
Abstract
Measurement of the changes in intracellular Ca2+ levels is an important assay for drug discovery. In this report, we describe a novel Ca2+ indicator, dCys-GCaMP, based on the green fluorescent protein and the development of a rapid and simple cell-based functional assay using this new Ca2+ indicator. We demonstrated the sensitivity and reliability of the assay by measuring the cellular responses to the agonists, antagonists, channel blockers, and modulators of the ionotropic N-methyl-D-aspartate (NMDA) subtype of glutamate receptors. HEK293 cells coexpressing the NMDA receptor and dCys-GCaMP displayed a strong increase in fluorescence intensity when stimulated with the agonist glutamate. This increase in the fluorescence signal was agonist concentration dependent and could be blocked by NMDAR antagonists and channel blockers. The pharmacological parameters measured with the dCys-GCaMP assay are in close agreement with those derived from conventional assays with synthetic dye fluo-4 and literature values. In addition, we showed that this assay could be used on G protein-coupled receptors as well, as exemplified by studies on the α1A adrenergic receptor. A limited scale evaluation of the assay performance in a 96-well compound screening format suggests that the dCys-GCaMP assay could be easily adapted to a high-throughput screening environment. The most important advantage of this new assay over the conventional fluo-4 and aequorin assays is the elimination of the dye-loading or substrate-loading process.
Introduction
Ca2+, serving as a second messenger or charge carrier, mediates the functional responses of a variety of membrane receptors and ion channels. Functional assays measuring the changes in the intracellular Ca2+ levels have been commonly used to screen for compounds that modulate the activities of target receptors or ion channels. Synthetic dyes, such as fluo-4 and fura-2, are the predominant calcium indicators used in these assays. 1,2 Aequorin, an enzyme from jellyfish, has also been used as a calcium indicator in cell-based functional assays. 3 In the presence of Ca2+, this enzyme catalyzes the oxidation of coelenterazine, which generates a luminescent signal.
Recently, several novel Ca2+ indicators that derived from fluorescent proteins have been developed, and they are generally named as genetically encoded calcium indicators (GECIs). These protein-based indicators are produced by translation of a nucleic acid sequence and are typically incorporated into cells by gene transfer techniques. GECIs comprised solely natural protein and do not require synthetic chemicals or cofactors for Ca2+ detection. Based on their overall architectures, GECIs are divided into two classes. The first class is based on single fluorescent proteins. They include the GCaMPs, camgaroos, pericams, and case sensors. 4 These indicators are composed of Ca2+-responsive elements, such as calmodulin (CaM), inserted into a fluorescent protein, such that Ca2+ binding alters the protonation state, and hence, the spectral properties of the chromophore. The second class of GECIs are the cameleon type, in which Ca2+-responsive elements are inserted between two fluorescent proteins such that upon Ca2+ binding, an alteration in the efficiency of fluorescence resonance energy transfer occurs. 4 Compared to synthetic dyes, GECIs have some unique advantages, such as low cytotoxicity, high fluorescence stability, and the possibility to target the indicators to subcellular compartments or specific cells in a tissue. 5 –7
GCaMP is superior to other GECIs due to its high fluorescence strength, thermal stability, rapid kinetics, and the suitable Kd range for measuring cytosolic Ca2+ levels. 8,9 The GCaMP protein is composed of a circularly permutated enhanced GFP (cpEGFP) moiety, a CaM domain, and a Ca2+-CaM-binding peptide referred to as M13. 9 Without Ca2+ binding, GCaMP is nonfluorescent due to a defect in the barrel structure caused by the circular permutation. Upon Ca2+ binding, the fluorescence intensity of GCaMP is drastically increased. The change in fluorescence signal is very rapid, reversible, and dependent on Ca2+ concentrations. GCaMP has been used as a Ca2+ indicator in a variety of cellular imaging studies, including the measurement of neuronal activity in different animal models as well as the detection of smooth muscle or heart activities. 8,10 –15 However, to date, neither GCaMP nor any of the other GECIs have been evaluated in a cell-based functional assay for drug screening or characterization of pharmacological agents. This may be due to the high background fluorescence from dead or damaged cells. To overcome this problem, we have engineered a new GCaMP by introducing cysteine pairs into the core structure. In dead or damaged cells, the cysteine pairs would form disulfide bonds, which distort the fluorescent protein structure and, hence, reduce the background fluorescence. Using this new dCys-GCaMP as a Ca2+ indicator, we have developed a cell-based functional assay in a 96-well format and demonstrated the sensitivity and reliability of the assay for drug screening and pharmacological studies on the agents acting on ion channels and G protein-coupled receptors (GPCRs).
Materials and Methods
Reagents
Dulbecco's modified Eagle's medium (DMEM), Opti-MEM, fetal bovine serum (FBS), 0.25% trypsin/EDTA, penicillin/streptomycin, and fluo-4-AM were purchased from Thermo Fisher Scientific. Polyethylenimine (PEI) was from Polysciences. Mianserin, clozapine, serotonin, MK801, (2R)-amino-5-phosphonopentanoate (AP5), Ro256981, and poly-D-lysine (PDL) were purchased from Sigma Aldrich. The His60 Ni Gravity Column Purification Kit was from Takara Bio. The PD-10 desalting column was purchased from GE Healthcare. The black-wall clear-bottom 96-well plate was from Corning.
Construction of Plasmids
The DNA for the GCaMP3 coding region was fully synthesized by GenScript China (Nanjing, China) based on the published sequence. 9 Two threonine to cysteine mutations were introduced by site-directed mutagenesis using the following primers: 5′-TATGCACCAAGGAG CTGGGGACGGTG-3′, 5′-TTGTCCCATCCCCGTCCTTG-3′, 5′-GCTGC ATCGACTTCCCTGAGTTCCTG-3′, and 5′-CATTACCGTCGGCATCTAC-3′. The final mutant protein was named as dCys-GCaMP. The dCys-GCaMP cDNA was cloned into the pEcoli-Cterm 6xHN vector for expression in Escherichia coli and into the pCDNA3.1/hyg vector for expression in mammalian cells. The cDNAs encoding human N-methyl-D-aspartate (NMDA) receptor subunit NR1 (NM_000832), NR2A (NM_000833), and NR2B (NM_000834), and the adrenergic receptor α1A (NM_000680) were cloned from a cDNA library and inserted into the pcDNA3.1/hyg vector.
