
Abstract
Abstract
The Adverse Outcome Pathway (AOP) framework provides a means to outline a knowledge-driven sequence of events from exposure to adverse outcome. As a concept, AOPs have been readily accepted by the toxicology community as a means to organize available mechanistic information linking exposures to toxic effects in a standardized way encompassing all organizational levels of a given biological system. However, there is also an inherent benefit in applying AOPs to health outcomes that are not necessarily linked to standard toxicological end points, for example, in the context of predicting disease risk or in drug development. In this study, we propose an AOP for decreased lung function that originates in oxidative stress-mediated epidermal growth factor receptor activation in the airway epithelium. This article provides an overview of the supporting evidence for key events (KEs) on the molecular, cellular, tissue/organ, and organism level and how they relate to each other through key event relationships (KERs). The essentiality of the identified KEs, as well as the biological plausibility of the KERs, is also assessed. Moreover, the envisioned application of the proposed AOP is discussed, highlighting the utility of the AOP framework for developing preclinical tests that could prove useful in public health risk assessment for long-term e-cigarette use.
Introduction
T
Adverse outcome pathways (AOPs) provide a tractable modular framework for integrating existing mechanistic knowledge that connects chemicals or other stressors to a defined molecular initiating event (MIE) on the cellular level, subsequent downstream key events (KEs) at ever-increasing levels of biological hierarchy, and finally to a population-level adverse outcome (AO) of regulatory relevance. 7 As such, structured assembly of the available mechanistic information into an AOP is expected to prove useful for predicting public health outcomes based on tests that can be carried out rapidly and cost-effectively and that do not necessarily involve animal experimentation. For example, understanding how chronic exposure to inhaled toxicants leads to mucus hypersecretion is relevant to risk assessment of airborne pollutant and cigarette smoke exposure and elucidating how these exposures contribute to the development and progression of the disease. In addition, understanding the molecular underpinnings of these processes can aid in informing on early KEs, before onset of disease, and hence, public health risk-reduction strategies.
The following article proposes an AOP for decreased lung function that arises from oxidative stress-mediated epidermal growth factor receptor (EGFR) activation in the airway epithelium and provides an overview and evaluation of the existing mechanistic evidence as outlined in the Organization for Economic Cooperation and Development (OECD) handbook 8 in support of such an AOP. While the AO in this study is not a typical toxicological end point, it is hoped that the proposed AOP will find its future application not only in human risk assessment but also in human health research, as initially introduced by Langley et al. as “repurposing” of the AOP concept. 9
Summary of KEs and Mechanisms
A schematic of the AOP is presented in Figure 1; the evidence collated to date is available on the AOP-Wiki (https://aopwiki.org/aops/148, accessed December 12, 2016). Mucus hypersecretion in the airways is a key characteristic of many lung diseases, including asthma, cystic fibrosis, and chronic bronchitis. 10 Mucus hypersecretion is characterized by an increase in the number of goblet cells, mucin synthesis, and mucus secretion, which can result in airway obstruction, decreased peak expiratory flow, and respiratory muscle weakness.10,11 EGFR-mediated signaling has been identified as the key pathway that leads to airway mucus hypersecretion 12 and redox signaling as the major initiator of receptor activation. 13 Therefore, we believe that the MIE of this AOP is oxidative stress leading to activation (phosphorylation) of EGFR (KE1) on the surface of lung epithelial cells.
