
Abstract
Adenosine monophosphate (AMP) is a key cellular metabolite regulating energy homeostasis and signal transduction. AMP is also a product of various enzymatic reactions, many of which are dysregulated during disease conditions. Thus, monitoring the activities of these enzymes is a primary goal for developing modulators for these enzymes. In this study, we demonstrate the versatility of an enzyme-coupled assay that quantifies the amount of AMP produced by any enzymatic reaction regardless of its substrates. We successfully implemented it to enzyme reactions that use adenosine triphosphate (ATP) as a substrate (aminoacyl tRNA synthetase and DNA ligase) by an elaborate strategy of removing residual ATP and converting AMP produced into ATP; so it can be detected using luciferase/luciferin and generating light. We also tested this assay to measure the activities of AMP-generating enzymes that do not require ATP as substrate, including phosphodiesterases (cyclic adenosine monophosphate) and Escherichia coli DNA ligases (nicotinamide adenine dinucleotide [NAD+]). In a further elaboration of the AMP-Glo platform, we coupled it to E. coli DNA ligase, enabling measurement of NAD+ and enzymes that use NAD+ like monoadenosine and polyadenosine diphosphate-ribosyltransferases. Sulfotransferases use 3′-phosphoadenosine-5′-phosphosulfate as the universal sulfo-group donor and phosphoadenosine-5′-phosphate (PAP) is the universal product. PAP can be quantified by converting PAP to AMP by a Golgi-resident PAP-specific phosphatase, IMPAD1. By coupling IMPAD1 to the AMP-Glo system, we can measure the activities of sulfotransferases. Thus, by utilizing the combinations of biochemical enzymatic conversion of various cellular metabolites to AMP, we were able to demonstrate the versatility of the AMP-Glo assay.
Introduction
Adenosine-based cellular metabolites play an extremely critical role in all aspects of cellular function. Adenosine acts as ligand for various purinergic receptors regulating memory, behavior, sleep, and cardiac functions, and immune regulation. 1 Adenosine triphosphate (ATP), adenosine diphosphate (ADP), and adenosine monophosphate (AMP) are used in intracellular energy transfer and signal transduction mediated by protein phosphorylation, and are considered the main currency for energy charge in the cell. Cyclic adenosine monophosphate (cAMP) is a key cellular second messenger involved in diverse arrays of signal transduction pathways. The breakdown of cAMP to AMP brings about multiple alterations in cellular phenotypes since AMP is central to all adenosine-based cellular metabolites. It is a product of numerous cellular enzymes, including ubiquitination, DNA ligases, aminoacyl tRNA synthetases (aaRS), and phosphodiesterases (PDE), and plays a role in regulating these enzymes by negative feedback mechanism.
AMP is a critical indicator of cell health and its level fluctuates under various pathological and environmental conditions like hypoxia and nutrient deprivation. AMP is a key signaling molecule on its own right since it modulates the activity of AMP-activated protein kinase (AMPK). AMPK acts as a master regulator of energy homeostasis by regulating glucose uptake, β-oxidation of fatty acids, cholesterol synthesis, and biogenesis of GLUT4 and mitochondria. 2 Other cellular metabolites like nicotinamide adenine dinucleotide (NAD+), S-adenosylmethionine (SAM), and 3′-phosphoadenosine-5′-phosphosulfate (PAPS) also have the adenosine moiety. Enzymes like synthetases could form carbon–oxygen bonds by joining their substrates (e.g., aaRS), carbon–sulfur bonds (e.g., acyl-coA synthetases), carbon–nitrogen bonds (e.g., ubiquitin ligases), carbon–carbon bonds (e.g., polyketide synthases), or phosphoric ester (e.g., DNA ligase). The term synthetase is used for enzymes that use energy from nucleotide triphosphates like ATP, otherwise they are termed synthases. Generation of AMP is often a recurring theme in many of these enzyme subclasses. Various other enzymes also generate AMP, like cAMP PDE that break the phosphodiester bond in cAMP. We used the AMP-Glo system to measure activities of both ATP-dependent and NAD+-dependent DNA ligases. Subsequently, we incorporated the specificity of NAD+-dependent DNA ligases for NAD+ to couple it to the AMP-Glo system to detect NAD+- and NAD+-dependent processes like ADP-ribosylation.
Like ADP-ribosylation, sulfation is yet another biological reaction for modification of small molecules, cellular metabolites, and proteins. Sulfation is carried on by a class of enzymes called sulfotransferase and is involved in a variety of biological processes, including detoxification to drugs, hormone and neurotransmitter regulation, cell signaling, and synthesis of glycosaminoglycans like heparin sulfate and chondroitin sulfate. 3 In this study, PAPS the common sulfo-group donor and phosphoadenosine-5′-phosphate (PAP) is the common product. PAP can be converted to AMP by a Golgi-resident PAP-specific phosphatase; thus by modifying the AMP-Glo system, we show that it can be used to measure the sulfotransferase activity.
Materials and Methods
Reagents
pBR322 plasmid was obtained from Roche, Escherichia coli DNA ligase was obtained from Takara Bio, Inc., DNA oligonucleotides were obtained from IDT, T4 DNA ligase, complete amino acid mix, EcoRI, and Ultrapure ATP were obtained from Promega Corp. (Madison, WI), E. coli aaRS, poly-ADP ribose polymerase 1 (PARP1), and ADP-ribosyltransferase C3 from Clostridium botulinum, 3-isobutyl-1-methylxanthine (IBMX), RO 20-1724, zardaverine, zaprinast, calf liver tRNA, E. coli tRNA, IBMX, cilostamide, and rolipram were obtained from Sigma-Aldrich, and PARP1 inhibitor 3,4-dihydro-5[4-(1-piperindinyl)butoxy]-1(2H)-isoquinoline (DPQ) was obtained from EMD Millipore. Human Asp and Thr aaRS were obtained from Prospec Bio (East Brunswick, NJ). PDE3B, PDE4B2, PDE7A, PDE8A, PDE10A1, and PDE11A4 were obtained from BPS Biosciences (San Diego, CA). All reactions were performed in Corning white 384-well plates (# 3674).
aaRS Activity Assay
E. coli and human aaRS (human AspRS and ThrRS) reaction were performed in a buffer containing 30 mM HEPES pH 7.5, 150 mM NaCl, 30 mM KCl, 40 mM MgCl2, 1 mM dithiothreitol (DTT), 50 μM ATP, 0.5 mg/mL calf liver tRNA (for human aaRS) or E. coli tRNA (for E. coli aaRS), and 0.5 mM amino acids (Asp or Thr, for human AspRS and ThrRS, respectively) or an amino acid mix (for E. coli aaRS) for 1 h at 37°C in a 5 μL reaction volume. AMP generated during the process was detected by addition of 5 μL of AMP-Glo reagent I and incubated for 1 h at room temperature. This was followed by the addition of 10 μL of AMP-Detection Solution (prepared by adding AMP-Glo Reagent II to Kinase Glo One Solution in a 1:100 ratio), and luminescence readings were recorded after 30 min.
cAMP-Phosphodiesterase Activity Assay
The cAMP-phosphodiesterase activity was measured by incubation of the enzymes (5 μL) with 5 μL of 100 μM cAMP in a buffer containing 40 mM Tris-HCl, pH 7.5, 20 mM MgCl2, and 0.1 mg/mL bovine serum albumin (BSA) for 60 min at room temperature. To each well, 10 μL AMP-Glo reagent I containing 0.5 mM IBMX/20 μM cilostamide/20 μM rolipram was added and incubated for 30 min at room temperature. This was followed by the addition of 20 μL AMP detection solution and incubated for 30 min, and the light output was measured in a luminescence reader.
