
Abstract
ADP-Glo™ is a novel bioluminescent, homogeneous assay for monitoring ADP producing biochemical reactions and thus it is an ideal assay for detecting enzyme activity using a wide variety of substrates. It is a universal assay that can be used with protein kinases, lipid kinases, sugar kinases, and many more kinases as well as ATPases. Because of its high sensitivity, it is suitable for monitoring enzyme activities at very early substrate conversions requiring very low amount of enzymes. Furthermore, as the assay is applicable to a broad range of ATP and substrate concentrations, it is optimal for enzymes that require high ATP and substrate concentrations. This is critical since inhibitor potency has to be demonstrated at the cellular level where ATP is present at millimolar concentrations. ADP-Glo is performed in 2 steps upon completion of kinase reaction: a combined termination of kinase reaction and depletion of remaining ATP in the first step, and conversion of generated ADP to ATP and the newly produced ATP to light output using luciferase/luciferin reaction in the second step. The luminescent signal generated is proportional to the ADP concentration produced and is correlated with the kinase activity. Due to its high signal to background and luminescent readout, this assay is less susceptible to generation of false hits and thus it is applicable to not only primary and secondary screening but also kinase profiling.
INTRODUCTION
Kinases encompass a wide range of enzymes that utilize diverse substrate chemistry such as proteins, lipids, or sugars.1 , 2 The phosphorylation by these kinases results in alteration of cellular signaling pathways leading to changes in cellular phenotypes. As the role of kinases in regulating normal and abnormal cell growth became well appreciated, kinases were recognized as ideal drug target candidates. Over 30 distinct kinases have been validated as drug targets and for the development of drug candidates in multiple phases of clinical trials.3 –5 The successes of kinase inhibitors in treatment of diverse forms of cancer have added momentum to exploration of more kinase activity modulators as drug candidates. The search for such drugs starts with identifying selective and potent enzyme inhibitors that have minimal effect on other enzymes and less toxicity. To achieve this goal, a kinase assay that is homogeneous, robust, and sensitive is essential. The assay should also be applicable to all kinases regardless of the chemical structure of the substrate and be flexible to be performed under a wide range of assay conditions. Furthermore, it is desirable to have one platform technology that can be used for compound screening, hit and lead identification, and enzyme profiling during the various stages of drug discovery prior to clinical investigations. Although current assays span a broad range of technologies,6 , 7 there is no one technology that provides or meet all the demands required. Thus, it is necessary to combine several formats to address all requirements.
The newly developed bioluminescent kinase assay is homogeneous, high-throughput formatted, robust, and covers a broad range of ATP and substrate concentrations. It is sensitive to 0.1 pmol of ADP and shows a significant signal to background at very early stages of substrate conversions. The assay has been tested at various plate densities and requires very low amount of enzymes. This assay can be used in early stages of primary screening, and since it is universal to any enzyme/substrate combinations, it is an ideal assay for kinase profiling.
MATERIALS AND METHODS
Kinases and Substrates
PI3 Kinase (p120γ) and sphingosine kinase 1 (SPHK1) were purchased from BPS Bioscience, Inc. (San Diego, CA). EGFR, VEGFR2, AKT2, IKKβ, ERK2, and PDGFRα were purchased from Invitrogen Corp. (Madison, WI). cAMP-dependent protein kinase catalytic subunit (PKA) and DNA-dependent protein kinase (DNA-PK) were obtained from Promega Corporation (Madison, WI). Glucokinase was obtained from Sigma-Aldrich (St. Louis, MO).
IKKtide peptide substrate and sphingosine substrate were purchased from ENZO Life Sciences International Inc. (Plymouth Meeting, PA). Crosstide peptide substrate was purchased from EMD Calbiochem (Gibbstown, NJ). Poly(Glu4, Tyr1) substrate was purchased from Sigma-Aldrich. Kemptide (PKA) peptide substrate and DNA-dependent protein kinase peptide substrate were obtained from Promega Corporation. The lipids phosphatidylinositol (PI) and phosphatidylserine (PS) were obtained from Avanti Polar lipids, Inc. (Alabaster, AL). Myelin basic protein (MBP) substrate was purchased from Millipore (Billerica, MA).
