
Abstract
The lipid second messengers phosphatidylinositol-3,4,5-trisphosphate (PtdIns(3,4,5)P3) and sphingosine 1-phosphate (S1P) are well recognized to play important roles in a variety of cellular processes, including cell proliferation, apoptosis, metabolism, and migration. Disruption of lipid signaling pathways often leads to human cancers, making lipid kinases attractive drug targets. In order to develop novel drugs against these enzymes, an assay that monitors their activity and amenable to high-throughput scale for screening large number of compounds is essential. The newly developed ADP-Glo™ assay is such an assay that measures kinase activity of lipid kinases by detecting the formation of ADP using a highly robust and sensitive bioluminescence approach. We evaluated this technology for studying lipid kinases, class I PI3 kinases, and sphingosine kinases and we show that the assay exhibits good tolerance to different lipids substrates. It generates kinetic parameters for substrates and inhibitors similar to those reported in the literature using other published assay formats. The sensitivity and robustness of this assay allow the detection of 5% of substrate conversion with Z′ values >0.7 making it attractive for high-throughput screening (HTS) applications. It is noteworthy that ADP-Glo assay addresses the need for a single integrated platform to comprehensively measure all classes of lipid and protein kinases. The selected inhibitors of lipid kinases can be screened against the panel of desired protein kinases, making ADP-Glo assay a simple, inexpensive platform for HTS and profiling of lipid kinases.
INTRODUCTION
Lipids have long been recognized as important signaling molecules regulating cell growth, proliferation, and survival.1 Phosphoinositides and sphingolipids are the 2 examples of bioactive membrane lipids, the imbalance of which is often associated with human diseases such as cancer, diabetes, and inflammation.2 , 3
In the phosphoinositides signaling pathway, phosphorylation of the 3-hyroxyl group of the inositol ring by class I PI3 kinases results in generation of the second messenger phosphatidylinositol-3,4,5-trisphosphate (PIP3) that promotes nutrient uptake, suppresses apoptosis, and drives cell proliferation.4 Sphingosines in resting state have proapoptotic and antiproliferative actions but when phosphorylated by lipid kinases they can promote cell growth and proliferation by a still poorly understood intracellular mechanisms.5
Understanding the balance and interconnection of the 2 pathways might provide novel opportunities to intervene and regulate proliferation and growth of tumor cells. However, the complexity, sometimes redundancy and high interconnection of lipid signaling pathways, brings the biggest challenge in targeting lipid kinases that is the development of class and isoform-specific inhibitors.6 –9 Inhibitors for class I PI3 kinases family such as wortmannin and LY294002 have been useful tools in delineating the role of these kinases in cellular responses but could not be used to discriminate between the action of distinct PI3 kinases.10 , 11 Similarly several inhibitors have been developed for sphingosine kinases (SPHKs) but none of them showed the desired specificity or selectivity.12 , 13 As more efforts are undertaken to look for potent lipid kinase inhibitors, there is a growing need for sensitive and high-throughput biochemical assays. However, high-throughput approaches commonly used for protein kinases, for example detection of substrate phosphorylation with phospho-specific antibodies, are not readily applicable to lipid kinases.
Traditional lipid kinase assays are based on monitoring phosphate transfer to lipids followed by lipid extraction and thin-layer chromatography (TLC) analysis.14 , 15 Radioactive assays are highly sensitive and reliable, but they are labor-intensive and not easily applicable for high-throughput screening (HTS) applications. To overcome those limitations, several assay formats have been developed based on the use of fluorescent or biotin-labeled synthetic lipid substrates. Biotinylated and fluorescently labeled sphingosine analogs have been used to measure activity of SPHKs.16 , 17 The activity of class I PI3 kinases has been successfully detected by measuring competitive binding of fluorescently labeled PIP3 probe and phosphorylated products to protein domains that have higher specificity for PIP3 vs. PIP2 lipids.18 , 19 However, this assay is restricted to class I PI3 kinases and not suitable for other classes of PI3 kinases or other lipid kinases.