Fluorescence Spectra and Ca2+ Titration of Purified dCys-GCaMP
For protein purification, the cDNA of dCys-GCaMP was subcloned into the pEcoli-Cterm 6xHN vector, an inducible bacterial expression system with a His-tag at the amino terminus, and was transformed to E. coli BL21. Protein expression was induced by 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) at the exponential growth phase of the bacteria in a lysogeny broth medium at 37°C. After cell harvest, the His60 Ni Gravity Column Purification Kit was used to purify the protein with His-tag according to the manufacturer's protocol. The purified protein was further desalted with a PD-10 desalting column, and the protein concentration was determined by the bicinchoninic acid method. 16
The fluorescence spectra of the purified dCys-GCaMP protein were analyzed on FlexStation-3 (Molecular Devices) under the spectrum mode. The protein at 1 μM was dissolved in the MOPS buffer (30 mM MOPS, 100 mM KCl, 100 mM DTT, pH 7.2) in the presence of 2 mM Ca2+ or 10 mM EGTA. For the excitation spectrum, the sample was excited from 300 to 490 nm with an interval of 1 nm, and the emission was measured at 520 nm. For the emission spectrum, the sample was excited at 485 nm, and the emission was measured at every nanometer from 490 to 600 nm.
For the calcium titration experiment, the purified protein was diluted in either a Ca2+-free buffer (10 mM EGTA, 100 mM KCl, 30 mM MOPS, pH 7.2) or a high Ca2+ buffer (10 mM Ca2+ EGTA, 100 mM KCl, 30 mM MOPS, pH 7.2) at a final concentration of 1 μM. DTT at 0.1 mM was used to prevent oxidation of the proteins. The Ca2+-free buffer and the high Ca2+ buffer containing the purified protein were mixed at different ratios to provide different concentrations of free Ca2+. The fluorescence intensity was measured on FlexStation-3 under the endpoint mode, with excitation at 485 nm and emission at 520 nm. The titration curve was fitted by the four-parameter model.
Cell Culture and Transfection
Stable HEK293 cell lines expressing human NMDA receptor subtypes NR2A and NR2B were maintained in DMEM supplemented with 10% FBS, 104 unit/mL penicillin/streptomycin, 10 μM hygromycin, and 1 μM puromycin at 37°C in a humidified CO2 incubator. To protect cells from glutamate excitotoxicity, 10 μM MK801 and 50 μM AP5 were added to the culture medium.
The dCys-GCaMP plasmid was transfected into HEK293 cells stably expressing NR2A and NR2B using PEI. Briefly, 30 μg plasmid and 90 μL PEI were well mixed in 3 mL Opti-MEM and were incubated for 5 min at room temperature. The DNA/PEI mixture was added to the cells grown on a 100-mm culture dish at 95% confluency. After incubation at 37°C for 8 h, the DNA/PEI mixture was removed and the cells were incubated in a fresh medium. Twenty-four hours before the assay, the cells were harvested with 0.25% trypsin/EDTA and plated onto a PDL-treated black-wall clear-bottom 96-well plate at a final density of 8×104 cells/well.
dCys-GCaMP-Based Ca2+ Flux Assay on NMDA Receptor
For the concentration-dependent agonist assay (Table 1), the transfected cells in the black-wall clear-bottom 96-well plate were washed thrice with a 200 μL assay buffer (2 mM CaCl2, 0.1% BSA, 20 mM HEPES in HBSS, pH 7.4) by Wellwash Versa (Thermo Fisher Scientific), and the final volume of buffer was adjusted to 50 μL/well by Cybi-SELMA (Cybio). The plate was transferred to the Functional Drug Screening System (FDSS/μCELL), which was set to excite at 480 nm and measure the fluorescence emission at 540 nm. After a 10 s baseline reading, the stimulus solution containing agonist glutamate at concentrations ranging from 5.1 to 100 μM and coagonist glycine at 50 μM was transferred to the cell plate, and the changes in fluorescence intensity were recorded by FDSS/μCELL for another 110 sec. Four wells were used for the negative control in each 96-well plate.
The Assay Procedure for Agonist Concentration Response
1. cDNA (μg):PEI (μL)=1:3 in a 100-mm culture dish.
2. Ninety-six-well clear-bottom black-wall Costar plates coated with PDL.
3. Wash cells three times with assay buffer, leave 50 μL/well.
4. C-R, two compounds/plate, 1/3 serial dilution.
5. Baseline reading before first addition.
6. Dissolved in assay buffer, four wells for each control.
7. See data analysis section.
C-R, concentration response; FDSS, functional drug screening system; PDL, poly-D-lysine.
For the concentration-dependent antagonist assay (Table 2), the transfected cells in a black-wall clear-bottom 96-well plate were washed thrice with the 200 μL assay buffer. Test compounds were dissolved and serially diluted in dimethyl sulfoxide (DMSO). After a 100-fold dilution in the assay buffer, the compound solution was added to the cells on the 96-well plates at 50 μL/well. After a 10-min incubation at 37°C, the cell plates were transferred to FDSS/μCELL for the assay. The program started with a 10-sec baseline reading. After the addition of 50 μL/well stimulation solution (100 μM glutamate/50 μM glycine in assay buffer), the fluorescence change was measured for 110 sec. Twelve wells were used for the positive and negative controls in each 96-well plate.
The Assay Procedure for Antagonist Concentration Response
1. cDNA (μg):PEI (μL)=1:3 in a 100-mm culture dish.
2. Ninety-six-well clear-bottom black-wall Costar plates coated with PDL.
3. Wash cells three times with assay buffer, leave 25 μL/well.
4. C-R, two compounds/plate, 1/3 serial dilution.
5. Dissolved in assay buffer, four wells for each control.
6. Baseline reading before second addition.
7. Ligand of saturated concentration was dissolved in assay buffer.
8. See data analysis section.
For the NR1/NR2B antagonist screen, a similar procedure was used except that the cells were incubated with 10 μM test compounds for 10 min at 37°C.