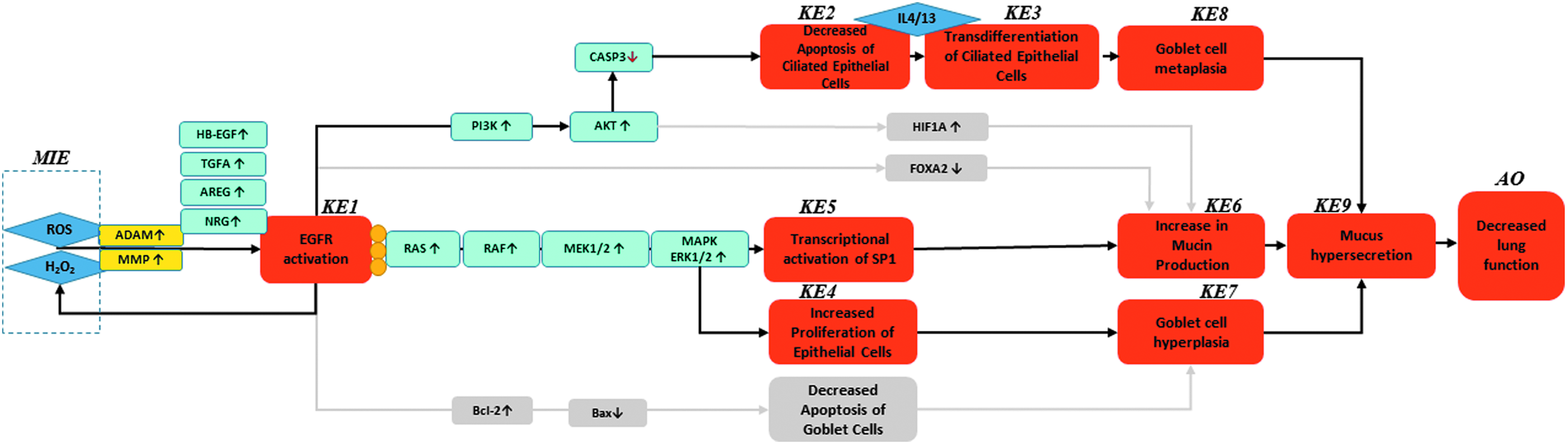
Schematic representation of the decreased lung function AOP. MIE, molecular initiating event; KE, key event; AO, adverse outcome; ROS, reactive oxygen species; H2O2, hydrogen peroxide; MMP, matrix metalloproteinase; HB-EGF, heparin-binding EGF-like growth factor; TGFA, transforming growth factor α; AREG, amphiregulin; NRG, neuregulin; EGFR, epidermal growth factor receptor; PI3K, phosphoinositide 3-kinase; CASP3, caspase 3; Bcl-2, B-cell lymphoma 2; Bax, bcl-2-like protein 4; IL4, interleukin 4; IL13, interleukin 13; HIF1A, hypoxia-inducible factor 1-alpha; FOXA2, forkhead box A2.
The EGFR family comprises four members: EGFR (also termed ErbB1/HER1), ErbB2/Neu/HER2, ErbB3/HER3, and ErbB4/HER4, all of which are transmembrane glycoproteins with an extracellular ligand-binding site and an intracellular tyrosine kinase domain. EGFR signaling is central to airway epithelial maintenance and mucin production, 14 and EGFR expression has been demonstrated in lung epithelial cells (although weakly) under physiological, as well as pathological, conditions in vitro and in vivo.15–18 Of note, lung epithelial EGFR phosphorylation was reported to increase under conditions of oxidative stress, including exposure to hydrogen peroxide (H2O2), 19 naphthalene, 20 cigarette smoke,21,22 and in the presence of neutrophils or neutrophil elastase.23–26 EGFR activation by oxidative stress may have a number of root causes: Reactive oxygen species (ROS) were shown to increase production of epidermal growth factor, the prime EGFR ligand, by lung epithelial cells. 27 Similarly, expression and secretion of transforming growth factor-alpha and amphiregulin (AREG), also EGFR ligands, were elevated in human bronchial epithelial cells in response to fine particulate matter (PM2.5), diesel particulate matter, and cigarette smoke exposure.28–30 Mechanistically, this process is dependent on ROS-mediated activation of metalloproteinases or members of the A disintegrin and metalloproteinase (ADAM) family of proteins, which cleave membrane-bound EGFR ligand precursors, making them locally available to bind to and transactivate EGFR in an autocrine manner.31–34 Importantly, ligand binding itself has been identified as a source of ROS, and specifically of H2O2, which function as second messengers potentially perpetuating the ensuing EGFR activation through chemical modification of the receptor.35,36 In addition, the presence of ROS may also contribute to EGFR activation by chemically modifying the receptor, thereby altering its structure and enhancing its kinase activity.35,37 While it is tempting to speculate that the increase in H2O2 would perpetuate EGFR activation through the continuous proteolytic shedding of pro-ligands in an autocrine loop, multiple lines of evidence suggest that oxidative modification, specifically EGFR sulfenylation, contributes to enhanced tyrosine phosphorylation of the receptor and downstream signaling.13,35,38–40
Downstream of EGFR activation, phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) signaling elicits an antiapoptotic response in ciliated cells, favoring their survival 41 (KE2: Decreased Apoptosis of Ciliated Epithelial Cells). Subsequent stimulation by pro-inflammatory stimuli such as the Th2 cytokines interleukin (IL)-4 and IL-13 then promotes transdifferentiation of ciliated cells into goblet cells, thereby increasing the number of goblet cells (“second hit hypothesis”) 42 (KE3: Transdifferentiation of ciliated epithelial cells). Alternatively, classical downstream activation of the mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) pathway, also