sequestering endogenous heterodimer binding partners (such as RXR).39,40 Nuclear receptor signaling via such nuclear pathways is essential for correct hormonal responses and interference may impact on both gonadal development and spermatogenesis that rely on the integrity of this signaling.41,42
Therefore, the potential differential impact of interactions of different chemicals with PPARs and their related coactivators or corepressors on the nuclear signaling pathways needs to be considered.
Selected chemicals known to interact with the PPARα are shown in Table 1. Although the evidence that PPARα activation is the MIE in this AOP is contradictory (see KER: the relationship between activation of PPARα and decreased TSPO levels), there are experimental data indicating that PPARα is implicated in regulating reproductive function. 43 The mechanistic link between PPARα activity and fertility or other reproductive impairment is correlative, but the role of PPARα in steroid metabolism and its activity in reproductive tissues allow us to infer that it is a plausible target for disruption of endocrine signaling and altered gametogenesis.
DBP, dibutyl phthalate; DEHP, bis(2-ethylhexyl) phthalate; MEHP, mono(2-ethylhexyl) phthalate; PFOA, perfluorooctanoic acid; PPARα, peroxisome proliferator-activated receptor α; RXR, retinoid X receptor; TBT, tributyltin; TSPO, translocator protein.
KER: the relationship between activation of PPARα and decreased TSPO levels
Activation of PPARα leads to decreased expression of the cholesterol transport (TSPO) gene in steroidogenic cells (e.g., Leydig cells). Consequently, the amount of cholesterol transported into mitochondria decreases, impacting on steroid production.
PPARs regulate genes involved in cholesterol uptake and transport.44–46 The indirect link between PPARs and regulation of cholesterol transport in mitochondria derives from studies demonstrating PPARα-dependent control of TSPO.45,46 PPARα is present in steroidogenic cells, for example, in testes during development as well as in adult testes31,47; and modulation of its activity has been shown to impact on TSPO transcriptional activity. 45
The exact nature of this relationship is not known, however, there is some evidence providing a basis for this hypothesis. Gazouli showed that PPARα activators (bezafibrate, MEHP) inhibited the transfer or loading of cholesterol to the inner mitochondrial membrane in MA-10 mouse Leydig tumor cells and decreased levels of TSPO protein. In addition, these chemicals also inhibited the formation of progesterone (a precursor of testosterone) in R2C cells (rat Leydig tumor cell line). In vivo, levels of the TSPO protein were decreased in testes of adult mice exposed to DEHP (although DEHP is not a PPAR activator in vitro, it is metabolized to MEHP, which is its metabolite) 45 and the messenger RNA (mRNA) levels were decreased in a dose-dependent manner in fetal testes. Bezafibrate and phthalates inhibited both hormone-induced and constitutively sustained steroidogenesis with similar IC50 values, indicating that they act on a common regulatory component of the steroidogenic pathway. 45
There are, however, some uncertainties related to the involvement of PPARα in regulation of TSPO. Treatment of adult mice with a PPARα activator (DEHP or WY-14,643) resulted in reduced levels of circulating testosterone and testis TSPO mRNA, consistent with the in vitro effects. 45 In contrast, liver TSPO mRNA levels have been increased, indicating a tissue-specific regulation of TSPO expression by PPARα activators. 45 In untreated PPARα-null mice, compared with the wild-type controls, circulating testosterone levels were decreased suggesting a positive constitutive role for PPARα in maintaining Leydig cell steroid formation. Surprisingly, treatment of the PPARα-null mice with PPARα activators (DEHP and WY-14,643) restored testosterone formation and TSPO mRNA returned to normal levels, suggesting PPARα-independent pathways might be involved in the regulation of TSPO genes and steroidogenesis. 45
In support of this hypothesis, other studies have demonstrated that part of the toxic effect of phthalate (DEHP) on testis was retained in PPARα-null mice unlike effects in the liver, which were absent. 48 However, because the null mice developed lesions in the testis, it indicated that DEHP can also act through PPARα-independent pathways in mediating testicular toxicity.
There is some evidence involving additional PPARs in transcriptional regulation of TSPO:
One genomic study in rat fetal testes at gestation day (GD) 14–18 does not support the hypothesis that activation of PPARα/γ pathways is involved in the effects of phthalates on sexual differentiation of the male rat, since the PPARγ isoform was not detected and, although PPARα was present, a PPARα activator (Wy-14,643) had no effect on testosterone production. 50 Also, differential patterns of TSPO expression in the fetal rat testis have been observed on phthalate (dibutyl phthalate [DBP]) treatment. In whole-testis expression of TSPO, mRNA was upregulated, whereas protein levels were decreased in interstitial cells. 51
KER: the relationship between activation of PPARα and decreased StAR levels
The direct link between PPARα regulation of cholesterol transport in mitochondria and hormone synthesis derives from studies demonstrating that PPARα may act as an indirect transrepressor of the key steroidogenic factor-1 (SF-1).52,53 SF-1 is a transcription factor essential for expression of genes involved in steroidogenesis (including StAR).