T4 DNA Ligase Assay
The T4 DNA ligase activity was measured by ligation of a linearized plasmid. Plasmid pBR322 was digested with EcoRI and the linearized plasmid DNA was purified. An aliquot containing 0.5 μg DNA was incubated with different amounts of T4-DNA ligase and 100 μM ATP in 30 mM Tris-HCl, pH 8.0, 10 mM MgCl2, and 1 mM DTT in a 10 μL ligase reaction and incubated for 30 min at room temperature. To each well, 10 μL AMP-Glo reagent I was added and incubated for 60 min at room temperature. This was followed by the addition of 20 μL AMP detection solution and incubation for 30 min, and the light output was measured in a luminescence reader.
E. coli DNA Ligase Activity
E. coli DNA ligase activity was measured by ligation of nicked duplex DNA. Nicked duplex DNA was prepared as described before. 4 Briefly, 100 μM of each of three oligonucleotides (5′-CGAATTCACTGGCCGTCGTTTTACAACGTCGTG ACTGGG-3′, 5′-phosphate-ACGGCCAGTGAATTCG-3′, and 5′-CCCAGTCACGACGTTGTAAAACG-3′) were mixed in 10 mM Tris-HCl, pH 8.0. The mixture was heated to 98°C for 10 min, followed by annealing at room temperature for 1 h to generate the nicked duplex DNA. The E. coli DNA ligase reaction was performed with 10 μM nicked DNA oligos and 10 μM NAD+, with different amounts of DNA ligase in a buffer containing 25 mM Tris-HCl, pH 8.0, 5 mM MgCl2, 10 mM (NH4)2SO4, 1 mM ethylenediaminetetraaceticacid, and 0.1 mg/mL BSA. The ligase reaction was incubated for 30 min. The ligase reaction detected using the AMP-Glo system as described above.
Detection of NAD+ and ADP-Ribosyltransferase
For detection of NAD+, the AMP-Glo system was modified such that the AMP-Glo Reagent I includes 20 μM nicked duplex DNA (as described above) and 0.6 U/μL (5 μg/mL) E. coli DNA ligase. This modified AMP-Glo reagent I would now be able to convert NAD+ in a sample to AMP and then to ADP. The ADP formed is then converted to ATP and detected as a bioluminescent readout using AMP Detection Solution.
NAD+ titration
NAD+ was titrated from 0 to 2.5 μM in 5 μL sample followed by 5 μL of modified AMP-Glo reagent I, and incubated for 1 h at room temperature. A 20 μL of AMP Detection Solution was then added to each well and incubated for an additional 30 min, and the light output is measured in a luminescence reader.
C3 ADP-ribosyltransferase activity
C3 activity was measured using 10 μM RhoA, 10 μM NAD+, and different amounts of C3 ADP-ribosyltransferase in 50 mM HEPES, pH 7.5, 2 mM MgCl2, and 1 mM DTT. The ADP-ribosylation reaction is allowed for 30 min and the remaining NAD+ was measured as described under the NAD+ titration protocol.
PARP1 activity
The PARP1 activity was measured using 50 μg/mL nucleosomes (Reaction Biology Corp.), 20 μM NAD+, and different amounts of PARP1 in 50 mM Tris-HCl, pH 8.0, 50 mM NaCl, 10 mM MgCl2, and 1 mM DTT. The ADP-ribosylation reaction was allowed for 60 min at room temperature, and the remaining NAD+ was measured as described above.
Detection of PAP and Sulfotransferases
For detection of PAP and sulfotransferase activity, the AMP-Glo system was modified such that the AMP-Glo Reagent I includes 10 μg/mL IMPAD1 (R&D Systems), a Golgi-resident PAP-specific phosphatase. This modified AMP-Glo reagent I would convert PAP in a test sample to AMP and then to ADP. The ADP formed was then converted to ATP and detected as a bioluminescent readout using the AMP Detection Solution.
PAP titration
PAP (Sigma) was titrated from 0 to 10 μM by the addition of 5 μL of the modified AMP-Glo reagent I, so the total volume was 10 μL and incubated for 1 h at room temperature. This was followed by the addition of 20 μL of AMP Detection Solution to each well and incubation for 30 min, and the light output is measured in a luminescence reader.
SULT3 activity
SULT 1A3 activity was measured using 100 μM PAPS (R&D Systems), 100 μM 1-napthol, and varying amounts of SULT 1A3 (R&D systems) in 50 mM Tris-HCl, pH 7.5, 15 mM MgCl2. The sulfation was carried out for 20 min at room temperature. The sulfation reaction was measured using the modified AMP-Glo system as described above.
Instrumentation
All enzymatic reactions were performed in a white multiwell plate (Corning). Luminescence was recorded using Tecan Infinite M1000 plate reader.
Results and Discussion
Assay Principle and Format
The AMP-Glo system is an enzyme-coupled bioluminescent assay platform for the detection of AMP producing enzymatic reactions. Upon completion of the enzyme reaction, the first step, requiring addition of AMP-Glo Reagent I, depletes the remaining ATP (for ATP requiring enzyme reactions, e.g., aaRS, T4 DNA ligase, and ubiquitin activating enzyme) and converts the AMP generated during the enzyme reaction to ADP. This step is completed in a 60-min incubation. As several AMP generating enzymatic reactions are dependent on ATP or cAMP as substrate and generate pyrophosphate (PPi) as a product in addition to AMP, which is a potent luciferase inhibitor. The AMP-Glo reagent I contains ATP adenyl cyclase and pyrophosphatase to eliminate the ATP and PPi, AMP generated during the enzyme reaction is also converted to ADP by polyphosphate-AMP phosphotransferase.
In the second step, AMP Detection Solution (prepared by combining AMP-Glo Reagent II and Kinase-Glo One solution in a 1:100 ratio) is added, which converts the ADP to ATP using adenosine kinase, which is concomitantly detected as a bioluminescent readout using a luciferase/luciferin system. The second step is completed within 30–60 min. The amount of light generated is proportional to the AMP produced and is a direct measure of the enzyme activity (Fig. 1A). Optimal AMP-Glo performance requires the addition of equal volume of AMP-Glo reagent I to the enzyme reaction, followed by twice the volume of AMP Detection Solution. Thus, volume ratios of 25:25:50 μL or 50:50:100 μL of ligase reaction: AMP-Glo reagent: AMP Detection Solution for a 96-well plate and volume ratios of 5:5:10 μL or 10:10:20 μL for a 384-well plate have been used in this study.