known as Raf/Ras/MAPK/ERK pathway, increases airway epithelial cell proliferation30,43,44 (KE4: Increased Proliferation of Epithelial Cells) and facilitates epithelial wound repair.12,20,45 While there is evidence that increased goblet cell proliferation may be the underlying cause of goblet cell hyperplasia (KE7: Goblet cell hyperplasia (GCH)), 46 the key players mediating an increase in airway goblet cell numbers following EGFR activation are still largely unexplored. Basal epithelial cells which exhibit stem cell-like properties have been postulated to function as a source of goblet cells in injured airways, utilizing cell fate pathways that favor secretory cells over other cell populations. 47 However, both in vitro studies and studies in mouse models of asthma, COPD, and viral infection indicate that transdifferentiation of ciliated or club cells into goblet cells, which is referred to as goblet cell metaplasia (KE8: Goblet cell metaplasia (GCM)), more likely contributes to the expansion of this cell population in the airways.41,48–53 Furthermore, such increases in the number of goblet cells are suppressed when EGFR activity is inhibited. 41
Once established, GCH and/or GCM are thought to be maintained by goblet cell overexpression of Bcl-2, an antiapoptotic protein, which may arise as a consequence of oxidative stress-mediated EGFR activation.54–57 Importantly, the increase in goblet cell numbers may directly contribute to increased mucin production (KE6: Increase in Mucin Production). Activation of EGFR by oxidative stress was shown to correlate with increased mucin mRNA and protein expression, involving classical EGFR signal transduction through the MAPK cascade to activate the Sp-1 (KE5: Sp-1 Activation) or, at least in mice, hypoxia-inducible factor-1 alpha (HIF-1α) transcription factors that govern MUC5AC gene expression.58–64 In addition, activation of EGFR can also downregulate FOXA2, a known transcriptional repressor of mucin genes, although the underlying mechanism is currently unknown.65,66 Alternatively, mucin production could be stimulated by other, concomitant oxidative stress-induced, but EGFR-independent, processes known to contribute to GCH/GCM and increased expression of mucins. For example, Gensch et al. reported MUC5AC upregulation in vitro and in vivo following cigarette smoke exposure and identified Src, ERK, c-Jun N-terminal kinase (JNK), and activator protein 1 (AP-1) activation as critical steps in mediating ROS effects in human airway tissues in vitro. 67 Zhou et al. reported increased mucin expression in response to autophagy caused by cigarette smoke exposure in vitro and in vivo. 68
Increased mucin production and mucus hypersecretion following acute exposure are thought to contribute to innate airway defenses and are most likely limited by anti-inflammatory mechanisms aimed at resolving the exposure-related stress.2,69 However, under chronic exposure conditions, airway remodeling will persist, leading to airway narrowing, and the elevated number of goblet cells results in higher basal mucus levels (KE9: Mucus Hypersecretion). 70 Eventually, increased mucin production and mucus hypersecretion may lead to airway obstruction and a progressive decline in lung function over time (AO: Decreased Lung Function).11,71,72
Biological Plausibility