Fluo-4-Based Ca2+ Flux Assay on NMDA Receptor
The stable cell lines of NR1/NR2A and NR1/NR2B were plated onto PDL-treated black-wall clear-bottom 96-well plates. After 24 h of culture, the cells were washed thrice with 200 μL of assay buffer, and the volume of buffer in each well was adjusted to 25 μL. An equal volume of staining solution (4 μM fluo-4-AM, 0.04% pluoronic F-127, 4 mM probenecid, and 100 μM AP5 in the assay buffer) was added to each well, and the cells were incubated for 60 min at 37°C. Subsequently, the extracellular dye was removed by washing for thrice with 200 μL of assay buffer. After incubation with test compounds for 10 min at 37°C, the cell plates were transferred to FDSS/μCELL for fluorescence reading.
dCys-GCaMP Assay on Cells Transiently Transfected with α1A Adrenergic Receptor
HEK293 cells were maintained in DMEM containing 10% FBS, 104 unit/mL penicillin/streptomycin, and 10 μM hygromycin. The dCys-GCaMP and ADRA1A plasmids were transfected into the cells with PEI, as described above. The transfected cells were plated onto a PDL-coated black-wall clear-bottom 96-well plate 1 day before the assay. The concentration-dependent responses to agonists and antagonists of the α1A adrenergic receptor (α1-AR) were detected using methods similar to those used for NMDA receptors, except that the stimulus buffer contained norepinephrine (Tables 1 and 2).
Data Analysis
The ratio of peak fluorescence to baseline in each well was captured as raw data. The data were subsequently normalized as follows:
Where I represents the signal from an individual well, I0 and Imax represent the mean values from buffer controls and agonist controls, respectively.
The quality of an assay was assessed by the Z′ factor, which was calculated by the following equation:
Where σp and σn are the standard deviation of the positive and negative control responses, respectively; while μp and μn are the mean values of the positive and negative responses, respectively. 17
For dose–response experiments, the value from each well of a 96-well plate was normalized to the maximum positive control response, and the entire set of the normalized values was subsequently fitted by four-parameter simulation to obtain EC50 or IC50 values for the agonist or antagonist, respectively.
Results and Discussion
Development and Characterization of dCys-GCaMP as a Fluorescent Ca2+ Indicator
A good fluorescent Ca2+ indicator allows monitoring Ca2+ signals in living cells in real time without perturbing cellular functions. To convert the Ca2+ signal to fluorescence change, the GCaMP protein uses three components: a Ca2+-binding CaM domain as a sensor, a Ca2+-CaM-binding peptide M13 as a transducer, and a circularly permutated EGFP (cpEGFP) moiety to deliver the signal. 9 The native EGFP has its chromophore located in the center of an 11-strand β-barrel-like structure that protects the chromophore from the bulk solvent. However, in the cpEGFP domain of the GCaMP protein, the native N- and C-termini of EGFP are joined together by a linker, whereas the new N- and C-termini are created by opening one of the β sheets at one side of the barrel, leading to exposure of the chromophore to a solvent and thereby rendering it nonfluorescent. The M13 and CaM domains are fused to the N- and C-termini of cpEGFP, respectively (Fig. 1A). When CaM binds Ca2+, the artificial opening on the barrel of the cpEGFP domain is blocked by the Ca2+-CaM/M13 complex, so that the chromophore is protected from the solvent and is stabilized in the fluorescent deprotonated form. 18 As a result, GCaMP is nonfluorescent without Ca2+, and upon Ca2+ binding to the CaM domain, it becomes strongly fluorescent.
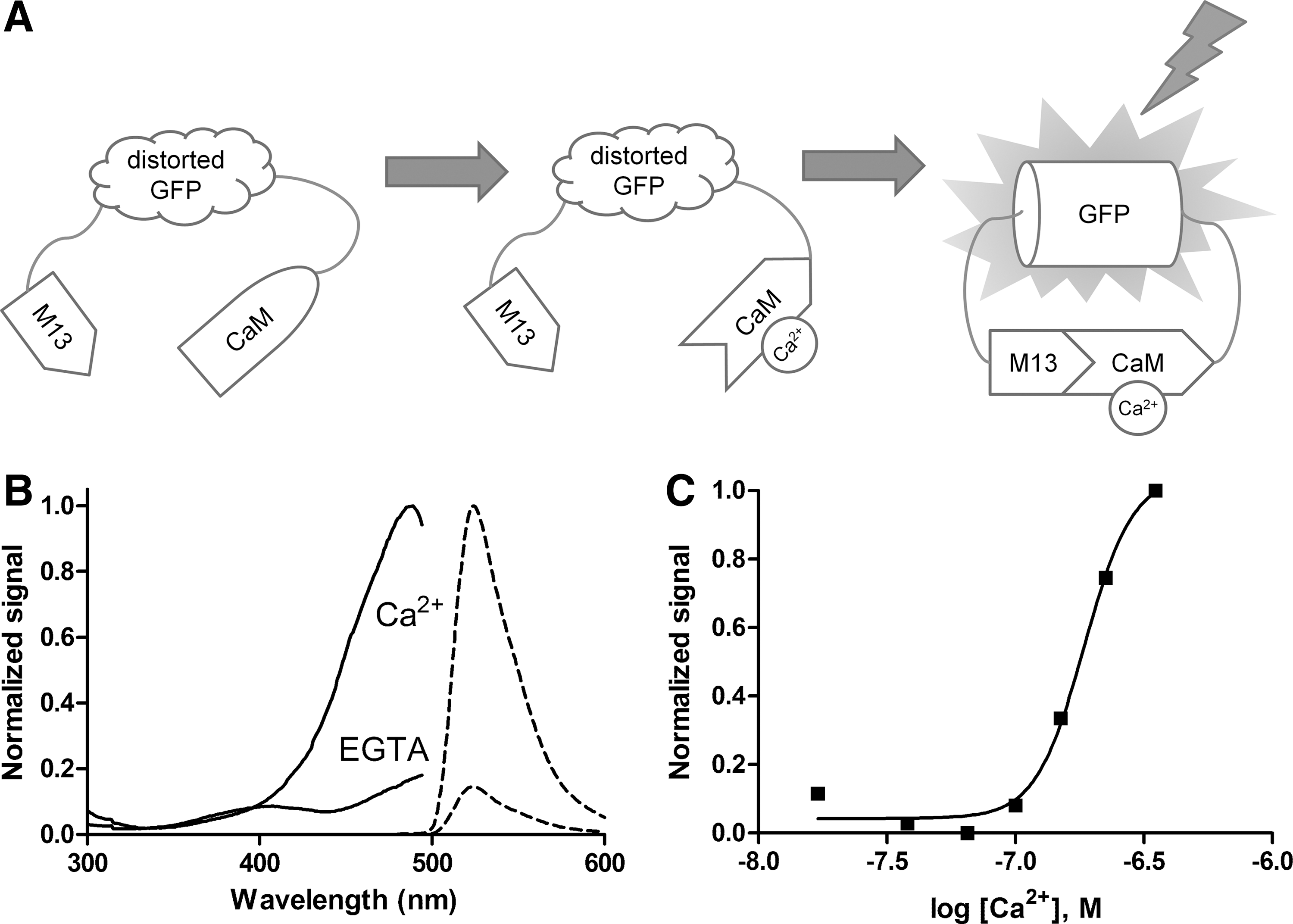
dCys-GCaMP as a Ca2+ indicator.