Chemicals and Assay Components
Staurosporine was purchased from LC Laboratories (Woburn, MA). H-89 and dimethylsulfoxide were purchased from Sigma-Aldrich. PD158780 was purchased from EMD Calbiochem. PKI and U0126 were obtained from Promega Corporation. White 96- or 384-well assay plates (Catalog # 3912 and 3673, respectively) were obtained from Corning Inc. (Corning NY).
The ADP-Glo™ kinase assay kit from Promega Corporation is composed of ADP-Glo reagent, kinase detection reagent (made by mixing kinase detection buffer with a lyophilized kinase detection substrate), Ultra Pure ATP and ADP. The Adapta™ kinase assay from Invitrogen contains Alexa Fluor® 647 ADP Tracer, Eu-anti-ADP antibody, ADP, and ATP.
ATP to ADP Conversion Curves
ATP to ADP standard curves were prepared in the kinase buffer to assess the linearity of the assay and during enzyme titrations in order to calculate the amount of ADP produced from each amount of enzyme used. The 1 mM ATP and ADP concentration range was created first by mixing proportionally an amount of 1 mM ADP and 1 mM ATP to always reach a concentration of 1 mM total nucleotides. For example, to form a stock solution of 100 µL of each of the 12 points of the 1 mM curve from 0% to 100% conversion, 100 µL ATP was used to form 0%, 1 µL ADP mixed with 99 µL ATP formed 1%, until the last one that contains 0 µL ATP and 100 µL ADP and corresponds to 100% conversion. The lowest concentration ranges described or used in this study were diluted in the corresponding kinase buffer from the 1 mM range to form the desired conversion curve range. Once the conversion curve samples are prepared, 5 µL of each of the 12 points was transferred to the assay plate and ADP-Glo assay reagents were added as follows: 5 µL ADP-Glo reagent was added then the plate was mixed for 30 s and incubated at room temperature for 40 min. Then, 10 µL kinase detection reagent was added and the plate was mixed again before incubating it for a minimum of 60 min to allow the highest concentrations of ADP to fully convert to ATP and to be used by luciferase/luciferin reaction. Luminescence is recorded.
Z′-Factor Determination
The Z′ factor values were determined according to published equation for 10, 100, and 500 µM ADP/ATP ranges using ADP-Glo assay.8 Forty-eight replicates of each of 5%, 10%, and 20% ADP in ADP/ATP mixtures were compared to 0% conversion replicates containing 10, 100, or 500 µM ATP. ADP-Glo component addition was as described above.
Kinase Assay Conditions
Generally, all kinase reactions were performed in Reaction Buffer A (40 mM Tris, pH 7.5, 20 mM MgCl2, and 0.1 mg/mL BSA) except the following: tyrosine kinase buffer for EGFR, VEGFR2, and PDGFRα and IKK buffer contained Buffer A supplemented with 100 µM sodium vanadate, 2 mM MnCl2 and 2 mM DTT; ERK2 buffer is Buffer A supplemented with 1 mM EGTA, 200 µM DTT, 200 µM sodium vanadate, and 5 mM β-glycerophosphate; Akt2 buffer contains Buffer A supplemented with 1 mM DTT; DNA-PK reaction was performed in 1× DNA-PK buffer (50 mM HEPES (KOH, pH 7.5), 100 mM KCl, 10 mM MgCl2, 0.2 mM EGTA, 0.1 mM EDTA, 1 mM DTT, 0.1 mg/mL BSA) supplemented with 100 µg calf thymus DNA for activation; and sphingosine kinase buffer was composed of Buffer A and 0.5 mM DTT.