EGFR activation by oxidative stress in humans, mice, and rats is well documented, and EGFR and its ligands are orthologous in these species. EGFR activation by ligand binding and ligand-independent mechanisms is supported by studies with EGFR inhibitors such as AG1478 and BIBX 1522, efficiently abrogating downstream signaling and, hence, cell proliferation and survival.41,56,73 However, evidence for a specific EGFR-mediated effect in airway goblet or ciliated cells is limited and partially correlative, so biological plausibility for EGFR activation increasing proliferation of goblet cells and decreasing apoptosis of ciliated cells is only moderate. Another gap in current knowledge pertains to how inhibition of ciliated cell apoptosis leads to transdifferentiation that eventually contributes to an increase in goblet cell numbers. The available evidence is indirect or correlative.41,46,49,50,74,75 It also is not in agreement with other studies, which show that ciliated cells do not give rise to goblet cells during airway remodeling in rodents and humans, and with studies that provide evidence for increased goblet cell proliferation.48,56,76–78 Therefore, we consider biological plausibility for this KER (KE2→KE3) to be moderate.
Transcriptional regulation of MUC5AC expression and MUC5AC protein expression in the airways has been directly linked to EGFR-mediated activation of Sp-1.58,60,62,64 Although limited, the evidence appears to be highly reproducible across several studies in human airway epithelial cells, thereby supporting moderate biological plausibility of KERs KE1→KE5: EGFR activation indirectly leading to Sp-1 activation and KE5→KE6: Sp-1 activation directly leading to increased mucin production.
Studies in airway epithelial cells and in rats have demonstrated that GCH/GCM and increased mucin production following infection with Mycoplasma pneumonia and exposure to PM2.5, acrolein, or cigarette smoke can be greatly diminished by (pre-)treatment with EGFR inhibitors,34,79–83 supporting biological plausibility for this KER. However, owing to the fact that there is only correlational evidence linking increased goblet cell numbers to increased mucin production that coincides with strong EGFR expression in human airways,84,85 plausibility for the KER KE7→KE9 is moderate.
Chronic mucus hypersecretion is a main feature of chronic lung diseases, and the presence of GCH or GCM in COPD, asthma, and CF has been inferred as cause for sustained mucus production.2,86 Clinical studies have shown that MUC5AC expression in bronchial epithelium is inversely correlated with FEV1 (% predicted) and with the FEV1/FVC ratio,87,88 two important diagnostic measures of lung function. Moreover, chronic mucus hypersecretion was reported to contribute to decreased lung function in a number of clinical studies.72,89,90 As a cause–effect relationship between GCH/GCM, increased mucin production, mucus hypersecretion, and airway obstruction cannot be conclusively proven; these findings support moderate biological plausibility.
Essentiality of KEs
EGFR signaling is considered critical for mucus hypersecretion and GCH/GCM, 42 and numerous studies indicate that inhibition of EGFR decreases mucin production or goblet cell numbers.25,41,57,61,73,78,79 EGFR blockade also was reported to prevent an increase in goblet cell numbers and cause activation of caspase-3 and loss of ciliated cells, indicating that EGFR is essential for decreased ciliated cell apoptosis. 41 However, there is also evidence supporting decreased apoptosis in airway goblet cells in vitro, in a mouse model of asthma, and in rats following intratracheal lipopolysaccharide instillation, as a result of EGFR activation.56,73,91 Whether the latter only occurs once GCH/GCM is established, as indicated by Harris et al., 54 or whether additional events are required to maintain GCH/GCM, is currently unclear.