To overcome the problems associated with high background fluorescence from dead and damaged cells, we introduced cysteine pairs to the neighboring β-sheets of the barrel structure of cpEGFP domain and identified a mutant GCaMP, which was sensitive to oxidation while maintaining the Ca2+ sensitivity and fluorescent properties. In dead or damaged cells, those cysteine pairs would form disulfide bonds due to the reduction of intracellular glutathione levels, which distort the barrel-like structure and result in reduced fluorescence even with Ca2+ binding. The mutant was named as dCys-GCaMP.
dCys-GCaMP was expressed in E. coli and purified for characterization. The excitation and emission spectra of purified dCys-GCaMP are shown in Figure 1B. In Ca2+-bound form, the excitation peak is at 489 nm and emission peak is at 524 nm. The fluorescence intensity dropped significantly from 1700 to 250 RFU when Ca2+ was removed by EGTA. These results are close to the values reported for earlier versions of GCaMP. 5,8,19 –21 Calcium titration showed that the purified protein had an approximately linear response to Ca2+ concentrations in the 100 to 300 nM range in vitro and reached peak fluorescence intensity at 1 μM. The Kd value was determined to be 185±5 nM (Fig. 1C). The Ca2+-dependent changes in fluorescence intensity were very rapid and reversible. These properties indicate that dCys-GCaMP is suitable as a fluorescent indicator for measuring intracellular Ca2+ signals.
Development of Ca2+ Flux Assay Using dCys-GCaMP
To test the feasibility of using dCys-GCaMP in a Ca2+ flux assay, dCys-GCaMP was transiently transfected into CHO and HEK293 cells. Ionomycin, a Ca2+ ionophore, was used to evoke increases in intracellular Ca2+, and the changes in fluorescence intensity were measured in real time with a fluorescence plate reader FDSS/μCELL. As shown in Figure 2A, the addition of 5 μM ionomycin triggered a rapid increase in fluorescence intensity in both HEK293 and CHO cell lines, although the signal was stronger in HEK293 cells, which was most likely due to higher expression levels of dCys-GaCaMP in those cells. In similar experiments using GCaMP3, the background fluorescence in the transfected cells was ∼30% higher, while the fluorescent signal from an ionomycin-stimulated response was significantly reduced. We further optimized the transfection conditions for HEK293 cells by varying the amount of plasmid DNA (Fig. 2B). In addition, we tested the feasibility of using cryopreserved cells for the assay, which is important for adapting the dCys-GCaMP-based Ca2+ flux assay in a high-throughput screening (HTS) environment. As shown in Figure 2C, the fluorescent signal in cryopreserved HEK293 cells was only slightly decreased as compared with the freshly transfected cells.
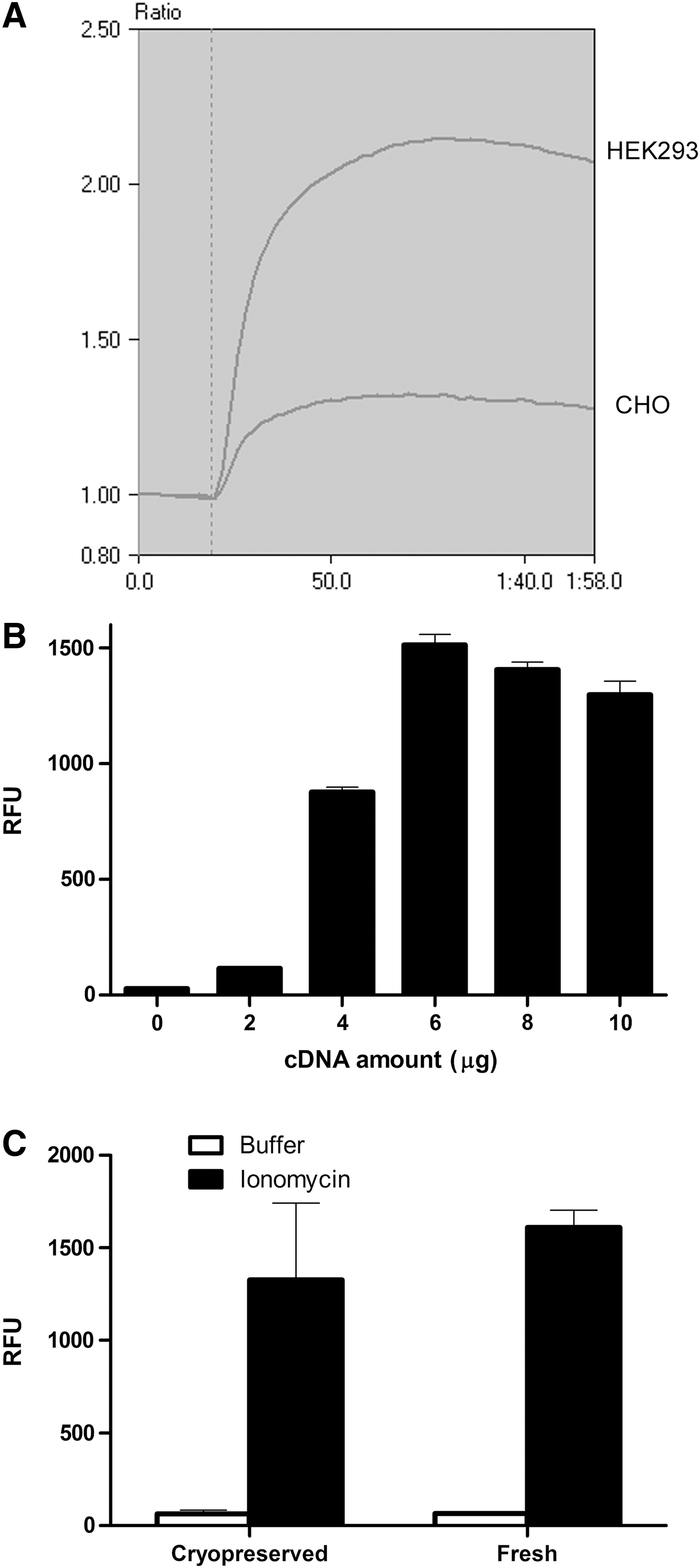
Expression and characterization of dCys-GCaMP in mammalian cells.