Recently, several formats of fluorescence polarization and time-resolved fluorescence resonance energy transfer (TR-FRET)-based assays that are based on anti-ADP antibody detection have been introduced.20 The universality of such assays offer unique advantages; however, the assays depend on the ability of monoclonal antibodies to discriminate between ATP and ADP. Therefore, it is limited to low ATP concentrations (with a maximum limit of 100 µM) and it has low dynamic range.21
Here, we describe the implementation of the recently developed ADP-Glo™ technology as universal approach for detection of various lipid kinases. The formation of ADP produced by kinase reaction is detected using sensitive and robust bioluminescence approach. The method relies on the fact that the ATP remaining after completion of the kinase reaction is depleted during the first incubation step. In the second step, the ADP produced in the kinase reaction is converted to ATP and is measured using luciferase/luciferin reaction. The amount of light generated is proportional to the ADP produced and the activity of the kinase. The assay is homogeneous and has been formatted to a variety of multi-well density formats and thus it is suitable for HTS. ADP-Glo detects the formation of ADP, the common product generated in all kinase reactions, but without the use of antibodies thus overcoming the ATP limitations of the antibody-based ADP detection assay. Due to the universal nature of ADP-Glo assay, the assay can be rapidly adapted to diverse lipid substrates, show high tolerance to different lipid preparations, and can be performed with natural or synthetic lipid substrates. Using this approach, the activity of lipid and protein kinases can be determined in a single format, offering flexibility and universality required for small molecules characterization across the entire kinome.
MATERIALS AND METHODS
Materials
Purified recombinant lipid kinases PI3 kinase isoforms γ and δ (p120γ, p110δ/p85α), sphingosine kinase 1 (SPHK1), and sphingosine kinase 2 (SPHK2) were purchased from BPS Bioscience, Inc. (San Diego, CA). PI3 kinase isoforms α and β (p110α/p85α, p110β/p85α) were purchased from SignalChem (Richmond, BC, Canada). Protein kinase C (PKC) and its substrate neurogranin were obtained from Promega Corporation (Madison, WI).
Adapta™ Universal Kinase Assay was purchased from Invitrogen (Carlsbad, CA). PI3 kinase homogenous time resolved fluorescence (HTRFTM) assay was from Millipore (Billerica, MA). The ADP-Glo Kinase Assay kit was obtained from Promega Corporation (Madison, WI).
Lipid Preparations
To prepare large multilamellar vesicles of PI or PIP2 lipids, lipids purchased as dry cake were rehydrated by agitation in buffer L (50 mM HEPES (pH 7.5), 1 mM EGTA) at a concentration of 500 µM. To prepare liposomes with mixed lipid composition, lipids were mixed in organic solvent. PI or PIP2 lipids were pre-mixed with DOPS at 4:1 weight ratio in a glass vial and dried under nitrogen flow in a chemical hood. The dried lipids were rehydrated in buffer L at 500 µM PI or PIP2 concentration. Prepared lipids were tested directly or were subjected to sonication for making small unilamellar vesicles. The lipid suspension was sonicated 3 times at room temperature for 5 min in a Branson 2510 bath sonicator. Tip sonicator Branson Sonifier 450 was also tested for liposome preparation. Both sonication conditions gave similar results and were used interchangeably. Prepared lipid solutions were stored at 4°C and did not show any loss of functionality as substrates for at least 2 weeks.
Kinase Assay Conditions
Generally, PI3 kinase and SPHK reactions were performed in 5 µL reaction volume using 384-well low volume plates. The assays were carried out in reaction Buffer A [(40 mM Tris, pH 7.5, 20 mM MgCl2, and 0.1 mg/mL BSA) supplemented with 1 mM DTT] with 50–100 µM of lipid substrate and at apparent ATP K m values. The reactions were stopped and processed as indicated in the ADP-Glo manual. In brief, to stop the reaction 5 µL of ADP-Glo reagent was added and the plate was incubated at room temperature for 40 min. Then, 10 µL of Kinase Detection Reagent was added and luminescence output was read following 45–60 min incubation at room temperature. Luminescence output was recorded using Tecan infinite F500 reader. The data presented in the figures are shown as relative light units (RLU).