PPARα is expressed in fetal rat Leydig cells47,53 and in adult rat Leydig cells. 31 Recent studies have shown that fetal testes contained PPARα protein-binding peaks in CYP11a, StAR, and CYP17a regulatory regions. 53 Binding of PPARα to the promoter region of steroidogenic genes occurs at binding sites different from those of SF-1, indicating that PPARα may be an indirect repressor of SF-1 binding. Moreover, it is possible that PPARα could act via sequestration of the shared coactivator CREB-binding protein (CBP). 53 PPARα and SF-1 share CBP, which is present in limited concentrations, 54 and PPARα and SF-1 may compete for binding to CBP. SF-1 controls transcription of the StAR gene. 55 StAR protein plays a critical role in the movement of cholesterol from the outer to the inner mitochondrial membrane. 56
Hence, it seems likely that the ability of PPARα to interfere with SF-1 binding/transactivation caused by exposure to chemicals (e.g., phthalates) could affect StAR expression and cholesterol transport in mitochondria. PPARα agonists have been shown to suppress Leydig cell steroidogenesis 45 and downregulate steroidogenic genes, including StAR.49,51,57
Moreover, PPARα agonists, which do not directly transrepress the StAR promoter, have been found to downregulate the expression of this gene in steroidogenic tissues (mouse ovaries). 58
KE 1a: Decreased TSPO Levels in Leydig Cells
TSPO (previously known as the peripheral benzodiazepine receptor [PBR]), is a mitochondrial outer membrane protein implicated in cholesterol import to the inner mitochondrial membrane. 59 It mediates the delivery of the substrate cholesterol to the inner mitochondrial side chain cleavage cytochrome P-450. 59 TSPO shows high sequence conservation from bacteria to mammals. It is expressed ubiquitously, but is most abundant in steroidogenic cells. 60
TSPO is present in virtually all mammalian peripheral tissues. 61 It has been implicated in many cellular processes; among these are steroid biosynthesis, protein import, heme biosynthesis, immunomodulation, cellular respiration, and oxidative processes. Although TSPO is present in virtually all mammalian peripheral tissues, TSPO protein expression is highly prominent in steroidogenic tissues.62,63 The presence of TSPO has been confirmed in Leydig and Sertoli cells, 64 granulosa cells, 65 and to a lesser extent in thecal cells. 64
In subcellular fractions, binding sites for the TSPO have been identified to be present in the outer mitochondrial membrane.62,66 Transcriptional regulation of the TSPO gene has been reviewed recently. 64
There are some contradictions with regard to function of TSPO protein. TSPO knockout mice showed embryonic lethality, 67 however, recent findings have shown no effect on viability. 68
Aberrant TSPO levels have been linked to multiple diseases, including cancer, endocrine disorders, brain injury, neurodegeneration, ischemia–reperfusion injury, and inflammatory diseases. 63 However, recent studies have shown some contradictory results as PBR/TSPO global knockout mice were viable and there were no effects on steroid hormone biosynthesis.68,69 As stated in the recent review, “At this point in time, a functional designation for TSPO is still actively being sought.” 70
KER: decreased TSPO levels lead to a reduction in cholesterol transport
TSPO ligands stimulate steroidogenesis and induce cholesterol movement from the outer mitochondrial membrane to the inner mitochondrial membrane. 59 Therefore, it follows that reduced activity of the TSPO will impair the delivery of cholesterol necessary for hormone biosynthesis.
Decreased TSPO protein levels have been shown to reduce cholesterol transport into mitochondria of mouse Leydig cells.45,49 Moreover, the uptake of cholesterol was decreased in Leydig cell mitochondria on exposure to phthalates. 71
There are some uncertainties reported in the literature concerning this KER as reported under KE1a above. Whereas targeted disruption of TSPO in rat Leydig R2C cells led to reduced steroidogenesis, 72 recent experiments involving TSPO knockdown in steroidogenic cells did not affect steroid hormone biosynthesis 68 nor did specific deletion of TSPO in Leydig cells impair their synthesis of testosterone. 69
KE1b: Decreased StAR Levels in Leydig Cells
StAR protein functions as a cholesterol transfer protein and acts directly on lipids of the outer mitochondrial membrane to promote cholesterol translocation. 56 Reduction of the StAR impacts on the amount of substrate available for steroidogenesis. StAR is expressed principally in steroidogenic tissues. 73 It has been cloned from many species and is highly conserved among mammals, birds, amphibians, and fish. 73 StAR appears to regulate acute steroid production. 74 Transcriptional or translational inhibition of StAR expression results in a dramatic decrease in steroid biosynthesis, whereas approximately 10%–15% of steroid synthesis appears to be mediated through StAR-independent mechanisms.74,75 In contrast, normal regulation of steroid production appears to be largely mediated by increased transcription of steroidogenic enzymes. 76
KER: decreased StAR levels lead to a reduction in cholesterol transport
StAR mediates the delivery of cholesterol from the outer to the inner mitochondrial membrane, where it undergoes side chain cleavage by a cytochrome P-450 enzyme (P450scc) that yields the steroid precursor, pregnenolone. 59 The cholesterol transfer within the mitochondria is the rate-limiting step in the production of steroid hormones (reviewed in Miller and Auchus 77 ). Therefore, reduced amount/activity of the StAR will impair the cholesterol delivery necessary for the hormone biosynthesis. StAR is primarily present in steroid-producing cells, including theca cells and luteal cells in the ovary, Leydig cells in the testis, and some cell types in the adrenal cortex.
Downregulation of StAR and impaired steroidogenesis were reported on exposure to phthalates, 49 and a correlation was observed between inhibition of steroidogenesis and reduction of cholesterol synthesis in the fetal testis, including total testis cholesterol levels. 78 It is worth noting that some steroidogenesis is independent of StAR, however, the mechanism of StAR-independent steroidogenesis is unclear. 77 Johnson et al. 82 also proposed the involvement of sterol regulatory element-binding protein (SREBP) in phthalate-mediated disruption of steroidogenesis. Their study showed that lipid metabolism pathways transcriptionally regulated by SREBP were inhibited in the rat but induced in the mouse, and this differential species response corresponded with repression of the steroidogenic pathway.
KE2: Reduction of Cholesterol Transport in Leydig Cells
In mitochondria of steroidogenic tissues, there are two specialized mechanisms related to hormone synthesis: one by which cholesterol is delivered to the mitochondria and the other by which specialized intramitochondrial enzymes participate in the synthesis of hormonal steroids.