Sp-1 binding sites are required for active MUC5AC gene expression, 60 and Sp-1-mediated mucin expression can be blocked by the Sp-1 inhibitor mithramycin A.61,92 However, since the MUC5AC promoter has multiple transcription factor binding sites, it is likely that alternative pathways contribute to increased mucin production, such as activation of HIF-1α or decreased FOXA2 expression.65,93,94
Mucus hypersecretion is a physiological response to inhalation exposures such as pollutants or infectious agents. As such, it is typically of short duration and does not pose a major problem to normal lung function. However, in the presence of GCH, increased mucus production may decrease airflow. Since this may be accompanied by impaired mucociliary clearance and ineffective cough, 69 and owing to the lack of direct evidence, it is currently unclear whether chronic mucus hypersecretion alone is sufficient to affect a decrease in lung function.
Considering the gaps in the current knowledge, in particular, when transitioning to the organizational level of the organism, we propose that three major independent mechanisms leading to mucus hypersecretion (KE9) exist: (1) MIE→KE1→KE2→KE3→KE8, (2) MIE→KE5→KE6, and (3) MIE→KE4→KE7. Whether all three mechanisms are necessary to cause mucus hypersecretion is difficult to evaluate. At least some of the evidence suggests that they may be executed in parallel as a result of EGFR activation by oxidative stress.
Empirical Evidence
Table 1 presents an overview of supporting empirical evidence for the KERs. Selection criteria applied to this evidence were based, as far as possible, on the OECD guidance and handbook for the development of AOPs8,95 and considered fitness for purpose, direct relationship to KE, support for causality, and general acceptance by the scientific community. In brief, proof of EGFR activation (KE1) can be derived from western blots, for example, of untreated and treated cell or tissue lysates, using specific antibodies targeting the phosphorylated EGFR epitopes. Densitometry evaluation of the colorimetrically stained, chemiluminescent, or radioactive bands on the blot then permit a (semi-)quantitative measure of activation. Moreover, the addition of EGFR inhibitors or neutralizing antibodies is well suited to demonstrate causality.
AG1478, 4-(3′-chloroanilino)-6,7-dimethoxyquinazoline (tyrphostin); AO, adverse outcome; Bax, bcl-2-like protein 4; Bcl-2, B-cell lymphoma 2; BrdU, 5-bromo-2′-deoxyuridine; CCSP, Clara cell secretory protein; COPD, chronic obstructive pulmonary disease; EGFR, epidermal growth factor receptor; FEV1, forced expiratory volume in one second; FVC, forced vital capacity; GCH, goblet cell hyperplasia; GCM, goblet cell metaplasia; H2O2, hydrogen peroxide; IL, interleukin; KE, key event; KER, key event relationship; LPS, lipopolysaccharide; MIE, molecular initiating event; MUC5AC, mucin 5AC; NHBECs, normal human bronchial epithelial cells; OVA, ovalbumin; PMA, phorbol 12-myristate 13-acetate; ROS, reactive oxygen species; TCDD, 2,3,7,8-tetrachlorodibenzodioxin.
Numerous proliferation and apoptosis assays exist. Traditionally, cell proliferation is determined by monitoring cell viability, and various means and methods exist to accomplish this.96,97 Similarly, cell death assays range from simple cellular stains for use with platforms such as flow cytometry for detection of specific markers, for example, caspase, to more sophisticated multiplex assays.98,99 In any case, inclusion of appropriate controls allows for quantitation of surviving or proliferating cell populations, as well as the extent and mode of cell death. That said, the evidence necessary to support cell death in a specific subpopulation of the airway epithelium would require identifying the cell type of interest first. This could be achieved by either staining the cell with a population-specific marker in situ or by enriching the population of interest before monitoring cell proliferation and death. To the best of our knowledge, to date only the double immunohistochemical/immunofluorescent staining of airway epithelial samples has been used to demonstrate increased proliferation of goblet cells (KE4) and decreased apoptosis of goblet or ciliated cells (KE2).41,77,79 Similarly, transdifferentiation (KE3) in the airway epithelium is difficult to quantify. A trained pathologist may assign a score that reflects the extent of airway remodeling in animal and human lung tissues, but no standard exists, and the results are at best semiquantitative and study-specific. Some investigators view the appearance of cells bearing morphological characteristics of both ciliated and goblet cells, and colocalization of ciliated and goblet cell markers in situ, as sufficient evidence for transdifferentiation.100,101 However, there is currently no quantitative evaluation of these data; in future studies, this could potentially be conducted using image analysis software.