dCys-GCaMP-Based Ca2+ Flux Assay on NMDA Receptor
To demonstrate the utility of dCys-GCaMP in the functional screening of pharmacological agents that act on membrane receptors and ion channels, we developed a Ca2+ flux assay on NMDA receptors using dCys-GCaMP. Glutamatergic NMDA receptors are a family of nonselective cationic channels permeable to Na+, K+, and Ca2+ ions. They are composed of two NR1 and two NR2 subunits. 22 NMDA receptors mediate excitatory neurotransmission in the central nervous system. As they are critically involved in Alzheimer's disease, Parkinson's disease, Huntington's disease, schizophrenia, depression, and epilepsy, NMDA receptors are considered as important drug targets. 23 –25
dCys-GCaMP was transiently transfected into cell lines stably expressing two different subtypes of the NMDA receptor, NR2A and NR2B, which are made up of the NR1/NR2A and NR1/NR2B subunits, respectively. After transfection, the cells were grown on 96-well plates, and the fluorescence signals generated in response to agonist stimulation were monitored on the FDSS/μCELL plate reader. Figure 3A shows the traces of fluorescence changes in dCys-GCaMP-transfected cells in response to NR2B receptor activation. The strong fluorescent signals provided a signal-to-noise-ratio (S/N) of 20 and 14 for NR2A and NR2B receptors, respectively. The calculated Z′ factors of >0.7 for both receptors also indicated excellent assay sensitivity (Fig. 3B). We also tested the DMSO tolerance of the assay. As shown in Figure 3C, there was no significant change in the fluorescence signal in the presence of DMSO at concentrations up to 1%.
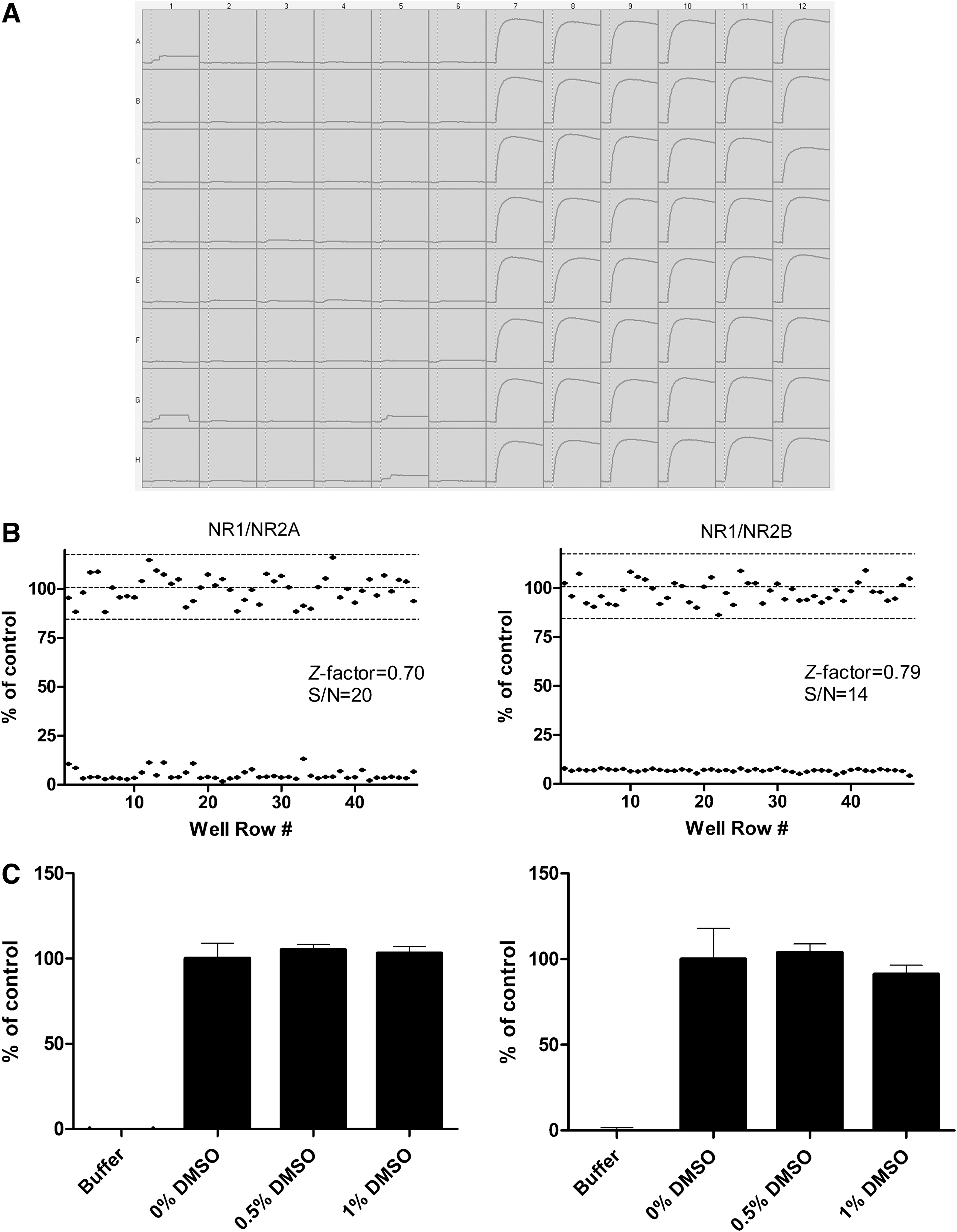
Evaluation of the dCys-GCaMP-based Ca2+ flux assay on NMDA receptors expressed in HEK293 cells.