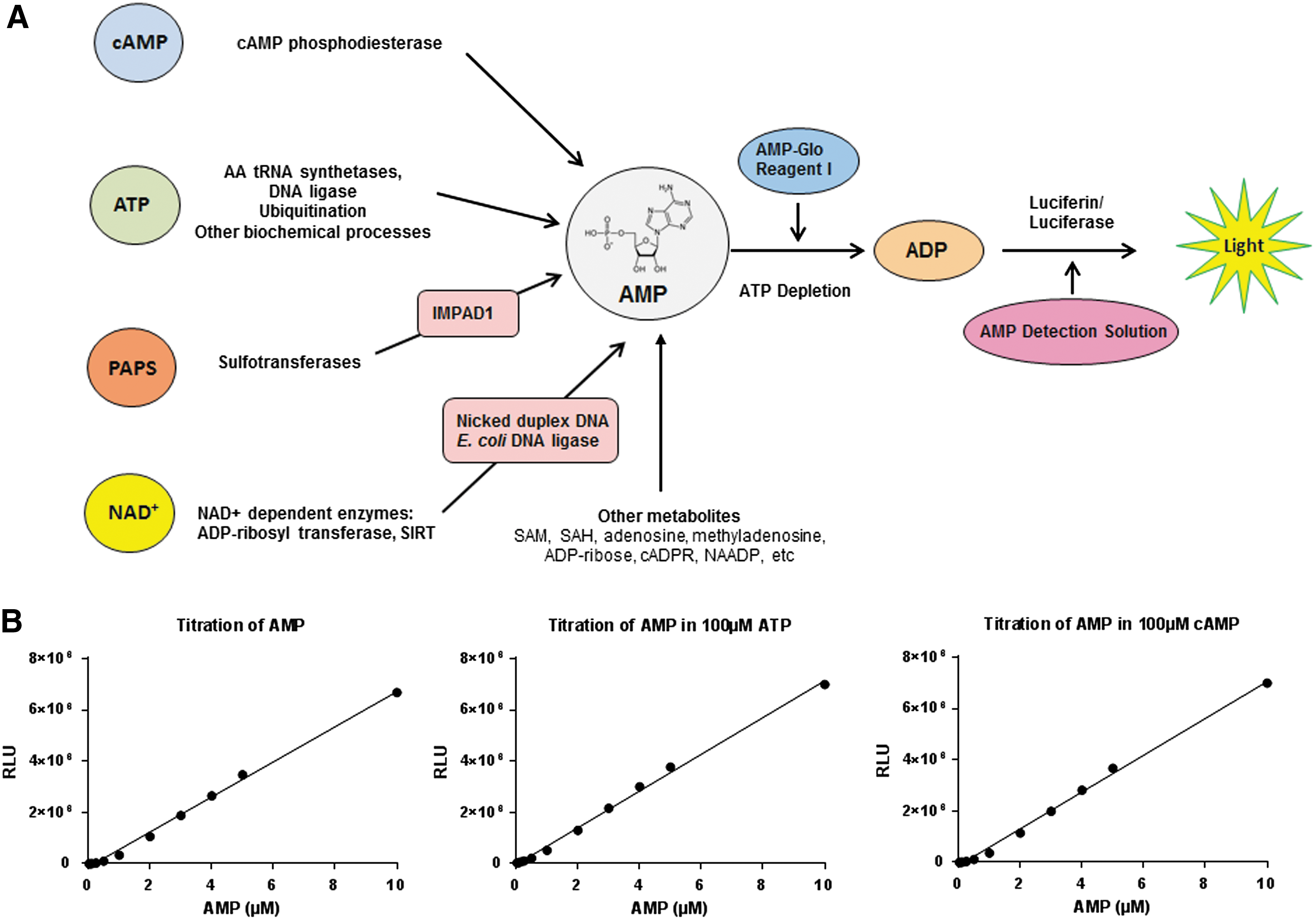
We tested the sensitivity of the AMP-Glo assay in detecting AMP in the presence or absence of ATP and cAMP. ATP used for all experiments is prepared in-house to eliminate contaminating nucleotides that may affect the assay performance (Ultrapure ATP; Promega). Both in the absence of ATP and in the presence of cAMP, the limit of detection (LOD) for AMP is 10 nM (concentration of AMP where signal/noise ratio [S/N] >3, S/N: [(signal) − (background)/3 × StdDevbackground]). In the presence of 100 μM ATP, the LOD for AMP is 100 nM (Fig. 1B). It is noteworthy that signal to background increases significantly when purified enzymes were nucleotide free as possible using dialysis or gel filtration for nucleotide removal.
Measuring aaRS Activity
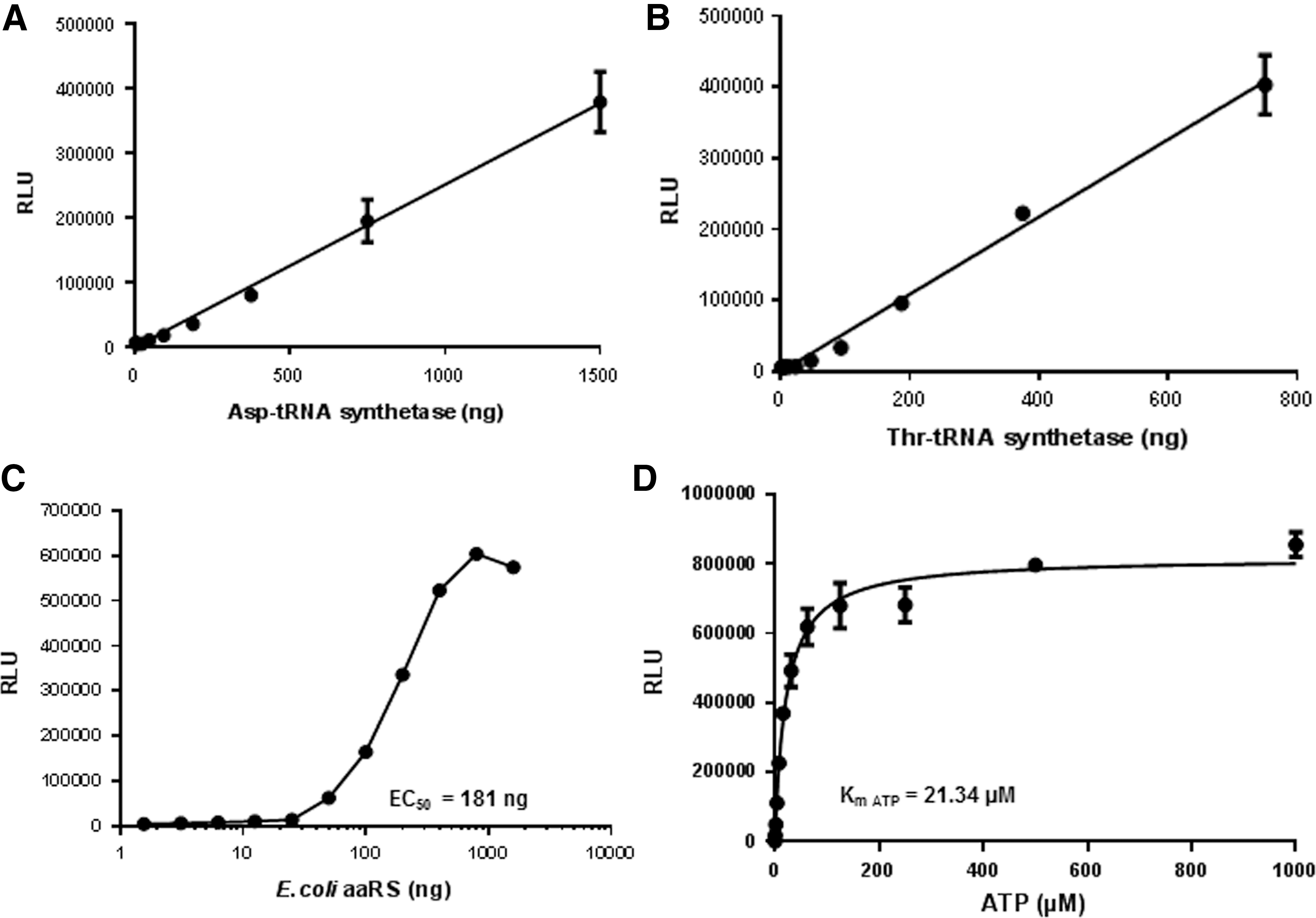
aaRS are enzymes that attach amino acids with their cognate tRNAs during protein translation. The reaction occurs in two steps: in the first step, aaRS hydrolyses ATP to activate an amino acid forming an aminoacyl-adenylate intermediate releasing PPi. In the second step, the amino acid is transferred to the tRNA resulting in the formation of aminoacyl-tRNA with the release of AMP. These enzymes are found in all living organisms and contain at least 20 different aaRS, one for each amino acid. Many aaRS are considered potential drug targets, especially for development of novel antibiotics because they are essential for growth and survival of microorganisms, broadly conserved, and distinct enough from human orthologs to facilitate identification of selective inhibitors. 5 Mupirocin (Bactroban), which inhibits the isoleucyl t-RNA synthetase in gram-positive bacteria resulting in an inhibition of protein synthesis, is used as a topical antibiotic against superficial skin infections. 6 A boron-based antimicrobial GSK2251052 that inhibits Leu-RS was developed for inhibiting gram-negative bacteria. It is disappointing to note that bacterial pathogens resistant to these inhibitors constantly emerge. Targeting eukaryotic aaRS from organisms like fungi (Candida spp.) or apicomplexan parasites (Trypanosoma spp., Plasmodium spp. or Toxoplasma spp.) is a major challenge in drug discovery as they have a higher degree of conservation to human aaRS. However, certain aaRS inhibitors targeting these infectious agents have shown promise. The fungal secondary metabolite cladosporin selectively inhibits Plasmodium falciparum LysRS and showed targeting aaRS as a valid drug target for antimalarial agents. 7 Selective Methionyl-tRNA synthetase inhibitors have also been developed against Trypanosoma infections. 8,9 Furthermore, mutations associated with aaRS (GlyRS, TyrRS, and LysRS) in humans lead to Charcot-Marie-Tooth disease and related neuropathies. 10 Using the AMP-Glo system, we first analyzed the activity of human AspRS and ThrRS. We observed an increase in AMP production proportional to the increase in the amount of aaRS (Fig. 2A, B).