Kinase Activity Determination
The enzymatic activities of PI3 kinases and SPHKs were characterized by measuring kinase activity at variable enzyme concentrations and time. The reactions were carried out at saturated substrate concentrations (100 µM PI/PS) and ATP K m values shown in Table 1. ATP to ADP conversion curves were performed in parallel under the same reaction conditions as described in ADP-Glo manual. The conversion curves were used to determine the percent of ATP converted to ADP and to calculate the amount of ADP produced with each amount of enzyme. In addition, signal to background values (luminescence ratio in the presence and absence of an enzyme) were determined and were used to compare the activities of different lipid kinases.
Comparison of ADP-Glo With Other Kinase Activity Assays
The performance of the ADP-Glo assay was compared with 3 other assay formats: radiometric, antibody-based ADP detection assay (Adapta™ from Invitrogen), and PH domain-based HTRF detection assay (PI3 kinase HTRF™ assay from Millipore).
To compare ADP-Glo assay with standard radioactivity assay, the assays were run in 25 µL final volume for 10 min at 30°C with 100 µM PI/PS lipid substrate and 50 µM ATP. Where needed, 1 µCi of [γ-32P] ATP per reaction was added to the samples. For the ADP-Glo approach, the reactions were stopped with 25 µL of the ADP-Glo Reagent and processed as described in ADP-Glo manual. For the radiometric assay, the labeled radioactive phospholipids were extracted manually with 100 µL of 1 N HCl and 160 µL of 1:1 chloroform/methanol mixture. The organic and aqueous phases were separated by a 30-s centrifugation in a bench-top centrifuge. The aqueous phase was removed carefully. The organic phase was washed twice with artificial upper phase to ensure the complete removal of unreacted radioactive ATP. Then, 10 µL of the organic phase containing the extracted 32P–PIP3 lipids were mixed with scintillant cocktail and were counted on the Beckman LC6500 scintillation counter.
To compare the ADP-Glo assay with Adapta™ and PI3 kinase HTRF™ assay, the kinase assays were performed with the increasing amounts of enzyme in 10 µL reaction volume following protocols provided by the suppliers. For HTRF™ assay, the buffer and PIP2 substrate (10 µM final concentration) provided with the kit were used. Adapta™ assays were run in kinase buffer supplied with the kit. As substrate, the suggested PIP2:PS lipid preparation was purchased from Invitrogen and was used at the recommended 50 µM concentration. The ADP-Glo assay was performed in reaction buffer A with 1 mM DTT and 50 µM PIP2 substrate prepared as described above. Since the tracer reagent provided in the Adapta™ kit was not sufficient to run reactions at higher ATP (100 µM) concentrations, we compared the performance of the 3 technologies using 10 µM ATP. Following kinase reactions, all samples were processed and kinase activity was calculated as recommended in the provided protocols.
Apparent K m Determination
For the substrate K m determination, the ATP concentration was fixed at 100 µM, whereas the lipid substrate concentration was titrated from 0 to 250 µM. For ATP K m determination, the reaction was done with increasing amounts of ATP (0–500 µM) at a constant lipid substrate concentration of 100 µM in the presence or absence of an enzyme. The luminescence without the enzyme was used as a background control and ATP K m was calculated based on net relative luminescence units (background subtracted). The apparent ATP and substrate K m values were calculated by fitting the data to Michaelis–Menten equation using SigmaPlot 9.0 software.
Inhibitor IC50 Determination
To determine inhibitor IC50 values, the tested compounds were diluted serially and added to wells prior to all other reaction components. Then, kinases to be tested were added to the wells and the reactions were initiated by substrate/ATP addition. For PI3 kinase inhibitor screening experiments, reactions were carried out with 1 ng α, 2.5 ng β, 0.2 ng γ, and 1 ng δ isoforms at saturated substrate concentrations of PI/PS (100 µM) or PIP2 (50 µM) and 50 µM ATP. The ADP formation was determined using ADP-Glo assay. When determined IC50 values using ADP-Glo were compared to the values obtained using standard radioactivity approach, both assays were run simultaneously under identical reaction conditions except using [γ-32P]-ATP and radioactive lipids were extracted and analyzed as described below.