To demonstrate the reliability of the dCys-GCaMP assay, we evaluated the pharmacological activities of an NMDA receptor agonist, an antagonist, a channel blocker, and an allosteric modulator in the assay. In parallel, we performed conventional Ca2+ flux assays on the same set of compounds using fluo-4. Figure 4 shows a direct comparison of the results from the dCys-GCaMP assay versus the conventional assay using fluo-4. The concentration-dependent response curves from the two assays were in close agreement. The EC50 value of the agonist glutamate and the IC50 values of the antagonist AP5, the channel blocker MK801, and the subtype-selective modulator Ro256981 derived from the assay were also consistent with values reported in the literature (Table 3). 26 The NR2B subtype selectivity of Ro256981 was also detected. As expected, Ro256981 was a potent modulator of NR2B, but inactive on NR-2A.
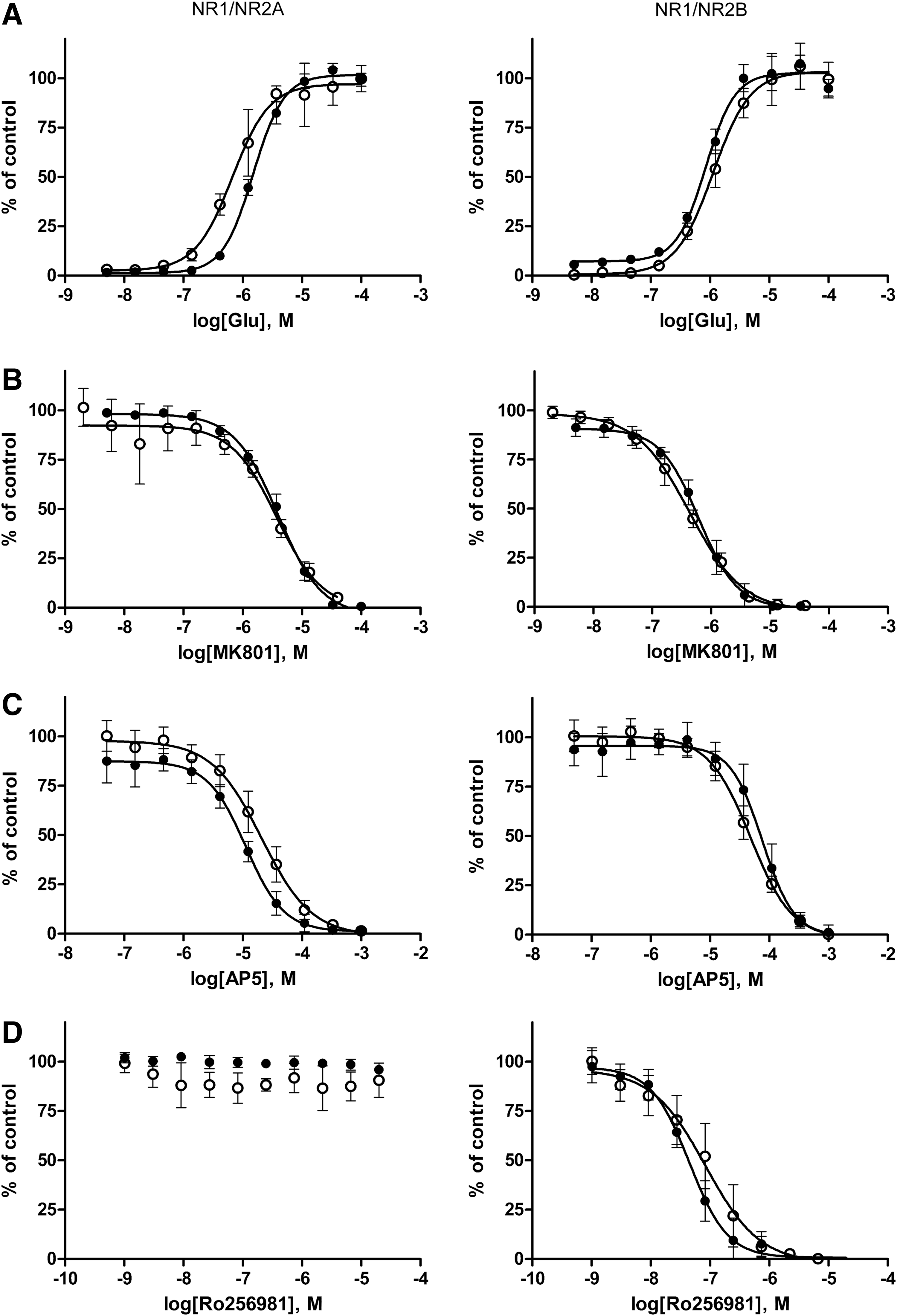
Comparison of the dCys-GCaMP assay (●) vs. conventional fluo-4 assay (○) for pharmacological evaluation of an NMDA receptor
Comparison Between the dCys-GCaMP and Fluo-4 Assays on NMDA Receptors Expressed in HEK293 cells
ND, not detectable.