Steroidogenesis begins with the transport of cholesterol from intracellular stores into mitochondria. This process involves a series of protein–protein interactions involving cytosolic and mitochondrial proteins located at both the outer and inner mitochondrial membranes. In steroidogenic cells, cholesterol import to the mitochondrial inner membrane is crucial for steroid synthesis. 79 This process is facilitated by the scavenger receptor class B type 1 (SR-B1; more relevant to rodents, than humans) that mediates the selective uptake of cholesterol esters from high-density lipoproteins. STAR and TSPO mediate the transport of cholesterol across the mitochondrial membrane.
The rate of formation of systemic steroid hormones depends on the rate of cholesterol transport from intracellular stores to the inner mitochondrial membrane and loading of P450scc with cholesterol. 77 Interference with one or more of these reactions leads to reduced efficiency of steroid production.
KER: reduction of cholesterol transport leads to a reduction of testosterone synthesis
Production of steroid hormones depends on the availability of cholesterol in the mitochondrial matrix. Decreased cholesterol inside the mitochondria (e.g., by decreased expression of StAR or TSPO) leads to diminished substrate for hormone (testosterone) production.
Steroid hormones play a critical role in sexual development, homeostasis, stress responses, carbohydrate metabolism, and reproduction. Within steroidogenic tissues they are synthesized from a common precursor, cholesterol. Mitochondria are a key control point for the regulation of steroid hormone biosynthesis. The first and rate-limiting step in steroidogenesis is the transfer of cholesterol from the outer mitochondrial membrane to the inner mitochondrial membrane, a process dependent on the action of StAR. 56 The subsequent transport across the inner mitochondrial space into the steroidogenic pathway is executed by TSPO. 80
Testosterone production by Leydig cells is primarily under the control of luteinizing hormone (LH). Stimulation of the Leydig cells results in the activation of StAR transcription and translation, facilitating the transfer of cholesterol into the mitochondrial matrix to P450scc (CYP11A), which converts cholesterol to pregnenolone. Pregnenolone diffuses to the smooth endoplasmic reticulum (ER) where it is further metabolized to testosterone via the actions of 3β-hydroxysteroid dehydrogenase Δ5-Δ4-isomerase (3β-HSD), 17α-hydroxylase/C17-20 lyase (P450c17, CYP17), and 17β-hydroxysteroid dehydrogenase type III (17HSD3). For review, see Payne and Hales. 81
Decreased expression of genes that are responsible for cholesterol transport and steroidogenic enzyme activity, in the Leydig cell, leads to decreased testosterone production.
There is evidence demonstrating a coordinated reduction in the expression of key genes and proteins involved in cholesterol transport and steroidogenesis, together with a corresponding reduction in testosterone in testes in response to phthalates. For example, exposure of rats to 500 mg/kg phthalate (DBP) caused a profound reduction in the expression of genes involved in lipid metabolism in fetal Leydig cells.82,83 Fetal Leydig cells normally exhibit a high rate of lipid metabolism, which is required for both synthesizing and importing the testosterone precursor cholesterol.
In addition, after phthalate exposure, testis cholesterol and cholesterol-containing lipid droplets in fetal Leydig cells are also reduced.51,78,82 Testosterone production may also be diminished due to reduction/inhibition of other genes than TSPO and StAR (e.g., P450scc).47,71
KE3: Reduction of Testosterone Synthesis in Leydig Cells
In humans and other mammals, testosterone is secreted primarily by the testes and, to a lesser extent, the ovaries. Small amounts are also secreted by the adrenal glands. Testosterone is synthesized by the gonads and other steroidogenic tissues (e.g., brain, adipose) acting locally or being transported to other tissues via blood circulation. Steroid synthesis takes place within the mitochondria of Leydig cells, the testosterone-producing cells of the testis.
Testosterone production depends on stimulation of these cells by LH that is secreted in pulses into the peripheral circulation by the pituitary gland in response to gonadotropin-releasing hormone (GnRH) from the hypothalamus. Testosterone and its aromatized product, estradiol, then feed back to the hypothalamus and pituitary to suppress transiently LH and thus testosterone production. In response to reduced testosterone, GnRH and LH are again produced. This negative feedback cycle results in pulsatile secretion of LH followed by pulsatile production of testosterone.84,85
Testosterone is the principal male sex hormone and an anabolic steroid. Male sexual differentiation in general depends on testosterone, dihydrotestosterone, and the expression of androgen receptors (ARs) by target cells. 86 During development, secretion of androgens by Leydig cells is essential for masculinization of the fetus. 87
The fetal Leydig cells develop in utero. These cells become competent to produce testosterone in rat by GD 15.5, with production increasing markedly thereafter. Peak steroidogenic activity is reached just before birth, on GD 19. Testosterone secreted by fetal Leydig cells is required for the differentiation of the male urogenital system late in gestation. 88 Fetal Leydig cells also play a role in the scrotal descent of the testis through their synthesis of insulin-like growth factor 3 (Insl3), for review, see Nef. 87
In humans, the first morphological sign of testicular differentiation is the formation of testicular cords, which can be seen between 6 and 7 weeks of gestation. Steroid-secreting Leydig cells can be seen in the testis at 8 weeks of gestation. At this period, the concentration of androgens in the testicular tissue and blood starts to rise, peaking at 14–16 weeks of gestation. At this time, the number of Leydig cells also increases (for review, see Rouiller-Fabre et al. 89 ).