To compare the selectivity profile of known SPHK inhibitor
The percent inhibition was calculated relative to enzyme control without inhibitor. Data were plotted using the sigmoidal dose–response, variable slope model supplied with SigmaPlot 9.0 software.
Z′ Determination
To determine the robustness of the assay, Z′ factor was calculated using PI3 kinase α as an example. The kinase reaction was carried out in the presence and absence of 100 nM kinase inhibitor wortmannin. Z′ value was calculated as described previously.22
RESULTS
Lipid Substrate Optimization for PI3 Kinases
The source as well as the method of preparing lipid substrates are among the most important considerations in developing successful lipid kinase assays. Natural lipids assembled into liposomes are preferred form of lipid substrates since it best mimic the native presentation of substrate on cellular membranes.
The natural lipid substrate for class I PI3 kinases in vivo is PIP2. However, for in vitro assays, either PI or PIP2 can be phosphorylated by those kinases, and depending on assay formats, both forms of lipids have been used. For example, assays that detect formation of PIP3 using the GRP1-derived PH domain can only use PIP2 as lipid kinase substrate.18 However, traditional lipid kinase assays where kinase activity is measured by monitoring phosphate transfer to lipid via lipid extraction or irreversible immobilization to nitrocellulose membranes offer more flexibility. In those assays, both PI and PIP2 substrates can be utilized with PI often being a preferred one.23 , 24
To determine if ADP-Glo provides flexibility toward the selection of lipid substrates, 4 isoforms of class I PI3 kinases were assayed against a panel of phosphoinositides. Long-chain native lipid substrates PI and PIP2, since they are poorly soluble in aqueous solutions, were assembled into liposomes by sonication. Short-chain synthetic diC8PIP2 lipids were directly dissolved in aqueous solution. Also since phospholipid composition of the liposomes can have an effect on substrate utilization,14 , 25 phosphatidylserine and other phospholipids have been tested as carriers during liposome assemble.
The performance of lipid substrates was tested with all four class I PI3 kinases and the kinase activity was determined based on ADP production. The kinase activity with natural PIP2 lipid was set as 100%. As shown in Figure 1A, all tested substrates were phosphorylated by class I PI3 kinases, although with variable specificity. PI3 kinase α and β isoforms showed higher activity with natural long-chain PIP2 substrate, while PI was the preferred substrate for γ and δ isoforms. Addition of DOPS increased the rate of PI phosphorylation between 15% and 50% depending on the isoform. Interestingly, under our experimental conditions, addition of DOPS to PIP2 vesicles had no effect on the rate of PIP2 phosphorylation (data not shown). Short-chain PIP2 substrate was poorly recognized by PI3 kinase γ isoform (Fig. 1 light gray bars). It also showed lower activity for α and δ isoforms. However, unexpectedly diC8PIP2 substrate was well recognized and phosphorylated by the β isoform.
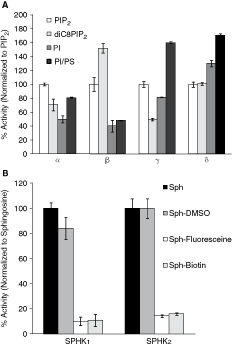
Comparison of substrate specificity for class I PI3 kinases and sphingosine kinases (SPHKs). (
These data clearly indicate that, in general, all four PI3 kinase isoforms prefer long-chain lipids and can efficiently utilize PI and PIP2 as lipid substrates in vitro. However, natural PI substrate is about 20-fold less expensive in comparison with PIP2 and can provide an important advantage for high-throughput applications. Therefore, we have chosen PI as a primary substrate for further assay characterization.
Lipid Substrate Optimization for Sphingosine Kinases
We also evaluated ADP-Glo assay to determine substrate specificity for another important class of lipid kinases, SPHKs. We compared the ability of purified recombinant SPHKs to phosphorylate natural substrate
Therefore, our results show that phosphorylation of commercially available natural and synthetic lipid substrates by 2 distinct lipid kinase families can be detected using the ADP-Glo assay. However, examining lipid substrate specificity and choosing the preferred lipid substrates is important in designing simple and robust in vitro lipid kinase assays and the ADP-Glo assay provides flexibility and compatibility needed for substrate evaluation and assay design.