Measurement of aaRS activity. Activities of human AspRS
We next tested the activity of E. coli aaRS complex using the AMP-Glo assay system. Like human aaRS, the E. coli aaRS also showed an increase in AMP production proportional to the amount of aaRS. Bioluminescent assays have the advantage of extremely low background noise, allowing high signal:background ratios (Fig. 2C). The large dynamic range is extremely useful for inhibitor studies.
Since the AMP-Glo assay is extremely sensitive in detecting AMP generated from biochemical reactions, use of very high ATP (substrate) concentrations may lead to poor assay performance. We observed a Km for ATP of 21.34 μM (Fig. 2D). Often radioactive assays for analyzing tRNA synthetase rely on the use of γ-[P 32 ] ATP where the generation of the PPi is measured, such assays often use very high ATP concentrations in the millimolar range. Use of such high ATP concentrations is not recommended when using the AMP-Glo assay.
Measuring cAMP Phosphodiesterase Activity
PDE hydrolyze the phosphodiester bond in cyclic nucleotide cAMP and cyclic guanosine monophosphate (cGMP), which are both critical cellular second messengers, thereby lowering their concentration resulting in significant physiological changes. PDE comprise 11 enzymes (PDE 1–11) with over 55 isoforms, and they are either selective for cAMP or cGMP or nonselective for either cyclic nucleotide. Thus, PDE 4, 7, and 8 are selective toward cAMP, while PDE 5, 6, and 9 are selective for cGMP as substrate, while others PDE1, 2, 3, 10, and 11 have a dual specificity. These enzymes are expressed by all cell types in the human body and have great clinical significance. 11 PDE3 inhibitors, including Inamrinone and Enoximon, are used for short-term treatment of cardiac failure 12 ; PDE4 inhibitor apremilast is used for treatment of psoriasis and crisaborole for atopic dermatitis. 13,14 We tested the enzymatic activities of PDE (PDE3B, PDE7A, PDE8A1, and PDE4B2) using the AMP-Glo system. As shown in Figure 3A, increase in the amount of PDE resulted in increasing amounts of AMP production linearly up to 0.4 ng/reaction. We then measured the Km for cAMP for various PDE, the observed Km values were similar to published data (Fig. 3B). Next, we used the system to determine the IC50 value for PDE inhibitors for the cAMP-specific PDE3B. The IC50 value determined for cilostamide, zardaverine, and IBMX for PDE3B was 2.6, 29.4, and 429 μM respectively, which is similar to the published IC50 value. Inhibitors that were specific for cGMP0dependent PDE (Zaprinast) did not inhibit PDE3B activity (Fig. 3C).
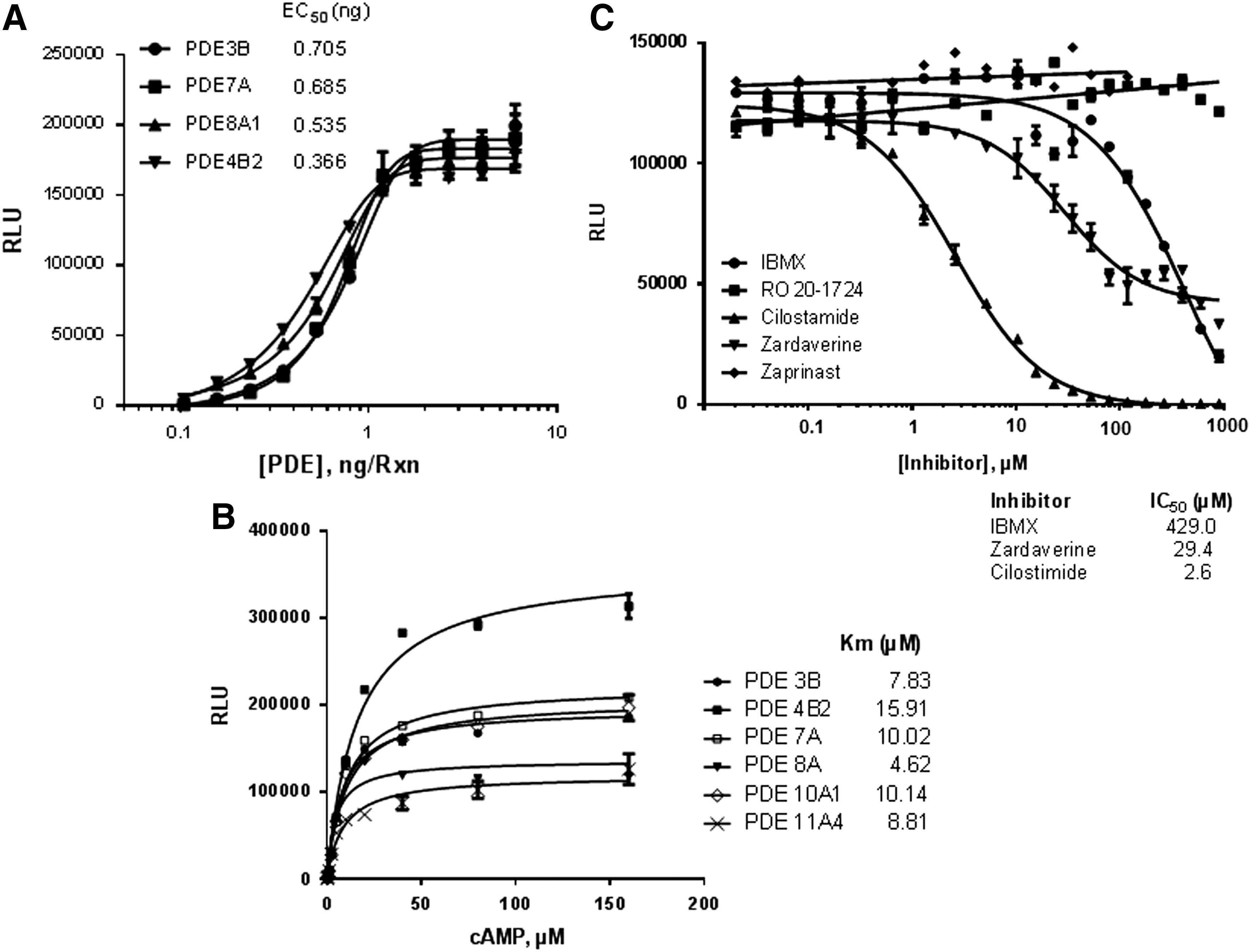
Measurement of cAMP phosphodiesterase activity.