Adult Leydig cells, which are distinct from the fetal Leydig cells, form during puberty and supply the testosterone required for the onset of spermatogenesis, among other functions. Distinct stages of adult Leydig cell development have been identified and characterized. The Leydig stem cells are undifferentiated cells that are not only capable of indefinite self-renewal but also of differentiation to steroidogenic cells. These cells give rise to progenitor Leydig cells, which proliferate, continue to differentiate, and give rise to the immature Leydig cells. The immature Leydig cells synthesize high levels of testosterone metabolites. The adult Leydig cells are terminally differentiated cells that are derived from immature Leydig cells. These cells are characterized by their production of high levels of testosterone. With aging, both serum and testicular testosterone concentrations progressively decline, for review see Nef. 87
Androgens play a crucial role in the development and maintenance of male reproductive and sexual functions. Therefore, low levels of circulating androgens can cause disturbances in male sexual development, resulting in congenital abnormalities of the male reproductive tract. Later in life, this may cause reduced fertility, sexual dysfunction, decreased muscle formation and bone mineralization, disturbances of fat metabolism, and cognitive dysfunction. Testosterone levels decrease as part of the aging process: signs and symptoms caused by this decline can be considered a normal part of aging.
KER: reduction of testosterone synthesis leads directly to reduction of testosterone levels
Impairment of testosterone production in Leydig cells within the testes directly impacts on testosterone levels. The testosterone synthesized is used locally but also excreted into the circulation, causing reduced testosterone levels at other sites.
Rats administered DEHP or DINP during the gestational period showed a coordinated reduction in the production of testosterone followed by reduction of testosterone levels in fetal testes and in serum.49,90
KE4: Reduction of Testosterone in the Testes
Androgens (principally testosterone and 5α-dihydrotestosterone) are critical for the development of the male phenotype during embryogenesis and for the achievement of sexual maturation at puberty. The decreased production and consequently reduced levels of testosterone in the fetal period lead to disrupted development of the male reproductive tract. In adulthood, androgens remain essential for the maintenance of male reproductive function and behavior. Apart from their effects on reproduction, androgens affect a wide variety of nonreproductive tissues such as skin, bone, muscle, and brain. 91 Androgens exert most of their effects by interacting with the AR, for review see Murashima et al. 92 Testosterone may be metabolized by 5α-reductase to produce 5α dihydrotestosterone or aromatized to generate estrogens.
KER: reduction of testosterone levels leads indirectly to male reproductive tract malformations
Male sexual differentiation in general depends on levels of testosterone, dihydrotestosterone, and the expression of ARs by target cells. 86 Disturbances in the balance of this endocrine system by either endogenous or exogenous factors may lead to male reproductive tract malformations (e.g., hypospadias, cryptorchidism).
Fetal androgens are crucial for the development of the male reproductive tract especially during the first trimester of pregnancy. Androgens regulate masculinization of external genitalia. Testosterone is necessary for stabilization and differentiation of the Wolffian structures (e.g., the epididymis, vas deferens, and seminal vesicles) and also for normal development of the fetal testes. Dihydrotestosterone, produced locally from testosterone, is required for normal development of the genital tubercle and urogenital sinus into the external genitalia and prostate. 92 Therefore, any defects in androgen biosynthesis, metabolism, or action during development can cause hypospadias. 93 Substances with antiandrogenic activity may alter the complex regulation of male sex differentiation during fetal life. 94 Although the cause in most cases is unknown, hypospadias has been associated with aberrant androgen signaling during development. 95
The etiology of this frequent malformation has not been elucidated despite intensive investigation. 96 Hypospadias thus appears at the crossroads of genetic, endocrine, and environmental mechanisms. 96
Reduced testosterone production during male rat development was seen after exposure of rats to di-butyl phthalate,19,21 butyl benzyl phthalate, and DEHP, 20 this led to hypospadias in the newborn pups.
Epidemiological studies have demonstrated an association between fetal estrogen exposure and hypospadias.97,98 However, the molecular mechanism underlying this association is unknown.99,100 Maternal exposure to estrogenic and antiandrogenic endocrine disrupting compounds has been implicated in increased risk of cryptorchidism and hypospadias in human male offspring although the apparent association was not statistically significant. 101
Reduction in testosterone levels during fetal development and subsequent lower levels of its metabolite dihydrotestosterone lead also to impaired growth of the perineum. 102 This in turn may lead to reduced anogential distance (AGD). 102 AGD is a sexual dimorphism that results from the sex difference in fetal androgen dihydrotestosterone levels. 103 The AGD is a marker of perineal growth and caudal migration of the genital tubercle. It is androgen dependent in male rodents. 102
During development, androgens stimulate the growth of the perineal region between the sex papilla and the anus, resulting in an increased AGD in male offspring. 102 The AGD is believed to be a biomarker of prenatal androgen exposure in many species, and in humans it has been associated with several adverse reproductive health outcomes in adults. AGD reflects fetal androgen exposure only within a discrete masculinization programming window (MPW), during which development of male reproductive organs is taking place.95,104
Decreased AGD has also been associated with the perturbation of androgen-mediated development of the reproductive tract in rat males, which were exposed to antiandrogens in utero.95,105,106 Several studies have indicated that exposure to phthalates resulted in decreased AGD in human males,107,108 presumably due to lowered testosterone levels.17,109,110
AO1: Male Reproductive Tract Malformation
Male reproductive tract malformations (congenital malformation of male genitalia) comprise any physical abnormality of the male internal or external genitalia present at birth. Some result from excessive or deficient androgen concentrations, others result from teratogenic effects, or are associated with anomalies of other parts of the body in a recognizable pattern (i.e., a syndrome). The cause of many of these birth defects is unknown.
Malformations are detected macroscopically for any structural abnormality or pathological change. Congenital malformation of the genitalia is a medical term referring to a broad category of conditions for humans, classified by the International Classification of Diseases (ICD) in the chapter “Congenital malformations of genital organs” (Q50–Q56), for example, Q54 Hypospadias, Q53 Undescended testicle. Hypospadias is usually diagnosed during the routine examination after birth and is within the category of “Congenital malformation of the genitalia.” Hypospadias is a malformation in which the urethra opens on the underside of the penis instead of the tip. It results from an incomplete closure of the urethral folds, leaving a split on the penis. 96 When the urethra opens to the glans or corona of the penis, it is called distal, whereas opening to the shaft or penoscrotal area defines hypospadias as proximal.