Determination of Kinetic Parameters
To determine if the ADP-Glo assay can be used for characterizing kinetic parameters of lipid kinases, we first compared performance of the ADP-Glo with standard lipid extraction approach. To compare the performance of 2 assay formats, increasing amounts of PI3 kinase α isoform were incubated with PI substrate presented in liposomes with DOPS as a carrier. After incubation, the 32P-labeled lipids were extracted and the amount of phosphorylated lipids was determined by scintillation counting (Fig. 2A). The amount of ADP formed under the same conditions was determined using the ADP-Glo (Fig. 2B). As shown in Figure 2, both methods detected kinase activity with high sensitivity and linearity required for lipid kinase characterization. Using both methods, the linear response was observed between 0 and 40 ng in 25 µL reaction volume. Using 5 ng (0.9 nM) of PI3 kinases α, a 5-fold increase in signal above the background was obtained with the ADP-Glo assay in comparison with a 7-fold increase measured with radioactive approach validating the ADP-Glo assay as alternative approach to radiometric assays.
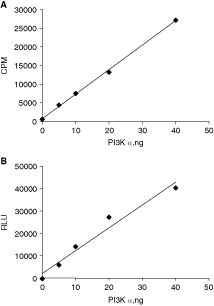
Enzyme titration of PI3 kinase α using radiometric and ADP-Glo assays. Enzyme reactions were carried out at 30°C for 10 min, and products were determined by measuring the formation of radioactive lipids (
We also examined the performance of the ADP-Glo assay in determining the kinetic parameters for PI3 and SPHKs. Since the ADP-Glo assay detects ADP formation, determining substrate K
m value is straightforward. As shown in Figure 3, the rate of ATP consumption by different isoforms of PI3 kinase increased with increasing concentrations of lipid substrate and the observed increase is commensurate with Michaelis–Menten kinetic profile. No ATP was consumed in the absence of lipid substrate, indicating that the substrate is required for PI3 kinase enzyme activity and class I PI3 kinases did not show any measurable ATPase activity under the experimental conditions used. The apparent K
m values for PI and PIP2 substrates are summarized in Table 1. Similar experiments were set up to determine substrate K
m values for SPHKs.
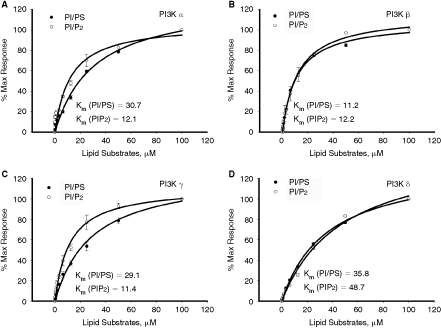
Determination of apparent substrate K
m. Assays were carried out at fixed ATP concentration (50 µM) while varying the amounts of lipid substrates. The K
m values were determined for 2 lipid substrates PI/DOPS or PIP2 for PI3 kinase α (
Determining apparent K m values for ATP using antibody-based ADP detection assays can be challenging since the measured activity is not linear at all concentrations of ATP. Thus, a standard curve of ATP concentrations needs to be established for each segment of ATP where the fluorescent output is linear.21 In contrast, ADP-Glo is linear up to 1 mM ATP as it is described in Zegzouti et al. 27 The apparent ATP K m values determined by ADP-Glo are summarized in Table 1 and are well within the K m ranges reported in the literature.28 –30
Assay Optimization
Optimizing reaction conditions to obtain robust and reliable signal with minimal amount of enzyme is an important step in designing high-throughput lipid kinase assays. ADP-Glo assay measures kinase activity by determining formation of ADP using luminescence approach. The sensitivity of ADP-Glo assay allows detection of 3%–5% of ATP conversion to ADP with signal to background higher than 5-fold. To determine minimal amount of class I PI3 kinases needed for setting up small molecule inhibitor screens, the enzyme titration curves were performed with all four class I PI3 kinase isoforms. Phosphorylation reactions containing varying enzyme concentrations were carried out using saturated amounts of lipid substrates and at K m values for ATP. As shown in Figure 4, luminescence signal increased with increasing amounts of enzyme indicating ADP formation and substrate phosphorylation and as expected, since the specific activity of different isoforms varies, the amount of enzyme required to obtain detectable signal varied depending on the isoform. For example, the use of 6.25 ng of α, β, γ, and δ isoforms resulted in 35%, 19%, 75%, and 32% of ATP conversion to ADP and showed signal to background of 43.7, 19, 81.7, and 31, respectively. Under our experimental conditions, PI3 kinase isoform β showed the lowest activity and 2.3 ng (2 nM) of enzyme was required to achieve 3.5% of ATP to ADP conversion with signal to background of 3.9-fold (Fig. 4B). On the other hand, 18% of ATP conversion rate was measured with only 0.19 ng (0.29 nM) of γ isoform with signal to background of 21.4.