To assess the performance of the dCys-GCaMP assay in a compound screening format, we screened a small compound library containing 66 known NR2B inhibitors, which have various potencies. Figure 5 shows the high reproducibility of results from two independent experiments (upper panel) and the good correlation with results from the conventional fluo-4 assay (lower panel). The strong fluorescence signal and the robust assay performance in the 96-well format suggest that the dCys-GCaMP assay could be easily adapted to the HTS environment.
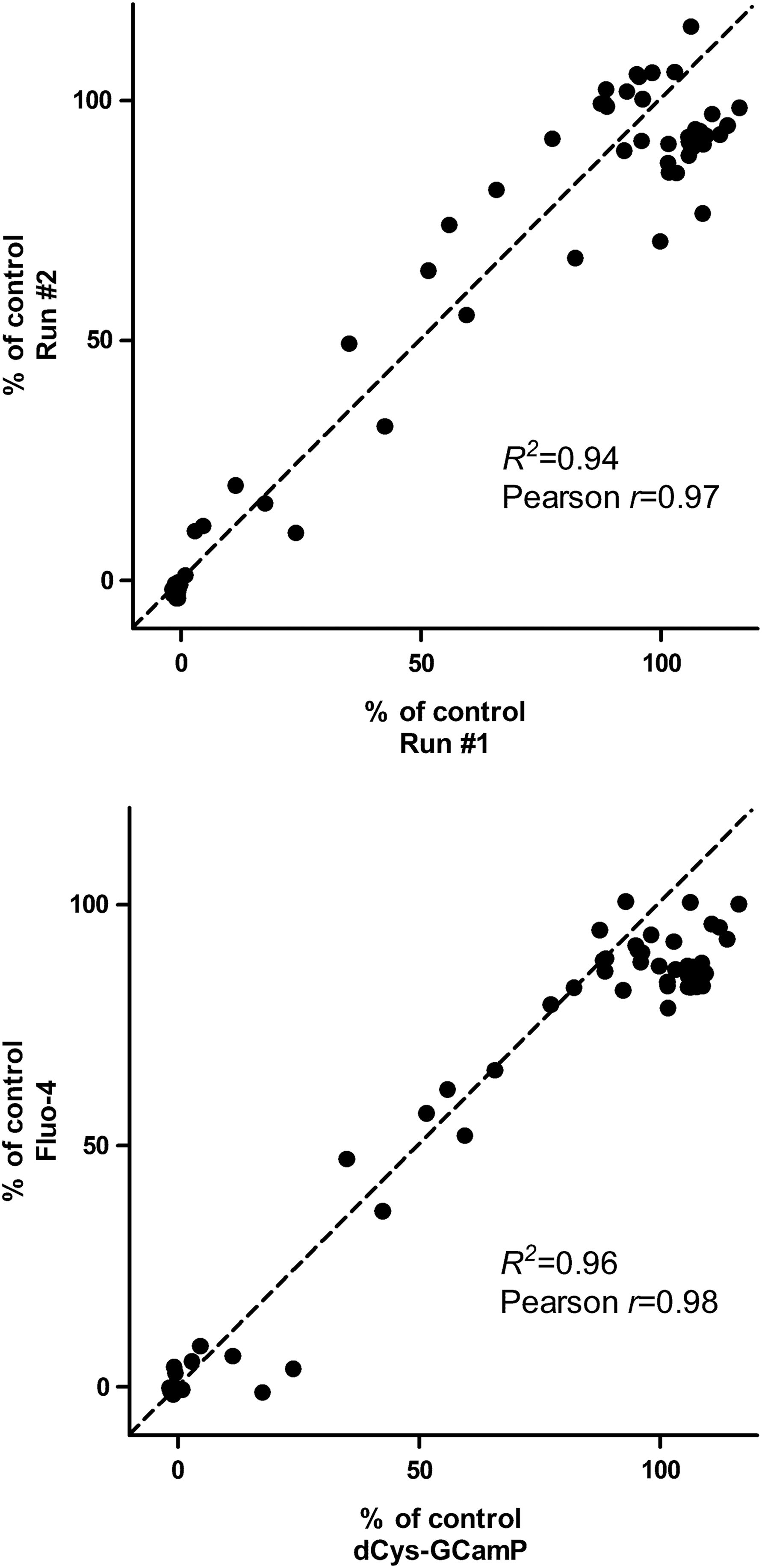
Assessment of the dCys-GCaMP assay performance for compound screening. (Upper panel) Reproducibility of the GCaMP assay. (Lower panel) Correlation of the dCys-GCaMP assay with fluo-4 assay. The activity of 66 antagonists at 1 μM on the NR1/NR2B receptor was tested in the HEK293 cell-based functional assay, using either dCys-GCaMP or fluo-4 as the Ca2+ indicator. The reproducibility of the assay was evaluated using linear regression (R2 ) and correlation (Pearson r) of two sets of data. R2 =0.94 and Pearson r=0.97 for (upper panel), and R2 =0.96 and Pearson r=0.98 for (lower panel).
dCys-GCaMP-Based Ca2+ Flux Assay on a GPCR
GPCRs are an important class of drug targets and many of them use Ca2+ as the second messenger in their signal transduction pathways. To evaluate the utility of dCys-GCaMP as a Ca2+ indicator in GPCR drug discovery, we developed a Ca2+ flux assay for the α1-AR, a prototypical Gq/11-coupled receptor. 27,28 In contrast to Ca2+ influx from the extracellular space through the ion channel of NMDA receptors, the release of Ca2+ from the endoplasmic reticulum is the main source of intracellular Ca2+ after GPCR activation.
dCys-GCaMP and the α1-AR were transiently cotransfected into HEK293 cells. Activation of α1-AR by norepinephrine produced a strong fluorescence signal (data not shown). On a 96-well plate, the dCys-GCaMP assay had an S/N ratio of 16 and a Z′ factor of 0.8 (Fig. 6A). The pharmacological properties of an α1-AR agonist and two antagonists were characterized in the dCys-GCaMP assay as well as the conventional fluo-4 assay. The EC50 value for norepinephrine was determined to be 7.5±0.7 nM in dCys-GCaMP assay and 2.6±0.2 nM in Fluo-4 assay (Fig. 6B). The IC50 values for antagonist haloperidol and prazosin were determined to be 14.3±0.8 nM and 2.0±0.1 nM, respectively, in the dCys-GCaMP assay, which were two- to threefold left shifted as compared to the values from the conventional fluo-4 assay (Fig. 6C, D). The reason for the differences in measured agonist and antagonist potencies between the two assays is unclear. Perhaps, the dCys-GCaMP assay has a better sensitivity.