Transcription factor activity can be evaluated by multiple means, including reporter gene assays, electrophoretic mobility shift assay, chromosomal immunoprecipitation, and array-based assays. Some of these assays are cumbersome and technically challenging, possess low sensitivity, and may even pose a financial burden to the investigator. 102 These may be among the reasons for the limited number of studies examining how EGFR activation impacts the transcription factor Sp-1 (KE5) directly and indirectly.60,62,64
In in vitro studies, a relative increase in MUC5AC gene and/or protein expression compared with control conditions is generally accepted as a surrogate measure of mucus hypersecretion (KE6). While this is true for some animal studies, mucus hypersecretion has also been concluded from increased Alcian blue/periodic acid–Schiff staining and the presence of goblet cells throughout the airways. Clinically, coughing and sputum production for >3 months in at least 2 consecutive years is defined as (chronic) mucus hypersecretion. 103 As such, diagnosis depends on the extent of medical examination, including questioning the patient and subject recall. Current clinical practice, however, does not include a quantitative measure of mucus hypersecretion, making this event an inferred one. This is quite different from lung function assessment, for which extensive and widely accepted clinical guidelines exist; abnormal lung function (AO) as a diagnostic criterion for COPD is well defined.104,105
In summary, while the quantitation of some KEs included in the proposed AOP meets the criteria outlined in the OECD handbook, evaluating others is more problematic. There is good quantitative understanding of how oxidative stress affects EGFR signaling and influences mucus production, epithelial cell proliferation, apoptosis, and transdifferentiation, individually assayed. In addition, in the majority of these studies, the summary evidence indicates dose–response relationships, time–response relationships, and causality for oxidative stress-induced EGFR activation leading to increased cell proliferation, lending strong support for the KERs MIE→KE1 and MIE→KE6. However, quantitative knowledge is lacking with respect to the identity of airway epithelial cells undergoing proliferation and apoptosis, processes purportedly forming a basis for subsequent transdifferentiation (KE2→KE3, KE3→KE8, KE4→KE8), which makes empirical support for these KERs weak. Furthermore, while cause–effect relationships can be derived from studies investigating Sp-1 activation, dose–response relationships are difficult to derive. Moreover, data in support of the KERs for increased mucin production (KE5→KE6) and mucus hypersecretion at the organism level (KE6→KE9, KE7→KE9, KE8→KE9) are mainly derived from surrogate measures, and while those may not adequately reflect quantitative mucus production, they are accepted in the clinical community as an indicator of chronic bronchitis. Taken together, we judge the quantitative evidence for the KEs and KERs on the tissue and organism level to be weak.
Domain of Applicability
Taxonomic applicability
The evidence presented in this study in support of the proposed AOP for mucus hypersecretion and decreased lung function is derived from both human and rodent biological systems. In vitro and in vivo studies in these systems have been performed to clarify the mechanisms of EGFR activation and mucus hypersecretion by studying the increase in goblet cells and subsequent increase in mucin transcript and protein expression, as well as mucus production.2,70 In summary, these evidences suggest that the majority of KEs are preserved across small rodents and humans. There are also several clinical studies on mucus hypersecretion and how it affects lung function in humans with chronic bronchitis, asthma, and other chronic lung diseases. However, the link between mucus hypersecretion and airflow obstruction is much less supported by studies in laboratory animals where the human disease phenotype cannot be modeled in its entirety, and traditional lung function measurements are difficult.106,107
Life stage applicability
EGFR activation leading to mucus hypersecretion is predominantly studied in adults, although it was also reported in pediatric asthma and bronchitis.108,109 In general, the exposures resulting in oxidative stress to subsequently induce EGFR activation apply to adults who are more likely to be exposed to these stressors. Moreover, until a considerable time of exposure or since exposure has elapsed, that is, an advanced life stage has been reached, decreased lung function may go unnoticed unless accompanied by other symptoms such as cough and, hence, not become clinically significant.