Measurement of ATP-Dependent T4 DNA Ligase and NAD+-Dependent E. coli DNA Ligase Activities
DNA ligases are primarily responsible for sealing nicks between adjacent 5′- phosphate and 3′-hydroxyl groups in duplex DNA that form during DNA replication, DNA repair, and recombination. DNA ligases are classified in two groups based on their substrate requirement: ATP-dependent DNA ligases from archaea, viruses, and eukaryotes or NAD+-dependent DNA ligases found in bacteria. 15 Although these two classes use different substrates, they both produce AMP as the common product of their reactions. Since DNA ligases generate AMP as a product, we tested the utility of using the AMP-Glo system for measuring the activities of DNA ligases. We first tested the enzymatic activity of ATP-dependent T4 DNA ligase using EcoRI-digested linear double-stranded DNA as the substrate in the presence of ATP. As shown in Figure 4A, increase in the amount of T4 DNA ligase resulted in a linear increase in light output up to 2,000 ng/reaction, reflecting the amount of AMP generated during the ligation process.
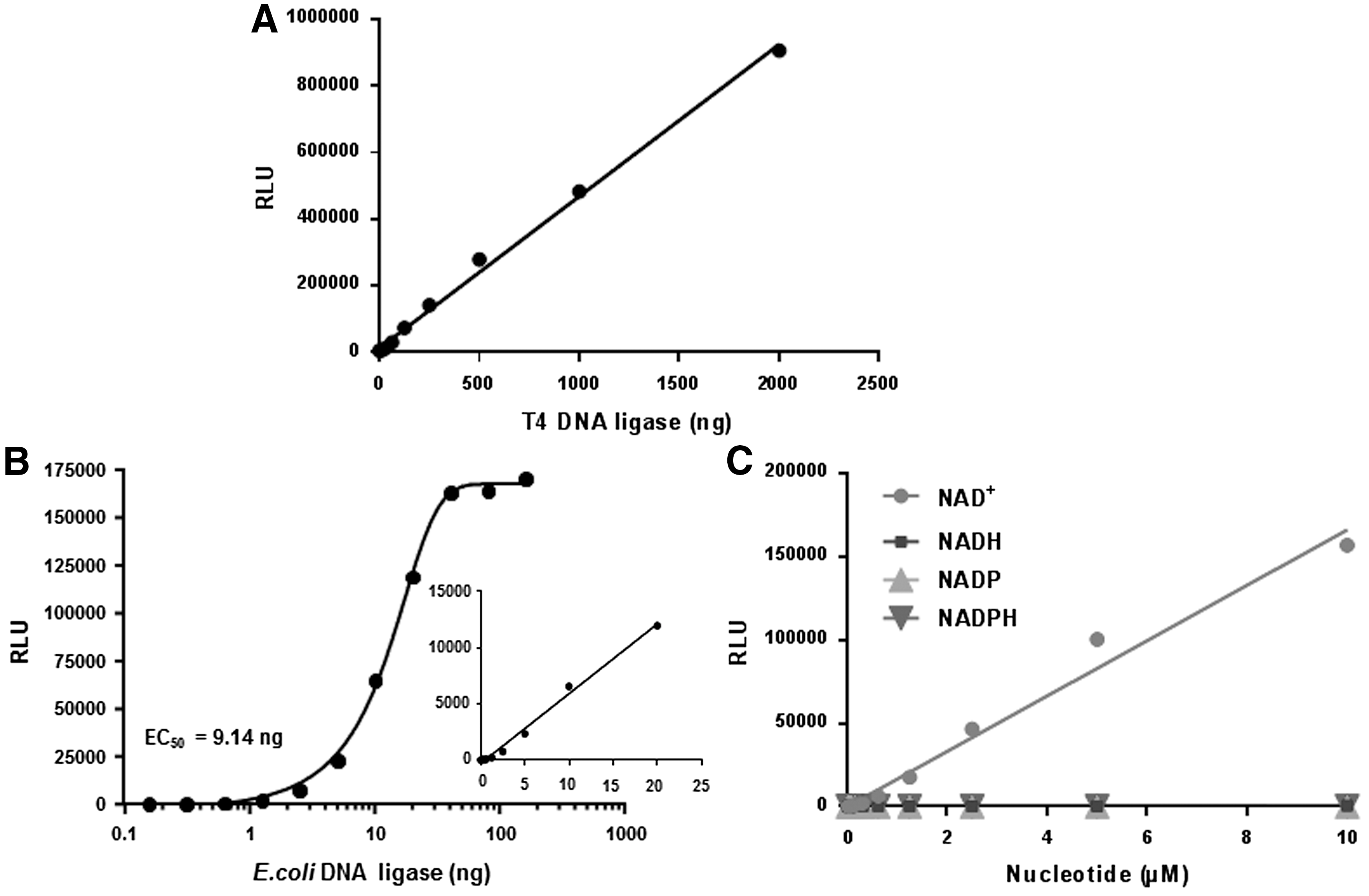
Measurement of T4 DNA ligase
Although the enzymatic reaction of NAD+-dependent DNA ligases and ATP-dependent DNA ligases are similar, they are structurally distinct. They are also indispensable for bacterial DNA replication and conserved among various bacterial pathogens. This has attracted significant interest in NAD+-dependent DNA ligases as a broad-spectrum antibacterial drug target. 16,17 Some potent inhibitors of NAD+-dependent DNA ligases have been identified and are being tested for in vivo efficacy in model systems. 18 We tested the activity of E. coli DNA ligase using the AMP-Glo system, using nicked duplex DNA and NAD+ as substrates. As shown in Figure 4B, increase in the amount of E. coli DNA ligase resulted in increasing amounts of AMP production linearly up to 20 ng/reaction. We also studied the substrate specificity of E. coli DNA ligase, using various nicotinamide nucleotides (NAD+, NADH, NADP+, and NADPH). As shown in Figure 4C, only NAD+ is used as a substrate by the E. coli DNA ligase. The light output increased linearly with an increase in NAD+ concentration up to 5 μM. The LOD for NAD+ was 1 nM.