All kinase reactions were performed at room temperature in 5 µL volume except when noted and were performed as follows. EGFR, VEGFR2, and PDGFRβ reactions contained 0.4 µg/µL poly(Glu4, Tyr1) substrate and 10, 100, and 100 µM ATP, respectively, and incubated for 60 min. DNA-PK reaction contained 2 mg/mL peptide substrate and 250 µM ATP for 60 min. IKKβ reaction contained 200 µM IKKtide substrate and 1 µM ATP for 60 min. Akt2 reaction contained 50 µM crosstide substrate and 300 µM ATP for 60 min. ERK2 reaction contained 1 µg/µL MBP substrate and 100 µM ATP for 120 min. PKA reaction contained 50 µM kemptide substrate and 10 µM ATP for 5 min. Hexokinase reaction contained 100 µM glucose and 100 µM ATP for 30 min. PI3 Kinase reaction contained 200 µg/mL phosphatidylinositol and 50 µg/mL phosphatidylserine and 10 µM ATP for 60 min. Sphingosine kinase reaction contained 200 µM sphingosine and 100 µM ATP for 60 min. For determining the K m for ATP and substrate in PKA assay, the reaction was performed in 25 µL and was incubated at room temperature for 20 min. For ATP titration, the reaction contained 0.2 U of PKA, 100 µM kemptide, and a serial dilution of ATP and for substrate titration, 0.5 U of PKA, 100 µM ATP, and a serial dilution of kemptide were used.
Inhibitor Studies Using ADP-Glo Assay
When the effect of different inhibitors on PKA kinase was monitored, reactions were performed at the same time in 25 µL mixture containing 1× Buffer A, 1% DMSO, 10 µM ATP, 50 µM kemptide, 0.5 U PKA, and serial dilution of the inhibitors. The reaction was incubated at room temperature for 5 min. For the determination of H-89 inhibitor mode of action, reactions were performed in 25 µL mixture containing 1× Buffer A; 1% DMSO; 10, 100, or 1,000 µM ATP; 50, 50, or 100 µM kemptide, respectively; 0.05, 0.05, or 0.1U PKA, respectively; and serial dilution of the H-89 inhibitor. The reactions were incubated at room temperature for 10 min for the 10 and 100 µM ATP reactions or 30 min for the 1,000 µM ATP reaction. For the determination of PKI inhibitor mode of action, the reactions were performed in 25 µL mixture containing 1× Buffer A; 1% DMSO; 10, 100, or 1,000 µM ATP; 50, 50, or 100 µM kemptide, respectively; 0.05, 0.1, or 0.1 U PKA, respectively; and serial dilution of the PKI inhibitor. The reactions were incubated at room temperature for 10, 15, or 30 min, respectively. After the indicated incubation times, 25 µL ADP-Glo reagent was added to the reactions and the plate was incubated at room temperature for 40 min. Then, 50 µL of kinase detection reagent was added and after an incubation time of 40 min, luminescence was recorded and IC50 values were determined.
Comparison Between ADP-Glo and Adapta Assays
To compare the performance of ADP-Glo assay to another ADP detection assay, Adapta™ from Invitrogen, EGFR kinase, a moderately active enzyme was chosen. Both assays were performed with the same conditions and at the same time to minimize variability. All the kinase reactions were performed in 8 µL using tyrosine kinase buffer with 0.4 µg/µL poly(Glu4, Tyr1) substrate, 20 µM ATP, and incubated at room temperature for 60 min. For both assays, during the enzyme titration, an ATP to ADP conversion curve for the 20 µM ATP/ADP range was performed at the same time in the same plate to calculate the percentage of conversion generated by each enzyme amount. When PD158780 inhibitor was titrated, EGFR amounts described in the figures were used and DMSO concentration was kept at 1% in all inhibitor concentrations.
To perform the Adapta assay according to the supplier protocol, upon completion of kinase reaction, 4 µL of a mixture containing 6 nM Adapta Eu-anti-ADP antibody, 30 mM EDTA, and 240 nM Alexa Fluor 647 ADP tracer was added and after 30 min incubation time, the emission ratio of 665/620 nm was calculated. For ADP-Glo assay, 8 µL ADP-Glo reagent was added to the completed kinase reaction and the plate was incubated at room temperature for 40 min. Then, 16 µL of kinase detection reagent was added and after an incubation time of 30 min, the luminescence was recorded.
Signal Detection and Data Analysis
All 96-well assay plates were read using a GloMax® 96 Microplate Luminometer from Promega. The instrument was set to 0.5 s integration time. The same setting was used for 384-well plates and luminescence was read in the Infinite F500 instrument from Tecan Ltd. (Mannedorf, Switzerland). When Adapta assay was used, the fluorescence emissions were recorded on the Pherastar plate reader from BMG LABTECH (Offenburg, Germany). To plot, analyze the data and calculate all kinase reaction biochemical values, both Microsoft Excel and Prism from GraphPad Software (La Jolla, CA) were used.