Androgens regulate the masculinization of external genitalia. Therefore, any defects in androgen biosynthesis, metabolism, or action during fetal development can cause hypospadias. Gene defects causing disorders of testicular differentiation, conversion of testosterone to dihydrotestosterone, or mutations in the AR can also result in hypospadias. 94 In about 20% of patients with isolated hypospadias, there are signs of endocrine abnormalities by the time of diagnosis. 93 The majority of hypospadias is believed to have a multifactorial etiology, although a small percentage results from single-gene mutations. 111 The only treatment of hypospadias is surgery, and thus, prevention is imperative.
The AGD is the distance from the anus to the genitals and the male/female differences result from the sex difference in fetal androgen (DHT) levels. 103 The AGD is widely used as biomarker of prenatal androgen exposure during a reproductive programming window.95,104,106 The AGD is a marker of perineal growth and caudal migration of the genital tubercle. It is androgen dependent in male rodents. 102 Measurement of AGD has also been proposed as a quantitative biomarker of fetal ED exposure in humans.112,113
Across numerous species, including humans, AGD is longer in males compared to females; for review see Barrett et al. 114 In males, a longer (more “masculine”) AGD is typically associated with favorable health outcomes, while a shorter AGD is associated with adverse health outcomes. The AGD in males is approximately double that of females. Less is known about clinical correlates of AGD in females, although one study found that in women a longer AGD was associated with increased odds of multifollicular ovaries. 115 AGD reflects the prenatal hormonal milieu and is a biomarker for the risk of reproductive health problems linked to that early hormonal environment. 114 In animal studies, AGD measured from the genital tubercle to the anus is a sensitive marker of in utero exposure to androgens and antiandrogens, and is used extensively in animal reproductive toxicology studies. 106 Decreased AGD in male rats is a hallmark of exposure to antiandrogenic substances. 116 A statistically significant change in AGD that cannot be explained by the size of the animal indicates an adverse effect of exposure and should be considered in setting the no-observed-adverse-effect-level (NOAEL). 117
KER: male reproductive tract malformation leads directly to impaired fertility
Impairment of the normal development of the male reproductive tract (e.g., genital abnormality and/or cryptorchidism) can impact on fertility later in life.
Hypospadias, followed by cryptorchidism, belongs to the most common male reproductive disorders that manifest at birth and may have a common origin in fetal life. 118 They are also associated with decreased fertility. 119 AGD reduction has also been associated with testicular dysfunction. 120 A study by Asklund et al. showed that semen quality was reduced in men with hypospadias and additional genital disorders, predominately cryptorchidism. 121 In another study by Bracka, 25% of 41 hypospadias patients, including 26 patients also with cryptorchidism, had a lower sperm density. 122
Men with a history of cryptorchidism have an increased risk of infertility. 119 Mendiola et al. examined healthy volunteers and found a positive relationship between semen parameters and anogenital distance. 123 Eisenberg et al. found shorter AGD among infertile men compared with fertile men. 120
In rodents, in utero exposure to substances (including phthalates) known to disrupt androgen-mediated pathways disrupts normal male genital development resulting in a decrease in genital length (i.e., phallus length, AGD) and impaired testosterone and sperm production.102,124,125
AO2: Impaired Fertility
Fertility is the capacity to conceive or induce conception. Impairment of fertility represents disorders of male or female reproductive functions or capacity. Fertility rate is measured by the number of offspring born per mating pair, expressed as individuals or populations.
Overall Assessment of the AOP
In the presented AOP, it is hypothesized that the KEs occur in a biologically plausible order before the development of AOs. The PPARα activators have been shown to alter steroidogenesis and impair reproduction (see reviews David, 8 Latini et al., 10 and Corton and Lapinska 35 ). The biochemistry of steroidogenesis and the predominant role of the gonad in synthesis of the sex steroids are well established. Steroidogenesis is a complex process that is dependent on the availability of cholesterol in mitochondria. Perturbation of genes responsible for cholesterol transport and steroidogenic enzyme activities in the Leydig cell may lead to a decrease in testicular testosterone production. As a consequence, androgen-dependent tissue differentiation and development will be adversely affected. The physical manifestation of this event may be reproductive tract malformation, which may possibly lead to impaired fertility.
This is a qualitative description of the pathway; the currently available studies only provide partial quantitative information on dose–response relationships. Experimental data are based on exposure to phthalates and indicate that KEs of this pathway occur at similar dose levels. Effects on the expression levels of genes that are responsible for the cholesterol transport into the Leydig cells were shown at >50 mg/kg/bw, a dose at which fetal testosterone was decreased and anatomical malformations (hypospadias) were produced.19,51,126,127
Currently, a more comprehensive quantitative assessment of the KERs in this AOP is not possible due to limited data. Figure 2 illustrates the available experimental data providing starting point for designing tailored experiments to further support the pathway. It can be seen that the data derive from different experimental conditions and measured endpoints and therefore are not intended to provide quantification of the dose–response relationships. The paucity of data shows the need for tailored experiments to interrogate this AOP further, that is, dose–response relationships between the KEs.