Sex applicability
At times, clinical evidence linked to occupational exposures is derived from a majority of male subjects, which could be related to a male predominance in certain professions.110,111 Similarly, in most Western countries, cigarette smoking is still more prevalent in men than in women, although this gap has been closing steadily over the past decades.112,113 Nevertheless, the available in vivo and clinical evidence in support of the proposed AOP suggests that there is no remarkable gender difference.
Application of the AOP
Ultimately, the future application of this AOP lies in its potential for predicting decreased lung function in humans exposed to potentially harmful inhaled substances. This becomes especially pertinent as impaired lung function carries a significant risk of morbidity and mortality. Given the long latency period between exposure and detectable decreases in lung function, together with the fact that lung function tests alone may not be sufficiently sensitive to account for early lung damage that remains asymptomatic, 114 means for early identification of potentially hazardous exposures are critical for the development of appropriate public health interventions. In this context, the proposed AOP could provide a starting point for developing simple cost-effective in vitro tests to screen compounds that may pose a risk to humans when inhaled and enable further classification based on the extent of harm they may cause. At the same time, biomarkers of effect/potential harm could be derived from the AOP, with the aim of developing reliable preclinical end points that are indicative of future disease or even mortality risk associated with impaired lung function. With this in mind, estimating the risk associated with regular e-cigarette use would be one example for the potential real-life applicability of this AOP. Long-term consequences of e-cigarette use with respect to lung health are currently unknown and are a topic of ongoing, and at times controversial, debate.115–118 The lack of chronic exposure data hampers a true assessment of how regular e-cigarette use might influence the mechanisms underlying mucus hypersecretion that ultimately lead to airway obstruction and, hence, a decline in lung function. Although e-cigarettes have now been in use for nearly a decade, those long-term data, including health outcomes of regular e-cigarette users, will not be available for a while. However, short-term observations, predominantly derived from in vitro assays, are beginning to accumulate. Multiple studies in lung epithelial cells exposed to e-cigarette aerosol showed, for example, increased ROS levels compared with air exposure (although those levels were still lower than those observed following cigarette smoke exposure).119–122 It would be of interest to confirm whether the subsequent event involves EGFR activation and downstream signaling, which is based on the AOP proposed in this study, an indicator that mucus hypersecretion and decreased lung function may be a long-term consequence of e-cigarette aerosol exposure. As such, this AOP could provide a framework for mapping out suitable in vitro models and tests allowing for the systematic evaluation of the KEs in the context of e-cigarette use. Moreover, such AOP-based in vitro tests may eventually also prove useful in substantiating the risk reduction potential of e-cigarettes as cessation aids or alternatives to cigarettes for smokers who cannot or do not want to quit.
Footnotes
Acknowledgments
The authors thank Ruth Dempsey and Christopher Proctor for their helpful discussions and critical review during the preparation of this article. The authors are also grateful to Edanz Editing for editorial assistance with this article.
Author Disclosure Statement
K.L., M.T., and J.H. are full-time employees of Philip Morris International (PMI). F.J.L., L.E.H., and M.G. are full-time employees of British American Tobacco (Investments) Ltd. J.P. was employed at Selventa at the time this project was undertaken, and Selventa received financial compensation from PMI for their services. The authors declare no further conflicts of interest with respect to the research, authorship, and/or publication of this article.