RESULTS
ADP-Glo Assay Principle and Assay Formats
ADP-Glo kinase assay is performed in 2 stages after the completion of kinase reaction; removal of remaining ATP and conversion of enzyme reaction product ADP to ATP combined with luciferase/luciferin reaction (Fig. 1). The kinase reaction can be carried out at room temperature or any other desired reaction condition. The assay encompasses 2-step additions after completion of the kinase reaction. Upon completion of the kinase reaction, a reagent (ADP-Glo reagent) is added that simultaneously terminates the enzyme reaction and removes remaining ATP. After 40-min incubation, additional reagent (kinase detection reagent) is added that converts ADP to ATP and simultaneously converts the generated ATP into light using luciferin/luciferase reaction. The amount of light generated is proportional to the ADP produced and the activity of the kinase. We have optimized the time required for depletion of ATP with ADP-Glo reagent so that all of remaining ATP is completely depleted resulting in a very low background (results not shown). Because of the broad range of ATP concentrations used in kinase assays, we found that 40 min for the ATP depletion step is sufficient to meet this requirement up to 1 mM ATP. We also optimized the second step for converting ADP to ATP and conversion of ATP into light for a wide range of enzyme activity and found that an average of 40 min is also sufficient to meet these requirements (data not shown). However, to convert 100% of 0.5–1 mM ADP requires an incubation of 60 min. Optimal ADP-Glo performance requires the addition of equal volume of ADP-Glo reagent to the kinase reaction and twice the volume of kinase detection reagent. The assay formats for various plate densities is shown in Table 1. Thus, volume ratios of 25:25:50 µL or 50:50:100 µL of kinase reaction:ADP-Glo reagent:kinase detection reagent have been used in 96-well plates. Similarly, volume ratios of 5:5:10 µL and 10:10:20 µL of kinase reaction: ADP-Glo reagent:kinase detection reagent have been used in 384-well plates. Lower volumes 2.5:2.5:5 µL of kinase reaction:ADP-Glo reagent:kinase detection reagent have been also used successfully in low volumes 384-well plates and 1,536-well plates.
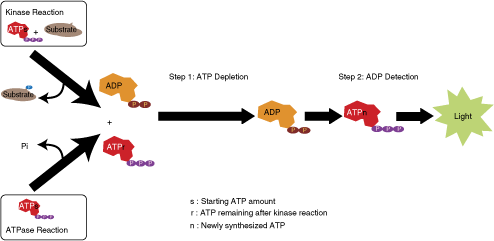
ADP-Glo™ assay principle. The assay is composed of 2 steps: after the kinase or ATPase reaction, a first step is performed with addition of a reagent that terminates the kinase reaction and depletes the remaining ATP, and then a second reagent is added to convert ADP to ATP and allow the newly synthesized ATP to be measured using a luciferase/luciferin reaction. The light generated is proportional to ADP present and kinase or ATPase activity.