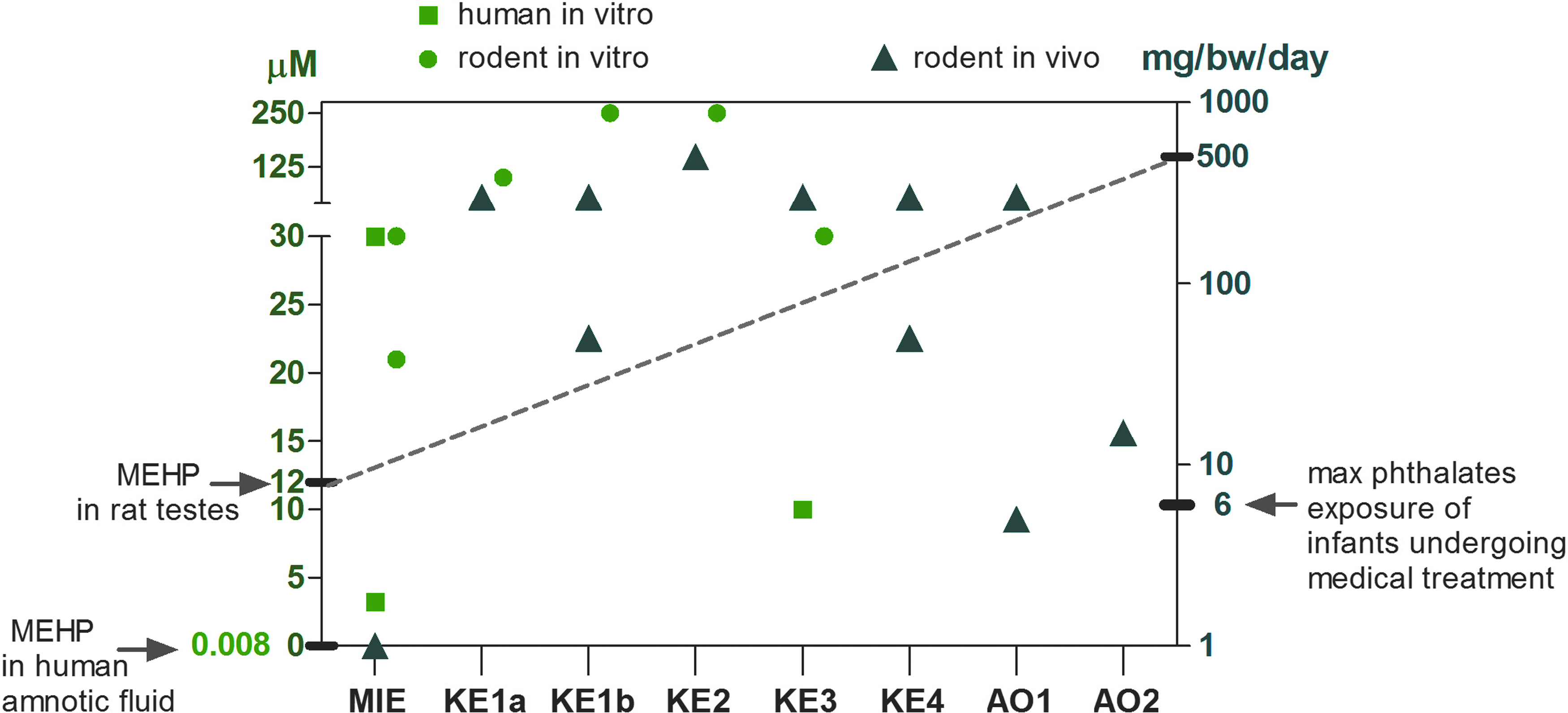
Illustration of quantitative data for described AOP. Doses/concentrations at which positive results for the KEs are shown. Green circles represent rodent—in vitro data, green squares—in vitro human, and blue triangles represent rat in vivo data. The points correspond to MIE (MEHP: EC50 = 3.2 μM, in vitro human cells, 161 DBP: EC50 = 30 μM in vitro human cells, 161 DBP: EC50 = 21 μM in vitro rodent cells, 161 MEHP: LOEC = 30 μM, 162 DEHP = 1 mg/kg/day in vivo rodent), KE1a (DBP:LOEC = 100 μM in vitro, MA-10 Leydig cells, 45 DEHP: LOEC = 300 mg/kg/day in vivo rat 49 ), KE1b (MEHP: LOEC = 250 μM in vitro rat, 163 DEHP: LOEL = 300 mg/kg/day in vivo rat, 49 DBP: LOEL = 300 mg/kg/day in vivo rat 78 ), KE2 (DBP: LOEL = 500 mg/kg/day in vivo rat 78 ), KE3 (MEHP:LOEC = 10 μM in vitro inhibition of testosterone production in human testicular explants, 164 DBP: LOEL = 500 mg/kg/day in vivo rat 78 ), KE4 (DBP: LOEL = 50 mg/kg/day in vivo rat, 165 DEHP: LOEL = 300 mg/kg/day in vivo rat 90 ), AO1 (DEHP: LOEL = 5 mg/kg/day in vivo rat 166 DEHP: LOEL = 50 mg/kg/day in vivo rat, 167 ) AO2 (DEHP: LOEL = 5 mg/kg/day in vivo rat, 168 DEHP: LOEL = 15 mg/kg/day in vivo rat 166 ). The mean testes concentrations of MEHP (12 μM) after an administered dose of 500 mg/kg/day in vivo to rats 169 are shown on the figure (dotted line). The concentration of MEHP in human amniotic fluid (0.0082 μM) 156 and the maximal exposure concentration of phthalates for infants undergoing medical treatment 125 are also shown. AOP, adverse outcome pathway; DBP, dibutyl phthalate; KE, key event; MEHP, mono(2-ethylhexyl) phthalate.
The AOs are results of the chemical exposure during a critical prenatal period of male development, the MPW, within which androgens must act to ensure the correct development of the male reproductive tract. 128 Therefore, the AOP focuses on the exposures within the MPW (15.5–18.5 GD days in rats).