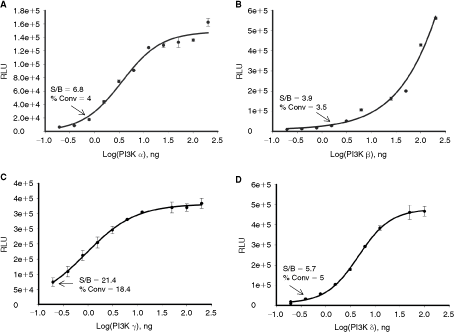
Generation of enzyme concentration curves for PI3 kinases. Enzyme dilutions of PI3 kinase α (
Comparison of ADP-Glo to Other Lipid Kinase Assays
Next, we tested how the sensitivity of ADP-Glo assay is compared to other lipid kinase approaches such as PI3 kinase HTRF™ assay and Adapta™ universal kinase assay. HTRF assay uses displacement approach to detect the PIP2 phosphorylation by class I PI3 kinases. Unlabeled PIP3 formed during kinase reaction displaces biotin–PIP3 from the energy transfer complex resulting in a loss of energy transfer and decrease in signal. Adapta™ assay is similar to ADP-Glo in principle where ADP formation is monitored but uses fluorescent-based immunoassays as detection method.
The assay comparison was done using PI3 kinase α as an example. All assays were performed using conditions recommended by the manufacturers. As shown in Figure 5, all 3 assay formats showed either an increase (Fig. 5C) or decrease (Fig. 5A and 5B) in signal with increasing PI3 kinases α enzyme concentrations indicating lipid phosphorylation by PI3 kinases α. However, the dynamic range and detection sensitivity varied depending on the method used. ADP-Glo assay showed wide dynamic range with signal to background ratios ranging from 2.9 with 0.4 ng of the enzyme to 41.6 with 12.5 ng. The signal to background for HTRF assay varied from 2.5 with 0.4 ng of enzyme to 14.5 with 12.5 ng. With Adapta™ assay, no signal increase above background was detected with 0.4 ng and only 3.6-fold increase was observed with 12.5 ng.
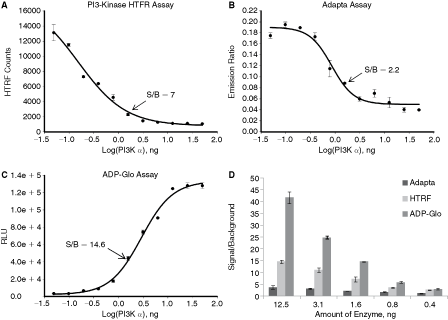
Comparison of ADP-Glo™ with other lipid kinase assay formats. Different kinase detection methods were compared using PI3 kinase α isoform. The reactions were carried out in 10 µL final volume with increasing amounts of enzyme for 60 min at room temperature. (
Thus, these data show that although the activity of class I PI3 kinases can be detected using all 3 methods, ADP-Glo has the widest dynamic range and allows the use of the least amount of enzyme required for setting up high-throughput screens.