Application of AMP-Glo System to Study NAD+-Dependent Processes
NAD+ is an important cellular metabolite acting as a coenzyme and substrate in various biological processes, including metabolism and signal transduction. As a coenzyme, NAD+ plays an important role as an electron carrier and a participant in metabolic redox reactions in which oxidoreductases interconvert NAD+ (oxidized form) to NADH (reduced form) to oxidize or reduce small molecule metabolites. NAD+ also acts as the ADP-ribosyl donor for ADP-ribosylation, an important posttranslational protein modification mediated by ADP-ribosyl transferase (ARTs) and sirtuins. NAD+ also acts as the substrate for ADP-ribosyl cyclases to produce cyclic ADP-ribose (cADPR), which is used as a second messenger in Ca2+ signaling. 19,20 ADP-ribosylation is an important posttranslational modification controlling DNA repair, apoptosis, inflammation, bacterial virulence, and pathogenicity, and thus medically relevant for the treatment of cancer, diabetes, and age-related diseases like Alzheimer, and for the identification of novel antibiotic strategies. 21,22 Both mono- and poly-ADP-ribosylation of proteins are observed in multicellular eukaryotic cells. In ADP-ribosylation, ADP-ribose units are added to Glu, Asp, Lys, Asn, Cys, phospho-Ser, and diphthamide residues of target proteins leading to consumption of NAD+ and production of nicotinamide. As E. coli DNA ligase uses only NAD+ as a cofactor, we modified the AMP-Glo assay system to include E. coli DNA ligase and nicked duplex DNA in the AMP-Glo Reagent I. This modified AMP-Glo Reagent I is capable of converting NAD+ in a test sample to ADP (Figs. 1 and 5A), which is then converted to ATP and detected using the AMP Detection Solution.
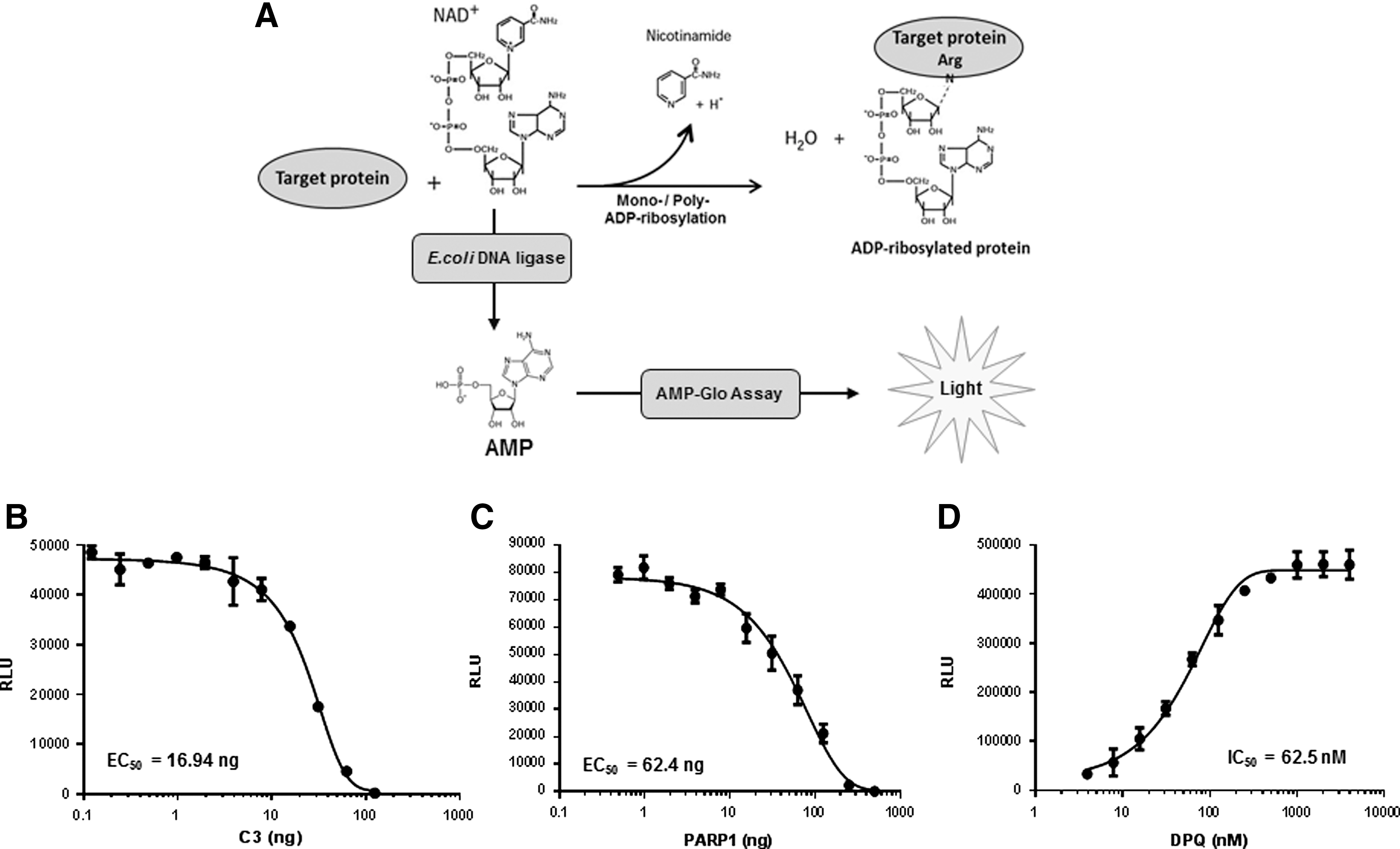
Measurement of ADP-ribosylation using the modified AMP-Glo assay.
ADP-ribosylation is responsible for the actions of several bacterial toxins, such as cholera toxin, diphtheria toxin, and pertussis toxin. 23,24 These toxin proteins act as mono-ARTs that modify target proteins, especially host GTPases. As an example, C. botulinum C3 toxin ADP-ribosylates Rho family guanosine triphosphatases (GTPases), thus affecting cytoskeletal functions in the host. 25,26 We evaluated the use of the modified AMP-Glo assay for measuring mono-ADP-ribosylation by C3 using RhoA as the substrate. We titrated C3 ADP-ribosyltransferase in the presence of 10 μM RhoA and 10 μM NAD+ and detected the amounts of remaining NAD+ upon reaction completion, as a surrogate readout for monitoring enzyme activity. As shown in Figure 5B, increasing the amount of C3 ADP-ribosyltransferase in the reaction resulted in a decrease in the light output, thus supporting NAD+ consumption in the ADP-ribosylation reaction.
In mammalian cells, the enzyme PARP1 is an abundant nuclear protein and one of the most NAD+-consuming enzyme. It transfers ADP-ribose groups from NAD+ to target protein, forming long branched chains of negatively charged poly-ADP-ribose. PARP1 is activated by DNA damage, including single strand breaks, during base-excision repair and for the repair of topoisomerase I cleavage complexes and DNA double-strand breaks. Auto-ADP-ribosylation of PARP1 and PARP2 and ADP-ribosylation of chromatin proteins promote recruitment of DNA repair factors, because of the highly negative charge of the poly-ADP ribose polymers. Cancer cells deficient in BCRA1 and BCRA2 result in extensive DNA damage and activation of PARP1. Inhibition of PARP1 has shown significant specificity to kill BCRA-negative cancer cells. Recent studies have shown that disruption of Fanconi anemia or other homologous recombination-related genes also sensitize cells to PARP inhibitors, providing a strong rational to develop PARP inhibitors for cancer treatment. 27 –29 We evaluated the use of the modified AMP-Glo assay for measuring PARP1 activity. As shown in Figure 5C, increasing the amount of PARP1 resulted in a decrease in the light output, thus reflecting consumption of NAD+ in the ADP-ribosylation reaction. Next, we examined the inhibition of PARP1 by a PARP1 inhibitor (DPQ). 30 DPQ exhibited a concentration-dependent inhibition of PARP1 with an IC50 of 62.5 nM (Fig. 5D). These results show clearly that the AMP-Glo system can be modified to monitor the activity of these enzymes and allow for high-throughput screening research searching for novel inhibitors.