The temporal relationship of exposure to GD has been investigated using phthalates and it has been demonstrated that the gestational timing of exposure is important for the production of adverse effects on the male reproductive tract (reviewed in Ema 129 ). Moreover, the temporal relationship between alterations of gene expression and changes in testosterone production has been investigated for phthalates (DBP).51,83 Initial increases in the expression of genes, which are associated with steroidogenesis, are followed by decreases in gene expression of the same genes. The observed decreased steroidogenesis and subsequent decrease in testosterone levels is a well-established precursor to anatomical changes in the developing male reproductive tract. Thus, those KEs of gene expression are temporally consistent with subsequent events, however, complete temporal concordance studies are missing.
The strength of the chosen chemical initiators as PPARα activators was shown to partially correlate with their ability to act as male reproductive toxicants. 35 The presented KEs leading to a decrease in steroidogenesis are plausible and consistent with the observed effects. There is also coherence between decreased testosterone synthesis and malformations.
Uncertainties, inconsistencies
The major uncertainty in this AOP is the functional relationship between the MIE: PPARα—activation and reduction of cholesterol transport (KE1a and KE1b): possible mechanisms have been proposed and investigated, however, dose–response data to support this relationship are lacking.
Studies exploring the role of PPARα using PPARα knockout mice showed that prenatal exposure to phthalates caused developmental malformations in both wild-type and PPARα knockout mice, thus suggesting a PPARα-independent mechanism. However, it is difficult to draw any conclusion on the role of PPARα in phthalate-related reproductive toxicity in this study since the intrauterine administration of phthalate (DEHP) occurred before the critical period of reproductive tract differentiation; moreover, the doses were high and caused maternal toxicity. 130 Intrauterine DEHP-treated PPARα-deficient mice developed delayed testicular, renal, and developmental toxicities, but no liver toxicity, compared to wild types, thus confirming the early observation by Lee et al. about the PPARα dependence of liver response 131 and, more importantly, indicating that DEHP may induce reproductive toxicity through both PPARα-dependent and PPARα-independent mechanisms. 48
PPARα-independent reproductive toxicity observed by Ward et al. may conceivably be mediated by other PPAR isoforms, 48 such as PPARβ and PPARγ, or by a nonreceptor-mediated organ-specific mechanism. 132 Other studies showed that the administration of DEHP resulted in less severe testis lesions and higher testosterone levels in PPARα-null mice than in wild-type mice. 45 A more recent report, investigating the role of PPARα, showed decreased testosterone levels in PPARα(−/−) null control mice, suggesting a positive constitutive role for PPARα in maintaining Leydig cell steroid formation. 18
Alternative mechanism(s) or MIE(s) described, which may contribute to or synergize with the postulated AOP
The inhibitory effect of PPARα activation seems to be attributable to an impairment of the multistep process of cholesterol mobilization, transport into mitochondria, and steroidogenesis leading to impaired androgen production. Therefore, it is plausible that several other mechanisms may contribute to or synergize with this AOP. For example, activation of other isoforms of PPARs (PPARβ/δ or/and γ) is hypothesized to be relevant for the pathway.133,134
Opposing effects of PPARγ ligands (thiazolidinediones, TZD) on androgen levels and/or production in male humans135–137 and animal models have been described.138–144 In rats no effects of the PPARγ ligand (rosiglitazone) on total circulating testosterone levels were seen in one study, 47 however, in another study a decrease in basal or induced testosterone production occurred in the Leydig cells of rosiglitazone-treated rats. 145 Confusingly, there are also contradicting reports as to the presence of PPARγ in the fetal testes. 50
Another mechanism leading to decreased fertility implicating the activation of PPARs may also be increased FA oxidation, and reduced capacity to cope with increased oxidative stress. 145
The involvement of epigenetics is also hypothesized. Changes in methylation patterns may be, in part, mediated by the PPAR pathway. DEHP affected the epigenome at doses from 1 mg/kg/day, doses that are relevant for human exposure. Epigenetic changes were observed in animals exposed to DEHP only during the fetal period, yet effects on steroid hormone levels were observed in adults. Moreover, the data suggest that high levels of DEHP affected key loci and deregulated gene expression beyond a point of compensation.147,148
Discussion
Endocrine disruption is an important health and policy issue with high priority for regulatory authorities globally. Environmental exposures to chemicals with endocrine disrupting properties may interfere with fertility in both male and female. Infertility, a disease characterized by the inability to have a child, affects one in six couples and will likely rise as the postponement of childbearing increases in developed regions of the world. 149 The associated healthcare cost of infertility is billions of dollars per year and does not include the tremendous physical and psychological burden for the couples. This highlights the public health importance of understanding risk factors that are connected with reproductive and developmental toxicity.
Adverse effects on the reproductive system may be induced directly, especially in adult males by damaging the semen producing epithelium or indirectly by interfering with sex hormone homeostasis. These processes might be disturbed by interfering agents (e.g., by antiandrogens), provided that the level of exposure is high enough. Effects on the reproductive system may involve perturbation of many biological pathways where critical events occur during well-defined periods of prenatal and early postnatal development. In the male, sexual differentiation depends on the presence of androgens during the male programming window and lack of this “imprinting” will result in irreversible defects or dysfunctions.
Human and experimental animal studies identify several classes of chemicals that adversely impact fertility and pregnancy. One class of environmental chemicals for which there is concern related to the risk of adverse reproductive and developmental effects are the phthalates. Human exposure to phthalates is widespread and occurs through multiple routes, including ingestion, inhalation, dermal contact, and parenteral exposure from medical devices containing phthalates. 150
Existing human data on phthalates concerning possible developmental effects derive from prospective epidemiological studies, lately reviewed by Lioy et al. 151 They are focused on association of maternal phthalate exposure with male reproductive tract developmental endpoints. The measurements of phthalate metabolites in the amniotic fluid would provide the best estimate of the actual internal dose for the fetus, taken during weeks 15–20 of fetal development (sensitive window). These measurements are, however, difficult to obtain and may pose some risk to the fetus, moreover, amniotic fluid has a continuous turn over. There are only few published studies on human amniotic fluid levels of phthalate metabolites.12,152,153 Therefore, maternal urinary levels may be the most appropriate surrogate of both maternal and fetal phthalate exposure.152,153
Reduced AGD has been reported in male infants in relation to higher maternal urinary concentrations of DEHP metabolites.17,107,154 The study by Huang et al. 155 also found similar associations of monoethyl phthalate and mono butyl phthalate with reduced AGD. This study did not find associations of any phthalate metabolite with reduced AGD in boys, but did so in girls.