ADP-Glo Assay of Sphingosine Kinases
To determine if ADP-Glo can measure activity of other lipid kinases with the same robustness and sensitivity as was shown for PI3 kinases, experiments were set up to measure activity of another important group of lipid kinases, SPHKs. As shown in Figure 6, the increase in ADP formation directly correlated with increasing amounts of SPHK1 and SPHK2 kinases. SPHK1 showed higher rate of substrate phosphorylation in comparison with SPHK2 and >8-fold signal to background was measured with only 0.5 ng of an enzyme. However, to achieve similar ATP conversion rate with SPHK2, about 3 times more enzyme was needed.
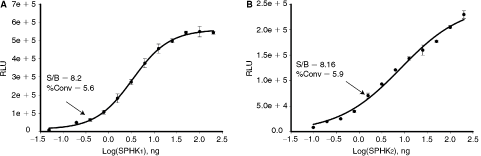
Generation of enzyme concentration curves for sphingosine kinases (SPHKs). Enzyme dilutions of SPHK1 (
Overall, the described data using 2 distinct groups of lipid kinases indicate that although optimal kinase concentrations required for measuring their activity using ADP-Glo might vary across lipid kinases depending on enzyme-specific activity, the activity of all tested kinases could be detected at low nM enzyme concentrations.
Assay Validation for Small Molecule Inhibitors
To further validate ADP-Glo assay for small molecule inhibitor screening and profiling, the performance of ADP-Glo assay was tested with known lipid kinase inhibitors. The amount of enzyme was chosen based on their ability to convert 5%–10% of ATP and the assays were performed with saturating amounts of lipid substrates and at ATP concentrations close to apparent K m values.
As shown in Figure 7A, LY294002, a known potent PI3 kinase inhibitor, results in inhibiting PI3 kinases α and γ isoforms in a dose-dependent manner; and the control compound LY303511 shows no inhibitory effect. Similarly, specific dose-dependent inhibition was also observed with β and δ isoforms (Table 2). In addition, the inhibitor IC50 values were similar for both natural PIP2 substrate and the alternative PI/PS. Using both lipids, IC50 values varied from 3 to 35 µM and is commensurate with their pharmacological values reported previously in the literature.14 , 18 , 20 The specific dose-dependent response was also obtained with another PI3 kinase class I inhibitor wortmannin (Fig. 7B). Also, it is worth mentioning that the profile of inhibitor response measured with ADP-Glo correlated well with data obtained using standard lipid extraction method (Fig. 7C). The wortmannin dose-dependent response curves for α and γ isoforms measured by 2 methods (ADP-Glo and radiometric) showed almost identical profile and thus validating that results obtained using ADP-Glo approach as an alternative to the gold standard radiometric assays.
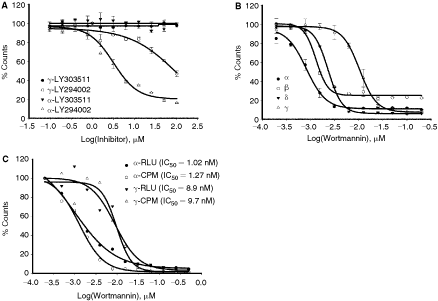
Determination of IC50 values for PI3 kinases. (
To demonstrate the universality of ADP-Glo assay for small molecules screening and profiling across entire kinome, the potency of known SPHK inhibitor
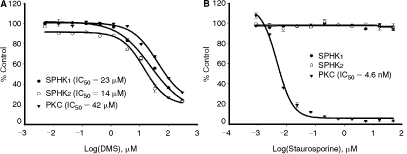
Determination of inhibitor’s IC50 values for sphingosine kinases (SPHKs) and protein kinase C. SPHK1, SPHK2, and protein kinase C (PKC) activities were determined in the presence of increasing amounts of
Robustness of ADP-Glo Assay
Finally, we tested the acceptability of ADP-Glo assay for lipid kinase high-throughput application by determining Z′ value. The Z′ factor is a standard parameter for evaluating the robustness of an assay and its suitability for HTS applications.22 The data presented in Figure 9 were generated using 0.5 ng of PI3 kinase α isoform. Under the experimental conditions used, 8% of ATP was converted to ADP with signal to background ratio of 5.5 and yielded a Z′ factor of >0.7. Since Z′ factor higher than 0.5 is considered suitable for HTS, based on our data 200 ng of PI3 kinase α should be sufficient to assay a 384-well plate for inhibitor screening using ADP-Glo as a read-out system.