Application of AMP-Glo System to Study Sulfotransferases
Sulfonation is an important reaction in the metabolism of xenobiotics to less active compounds and endogenous compounds like hormones and neurotransmitters. Sulfation is also involved in the synthesis of sulfonated glycosaminoglycans, such as heparin, heparan sulfate, chondroitin sulfate, and dermatan sulfate. Sulfotransferases are classified into two groups based on their localization: cytosolic and Golgi-resident sulfotransferases. Cytosolic sulfotransferases are responsible for Phase II drug metabolism of xenobiotics and Golgi-associated sulfotransferases are responsible for sulfonation of larger molecules like carbohydrates and proteins. 31,32 PAPS is the universal sulfo-donor leading to the production of PAP 3 . PAP can be converted to AMP by a Golgi-resident PAP-specific phosphatase (IMPAD1), 33 which can then be detected using the AMP-Glo assay (Figs. 1 and 6A). A sulfotransferase assay has been recently described that measures the inorganic phosphate using the malachite green reagent, 34 which is known to have a low sensitivity and low dynamic range than bioluminescent assays. To address that issue, we coupled IMPAD to the AMP-Glo system to detect PAP. Using this modified AMP-Glo system, we first tested its ability to detect PAP. PAP was titrated and detected as described in the Materials and Methods section. The light output increased linearly with an increase in PAP concentration (Fig. 6B). The LOD was 5 nM PAP (S/N >3). We then measured the activity of SULT 1A3. SULT 1A3 catalyzes sulfation of phenolic monoamines, including endogenous substrates such as dopamine, norepinephrine, and serotonin, and other phenolic and catecholic drugs. The PAP formed during the sulfotransferase reaction is then detected using the modified AMP-Glo reagent. As shown in Figure 6C, with an increase in the amount of SULT 1A3, the light output is increased, reflecting that more PAP is produced in the sulfation reaction.
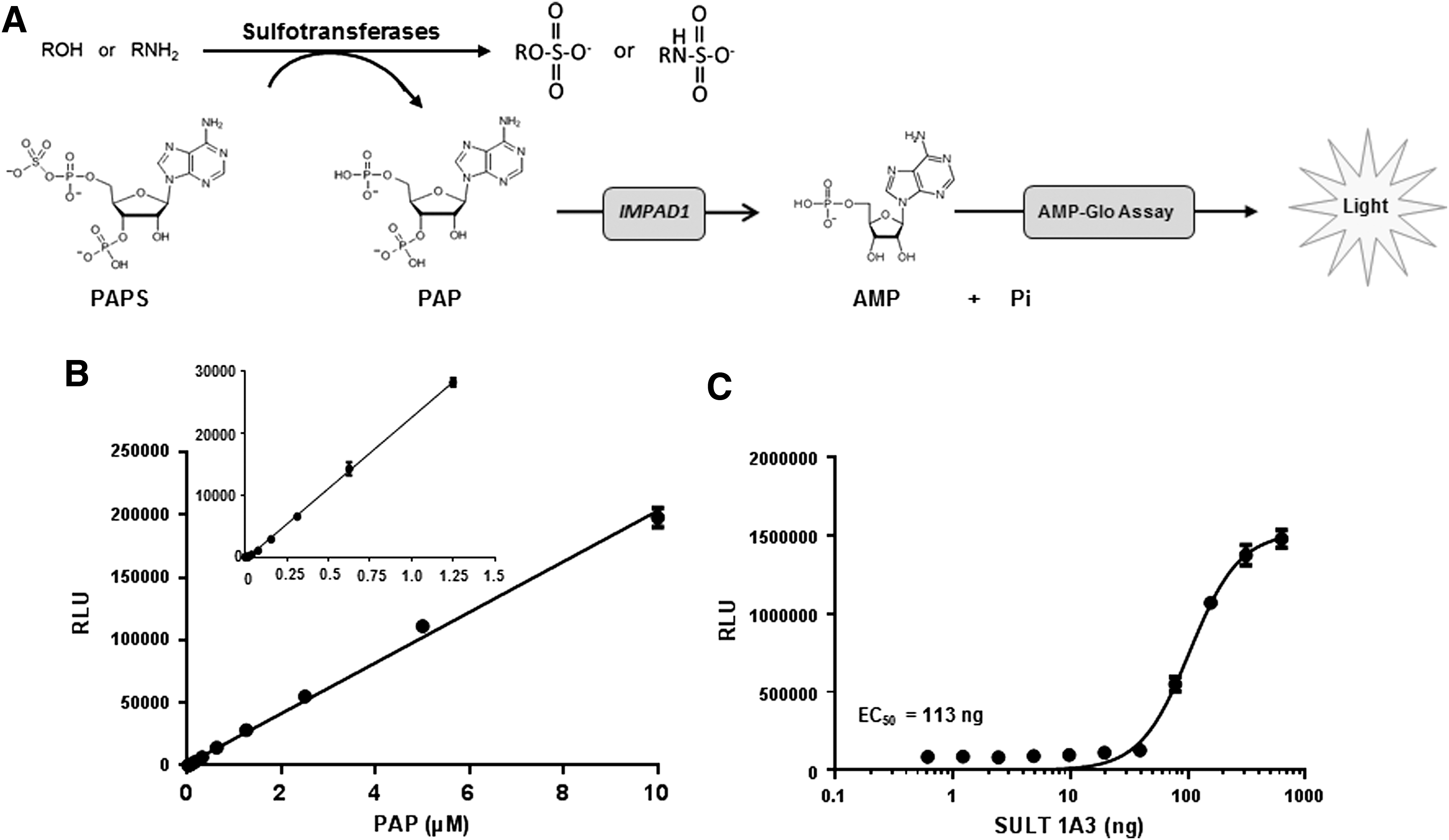
Measurement of sulfotransferase activity using the modified AMP-Glo assay.
z-Factor Analysis
z-Factor is a measure of assay quality and robustness in high-throughput screening, reflecting dynamic range, assay sensitivity, and reproducibility. 35 z-Factor is calculated as 1 − 3(σp + σn)/(μp − μn), where σ is the standard deviation, μ is the mean, p is positive control, and n is negative control. z′ factors >0.50 are indicative of a robust assay. To measure the z-factor for the AMP-Glo assay, we used PDE4B2 phosphodiesterase. Reaction containing 0.7 ng PDE4B2/reaction was performed in the presence of 20 μM cAMP for 60 min at room temperature in a low-volume 384-well plate. The AMP-Glo assay was performed as described above. Reactions without the enzyme PDE4B2, but including all other components, was used as control; these did not show any luminescent signal. Data analysis was performed using Sigma Plot/Sigma Stat, version 9.0. The calculated z-factor was 0.85, indicative of a highly robust assay system (Fig. 7).