Shortened AGD in males is regarded as a biomarker of insufficient androgen action in fetal life, 156 and there are relationships with other male reproductive disorders that are similar to those observed in rats with the phthalate syndrome. Thus, Hsieh et al. reported that boys with hypospadias had shorter AGD than boys with normal genitals. 157 Shorter AGD was associated with poorer semen quality 123 and infertility. 120 In addition, semen quality was reduced in men with hypospadias 122 and additional genital disorders, predominately cryptorchidism, 121 and men with a history of cryptorchidism have an increased risk of infertility. 119
In rodents, in utero exposure to agents known to disrupt androgen-mediated pathways disrupts normal male genital development with a decrease in genital length (phallus length, AGD) and impaired testosterone and sperm production,104,124 including exposure to phthalates. 125 Despite many years of research on phthalates, there is no scientifically agreed and established MoA of these chemicals for male reproductive toxicity. The exact molecular mechanism for the action of phthalates remains to be elucidated.
The presented AOP describes the scientific evidence and highlights data gaps. The major uncertainty in this AOP lies in the functional relationship between (MIE) PPARα and impaired cholesterol synthesis. Complete/pathway-driven studies to investigate the effects of PPARs and their role in male reproductive development are lacking. For establishing a solid quantitative and temporal coherent linkage, an MoA framework analysis for PPARα-mediated developmental toxicity is needed.
This approach has been applied for the involvement of PPARα in liver toxicity.158,159 The mechanistic basis for phthalate action needs structured well-designed experiments to further elucidate and substantiate the pathways and such experimental approaches may then be used to facilitate development of predictive models to be applied to other chemicals to evaluate if they have the potential to operate via a similar MoA.
Reproductive toxicity testing according to the present guidelines requires a high number of test animals. No single in vitro assay possesses the predictive power to identify reproductive toxicants to date. Therefore, structured approaches gathering relevant information across different types of studies can drive assessment in this field, describing connections and incorporating plausible mechanisms. Once the AOP is fully established and quantitative relationships are understood, then it may be possible to use in vitro mechanistic studies to help predict the adverse effects.
Future Perspectives and Conclusions
This AOP adds to the AOP network by mapping to MoAs for endocrine disruption other than via such well-established mechanisms of ER, AR (ant)agonism, and aromatase inhibition. It provides a framework supporting understanding of the mechanisms and the evidence for plausibility of the MoA through a combination of non/-testing approaches that can be practically integrated and used to support regulatory decisions. The AOP, along with other ED-relevant AOPs in the AOP-Wiki, was suggested to be used to support a plausible link between the available mechanistic data and the adverse effects in the screening methodology for identification of endocrine disrupters carried out in the context of a European Commission impact assessment on options for criteria to identify EDs 160 because it was considered that the pathway provides a basis for linking an endocrine MoA with an adverse effect, a prerequisite requirement for identification of endocrine disrupting chemicals (EDCs) according to the WHO definition. 2 Moreover, the AOP could inform the development of quantitative structure/activity relationships, r–ad-across models, and/or systems biology models to prioritize chemicals for further reproductive/developmental toxicity testing.
The current AOP framework uses a simplified depiction of a linear progression of events across levels of biological organization that translate a molecular perturbation to an AO. However, it is well recognized that AOPs actually operate as networks within a systems biology context and that various AOPs can share KEs and pathways may converge and lead to a variety of different outcomes within the umbrella term of reproductive toxicity.
Ultimately, understanding of responses to stressors to be able to effectively predict toxicity, in a systems biology context that operates across multiple levels of biological organization, will require the kind of network thinking that new approaches investigating interactions at the molecular and cellular level promote. As indicated substances may act on more than one MIE, it may be the combination of these interactions that is required to invoke a described AO. Further research is required to elucidate more precisely the MIE or MIEs that may lead to a decrease in steroidogenesis; however, it may not be necessary to establish a specific MIE to find a regulatory use for an AOP. Focusing on assay development for converging KEs in an AOP network may be one relevant use case not requiring knowledge of the individual MIEs.
AOP development is facilitated by the AOP knowledgebase—providing a platform for interdisciplinary teamwork between the scientists, facilitating the development of AOP networks. The success of the undertaking relies on the joint efforts of many researchers from various disciplines, including toxicology, biology, chemistry, clinical medicine, and computer modeling. Increasing contribution from AOP developers adding and connecting their AOPs through common KEs to build AOP networks will eventually better represent the complex biological processes and interactions in response to various chemical exposures.
The pathway-based approaches have the aim of improving the science of human and environmental safety assessment while ultimately reducing the reliance on animal toxicity testing. Many technical challenges still remain to be solved before this vision is realized. Central to these challenges is the need for AOPs to produce a quantitative output (i.e., response–response relationships). By structurally representing current knowledge of the pathway from PPARα activation to impaired fertility, this AOP may provide a basis for development (and interpretation) of strategies for Integrated Approaches to Testing and Assessment (IATA) to identify similar substances that may operate via the same MoA and lays a strong foundation for the development of other AOPs within the context of an AOP network.
Footnotes
Author Disclosure Statement
No competing financial interests exist.