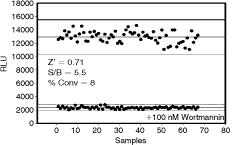
Z′ factor determination for PI3 kinase α. Reactions were carried out in 5 µL reaction for 60 min with 0.5 ng of enzyme at ATP K m values. Reactions with tested compound 100 nM wortmannin were compared to control reactions with enzyme only. Abbreviation: RLU, relative light unit.
DISCUSSION
Many of biologically relevant lipids in cells are present in hydrophobic membrane environment. Those lipids are insoluble in aqueous solutions and aggregate into macromolecular structures such as micelles or bilayer. Such aggregates, therefore, have unique biophysical properties and surface chemistries, which have important consequences for developing in vitro lipid kinase assays. Here, we demonstrate that ADP-Glo assay is compatible with diverse lipid preparations including lipids present in liposome form.
We demonstrate that ADP-Glo uses both natural and synthetic lipid substrates similar to radiometric method or assays based on irreversible binding of phosphorylated lipids to nitrocellulose membranes.23 , 24 , 30 The ability to use natural lipids provides more flexibility and is less costly. The data obtained with ADP-Glo assays are comparable to radioactive assays.24 However, in contrast to radiometric method, ADP-Glo is homogeneous, requires little hands-on time for assay optimization, is amenable to automation, and is not radioactive.
Since ADP-Glo assay eliminates the need for substrate modification that is required for non-radioactive assays such as scintillation proximity (SPA), α screen or TR-FRET18 , 30-based assays, it offers universal approach for all lipid kinases. Assays based on modified substrates are restricted to enzymes using the same substrate. For example, assays based on PIP3-binding pleckstrin homology domains18 , 33 can be applied only for class I PI3 kinases but are not suitable for other PI3 kinase classes that use PI or lipids other than PIP2 as substrates. In addition, replacement assays based on fluorescently labeled lipids are not applicable to other lipid kinases such as SPHKs supporting the need for universal lipid kinase assay.
The fluorescent antibody-based ADP assays offer universal approach, however due to cross reactivity of the anti-ADP antibody with ATP at high concentrations of ATP limits its utility to low concentrations of ATP (with a maximum of 100 µM ATP).21 In addition, nonlinear assay response and a narrow dynamic range makes assay development process more complex, in particular, using diverse groups of lipid substrates or assaying kinases with varying specific activities.
The universality of ADP-Glo assay offers unique advantages for lipid kinases characterization; however, to ensure maximum assay performance, high quality of purified kinases is required. The contamination of kinase preparations with ATPases or other ADP-producing enzymes can result in higher background that can be easily assessed using kinase inhibitors. Another important consideration related to the nature of detection reactions is the interference from screening compounds. However, to address this issue, it is noteworthy that ADP-Glo assay is based on the same detection chemistry as kinase-Glo whose performance has been tested in various HTS screens with minimal false hits rates.34 Moreover, the robustness of the ADP-Glo assay toward pharmacologically active compounds has been evaluated during assay development and is discussed by Zegzouti et al. 27 Since all of those approaches showed minimal compound interference with ADP-Glo detection system, the obtained results should be also applicable for lipid kinase assays.
In conclusion, ADP-Glo assay monitors kinase reactions using broad range of ATP concentration (up to 1 mM) with a high dynamic range and excellent Z′ value. Because of its high sensitivity, it also requires low amount of enzymes. As we presented here, all tested lipid kinases showed robust response at low nM range of enzymes. In addition, the universality of this assay provides excellent platform to profile compounds against a broad range of kinases without the need to use complementary technology or other kinase assay formats. Thus, ADP-Glo is an ideal alternative platform to radiometric method for all lipid kinases without the exposure to radioactivity hazards, homogeneous, and miniaturization to high-density plate formats.
AUTHOR DISCLOSURE STATEMENT
J.V., H.Z., and S.A.G. are employees of Promega Corporation. S.A.G. is also an employee of the University of Wisconsin School of Medicine and Public Health.