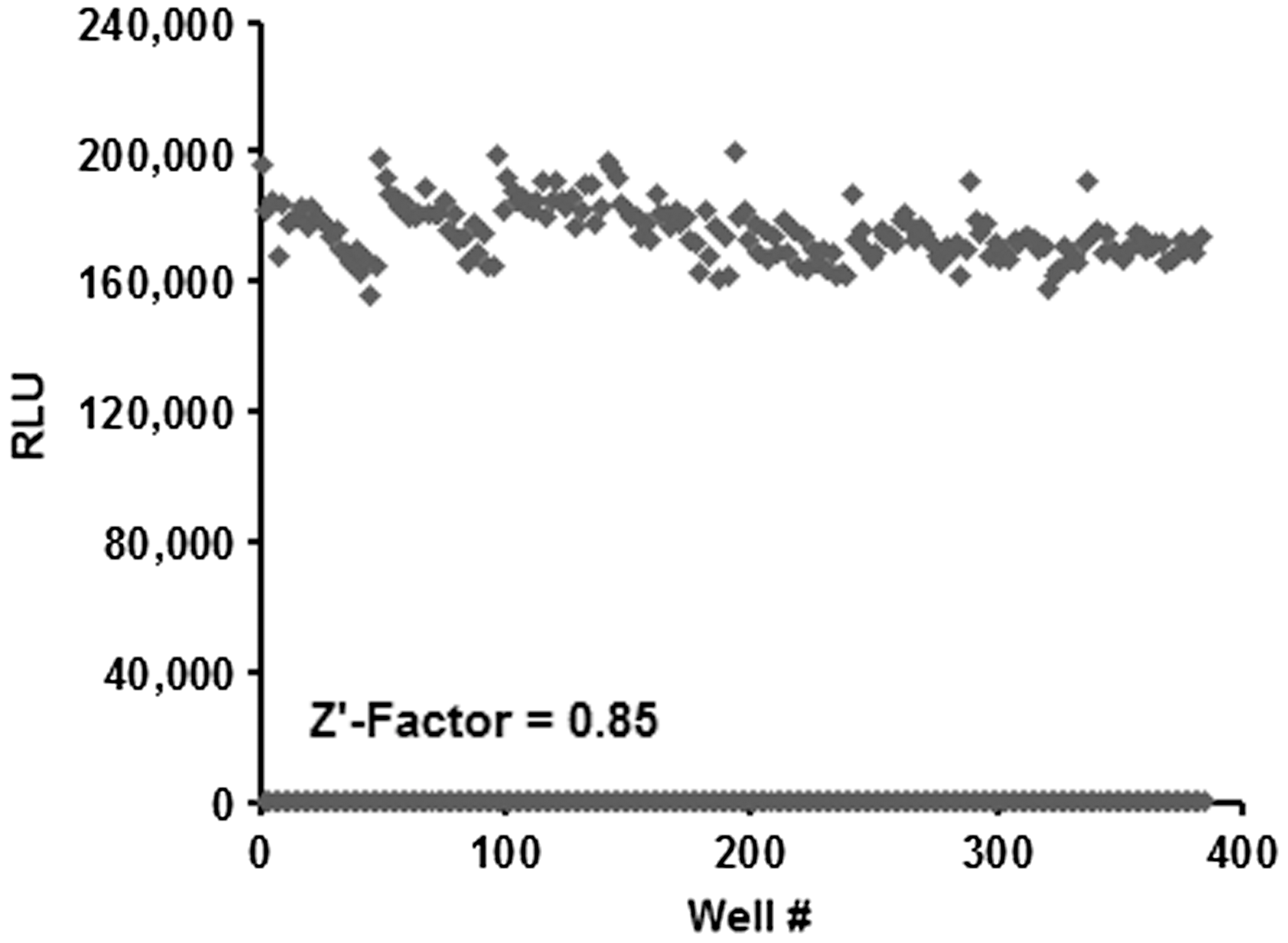
Determination of z′ factor of the AMP-Glo assay. PDE4B2 reaction at 0.7 ng/reaction performed in the presence of 20 μM cAMP for 60 min at room temperature in a low-volume 384-well plate. Reactions without the enzyme PDE4B2, but including all other components, were used as control. The AMP-Glo assay was performed as described in Materials and Methods section using the Deerac FluidicsR Equator HTS noncontact dispenser. Luminescence was measured using the PHERAstar high-end multiwell plate reader (BMG Labtech). Data analysis was performed using Sigma Plot/Sigma Stat, version 9.0.
Conclusion
This study exemplifies the critical role of the cellular metabolite adenosine monophosphate in various enzymatic processes. In addition to AMP being a product of various biochemical processes, it is also a ligand for activation of AMPK, critical for cellular energy homeostasis, glucose and fatty acid uptake, β-oxidation, and synthesis of proteins, fatty acids, cholesterol, and triglycerides. AMP also serves as a substrate for 5′ ectonucleotidase (CD73) generating adenosine, which acts through adenosine receptors on many immune cells causing immune suppression and regulating vasculature on smooth muscle cells. 36 The AMP-Glo assay is a homogenous bioluminescent assay, which offers several advantages over traditional radioactive and nonradioactive fluorescent assays. Radioactive assays need special facilities and personnel training; furthermore, radiolabeled γ-[P 32 ] has a short half-life and besides its health hazard-related issue, it also requires expensive waste disposal of radioactivity. Fluorescence resonance energy transfer (FRET) and time-resolved FRET-based assays 16,37 for DNA ligases have been designed and require labeled DNA oligos and significant optimization of assay conditions. Bioluminescent assays allow a large dynamic range, with high signal:background ratio and S/N ratio, making the assay highly sensitive, requiring low amounts of enzymes, and with no additional requirement for labeling, making these assays simple, homogenous, and amenable to high-throughput screening. In this study, we utilize the diverse range of enzymes that generate AMP directly or by sight modification can be converted to generation of AMP for monitoring their activities using AMP-Glo. We demonstrate the utility of this assay in detecting and monitoring the activity of various enzymes such as cAMP phosphodiesterase, aaRS, and DNA ligases, which produce AMP. Using the specificity of E. coli DNA ligase to use NAD+, we show that the system can be modified to detect NAD+ and NAD+-dependent processes like ADP-ribosylation. Furthermore, as PAPS, which is a derivative of AMP and serves as the universal sulfo-donor in the process of sulfation, we use the modified AMP-Glo system to detect the activity of sulfotransferases. We have previously shown the use of the AMP-Glo system to detect activities of enzymes involved in ubiquitin and ubiquitin-like protein conjugation. 38 We have also shown how the AMP-Glo system could be coupled with enzymes to convert S-adenosylhomocysteine (SAH) to AMP that allows measurement to methyltransferase activities. 39 Methyltransferases use SAM as the universal methyl donor and SAH is the common product. The AMP-Glo system could also be customized beyond what has been described in this work and elsewhere, to measure other adenosine containing metabolites or enzyme classes that regulate them. Metabolites, including, but not limited to adenosine, methyladenosine, deoxyadenosine, adenine, ADP-ribose, cADPR, nicotinic acid adenine dinucleotide phosphate, nicotinamide mononucleotide, homocysteine, methylthioadenosine, and polyamines, including spermine and spermidine that require SAM for synthesis, can all be coupled with enzyme/s to the AMP-Glo system to generate ATP, allowing detection by luciferase bioluminescence. These facts underline the critical role AMP plays as a cellular metabolite and how detection of AMP by using AMP-Glo assay platform can be used to study various biochemical enzymatic activities and facilitates the development of modulators of these enzymes for the purpose of developing next-generation drugs.
Footnotes
Disclosure Statement
S.M., K.H., and S.A.G. are employees of Promega Corporation. S.A.G. is also an employee of the University of Wisconsin School of Medicine and Public Health.