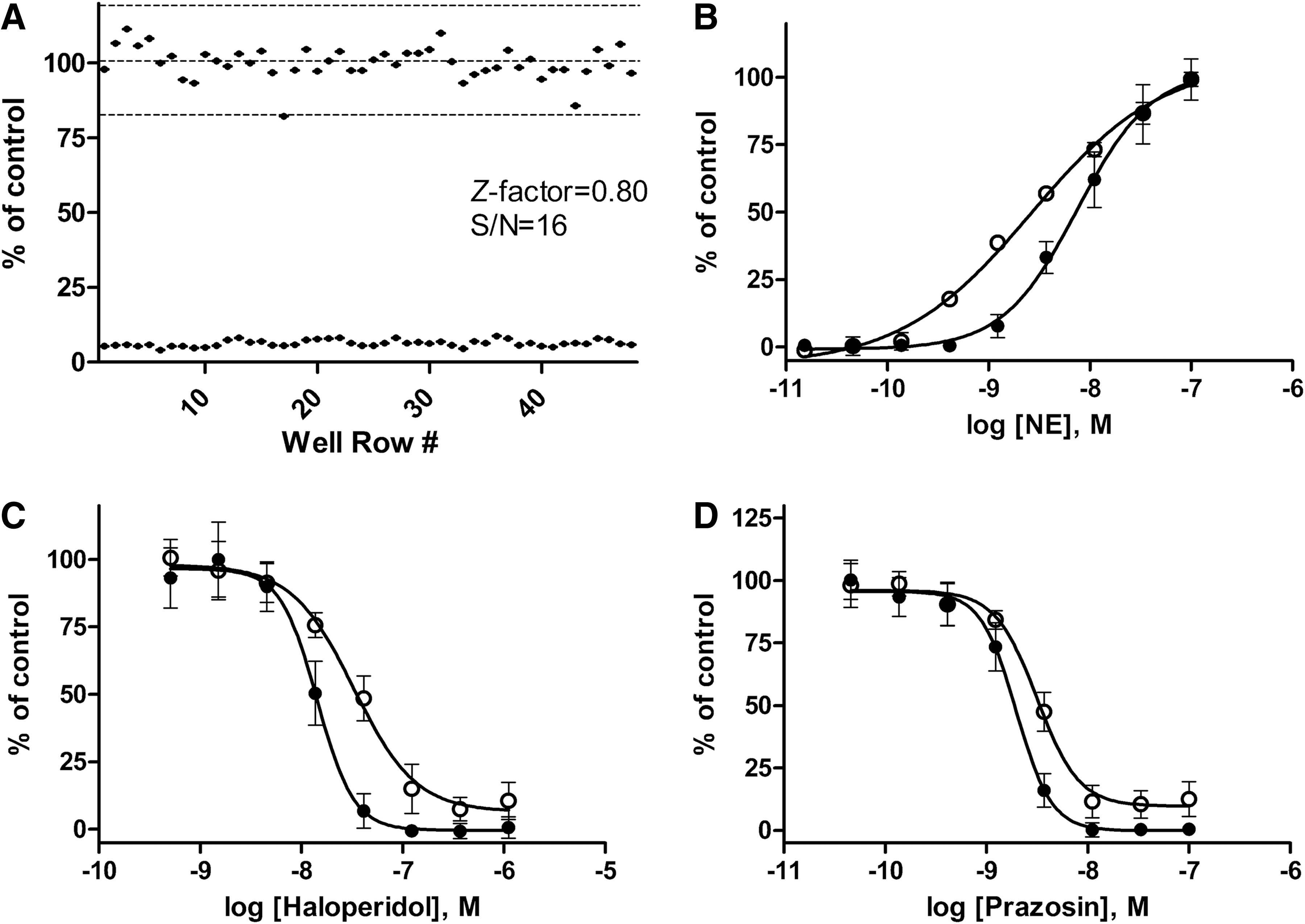
Ca2+ flux assay with dCys-GCaMP on the α1A adrenergic receptor, a GPCR, expressed in HEK293 cells.
Ca2+ flux assays using synthetic dyes, such as fluo-4, have been extensively used in HTS and pharmacological studies over the past two decades. The most common complaints about the assay are related to the requirement for the time-consuming dye-loading step. In addition to the high cost of the dye, the dye-loading procedure can be tricky since the dye is usually cytotoxic at high concentrations and is continuously pumped out by organic anion transporters on the cell surface and compartmentalized in an uncontrolled manner into cellular organelles. After dye loading, the fluorescence measurement has to be made within a limited time window. Since dye loading has to be done fresh every time, the variability between assays is usually high. The aequorin assay has similar problems since it also requires for a time-consuming loading step to get the substrate coelenterazine into the cells. By eliminating the dye-loading or substrate-loading step, the dCys-GCaMP assay significantly simplifies the assay procedure, reduces variability, and provides greater flexibility. In addition, the dCys-GCaMP assay allows for long-term monitoring of the cellular Ca2+ responses. Moreover, dCys-GCaMP can be targeted to specific cells in a tissue or a subcellular compartment for more detailed studies.
In summary, we have developed a Ca2+ flux assay using dCys-GCaMP, a novel fluorescent protein-based Ca2+ indicator, and demonstrated its utility in screening of pharmacologic agents acting on ion channels and GPCRs. The assay has a sensitivity, reliability, and performance similar to those of the conventional assay using fluo-4 as the Ca2+ indicator. Elimination of the dye-loading step represents a process and potential cost improvement over conventional assays.
Footnotes
Acknowledgments
The authors thank Dr. Guoping Feng, Dr. Zhigang He, and Dr. Katherine Zukor for critically reading the manuscript. The excellent technical assistance of Guozhen Tong and Min Xu at Rugen Therapeutics Ltd. is gratefully acknowledged.
Disclosure Statement
No conflict of interest for all the listed authors.