ADP-Glo Sensitivity and ADP Linearity
To determine the limit of detection of ADP produced and linear range of ATP concentrations that can be used in kinase reactions, we simulated kinase reactions by using a mixture of ADP and ATP to give a total concentration of ADP+ATP that equals to that used in the kinase reaction. Thus, for example, for a kinase reaction that was carried out in a 100 µM ATP, we constructed a 100 µM ATP–ADP conversion curve from 0% to 100% conversion where for example 0%, 1%, 2%, 5%, 10%, 20%, and 40% correspond to 0 µM ADP:100 µM ATP, 1 µM ADP:99 µM ATP, 2 µM ADP:98 µM ATP, 5 µM ADP:95 µM ATP, 10 µM ADP:90 µM ATP, 20 µM ADP:80 µM ATP, and 40 µM ADP:60 µM ATP, respectively. Then equal volumes of ADP-Glo reagent was added to each of these mixtures and incubated for 40 min at room temperature and then twice as much volume of kinase detection reagent was added and incubated for additional 40 min. Luminescence was then read and relative luminescence units (RLU) were plotted vs. the percentage of ADP in the mixture. Figure 2 shows the standard curves generated for 1 µM, 10 µM, 100 µM, and 1 mM total concentration of ATP–ADP in simulated reactions, and the signal-to-background ratios (SB) resulting from such mixtures of ADP and ATP at various concentration ranges are shown in Table 2. The assay is capable of detecting low ATP conversion down to 1% as the ratio of signal to background show 1.4, 2.1, 2.9, and 2.4 for 1, 10, 100, and 1 mM ATP concentration ratios, respectively. For 5% conversion, these ratios increase to 3.2, 7, 10.4, and 7.5 for 1 µM, 10 µM, 100 µM, and 1 mM ATP concentrations, respectively. During the development of this assay, we had to ensure complete depletion of remaining ATP upon completion of the kinase reaction. We have successfully achieved this goal by using the conditions described in Materials and Methods section. We found that the background level remaining is proportional to the amount of ATP used and it is estimated to be in the range of 0.5%–1% of the initial amount. Although there is always a residual light background generated by ADP-Glo assay, it is evident that signal to background is significant even at low ATP conversion. Higher SB ratios are observed for high ATP conversions (10%, 20%, etc.). The assay is sensitive to low ATP conversion and the signal readout (RLU) is linear with ADP present at various ATP–ADP concentration ranges as shown in Figure 2. We found that using this assay we can detect as low as 0.1 pmol of ADP generated in the enzyme reaction (Table 2), a sensitivity that is only matched by the radiometric assays using radioactive [γ-32P] ATP.
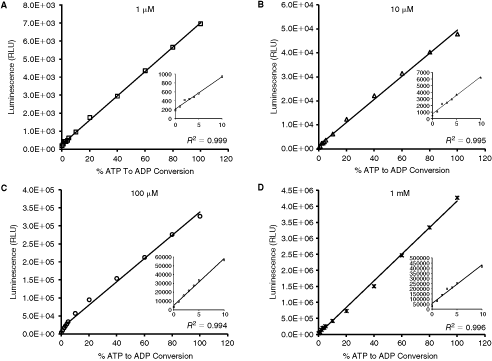
Linearity of the ADP-Glo™ kinase assay. ATP to ADP conversion curves were prepared at the indicated ATP+ADP concentration ranges in 5 µL of 1× Reaction Buffer A in a solid white 384-well plate. ADP-Glo kinase assay was performed using 5 µL of ADP-Glo reagent and 10 µL of kinase detection reagent at room temperature as described in Materials and Methods section. Luminescence values represent the mean of 2 replicates. Abbreviation: RLU, relative light unit.
The data in Table 3 show that this assay is robust as is indicated by the high values of Z′ at various ATP concentrations. Obtaining a Z′ value of over 0.6 at low ATP conversions with various ATP concentrations validates the use of this assay for high-throughput screening (HTS), where a good Z′ and high dynamic range are essential to perform a robust HTS of chemical libraries.
Universality of the ADP-Glo Assay
Because ADP-Glo detects ADP production, it is capable of monitoring enzyme activity for a wide range of enzymes and is not limited to classical protein kinases but also includes lipid kinases, and sugar kinases. Furthermore, many ATPases such as Na+/K+ ATPases, or proteins with ATPases activity such as heat shock proteins, helicases, topoisomerases, and ABC transporters can also be monitored using this assay (data not shown).
As shown in Figure 3, the assay was used to monitor protein tyrosine kinases using peptide substrate (EGFR), serine/threonine protein kinase using protein substrate (MAPK), and peptide substrate (DNA-PK). We also show that substrates other than peptides or proteins can be used with this assay such as lipids (PI3 kinase), alcohols (sphingosine kinases), and sugars (glucokinase). This demonstrates the universality of the assay and its capability in monitoring any enzyme reaction that produces ADP regardless of the chemical structure of the substrate. As a corollary, this assay offers a very important advantage as compared to any other nonradioactive assay, it is not necessary to modify or label either the substrate or the reagent needed to capture or monitor the product. These are not required for ADP-Glo and thus it saves not only time to make customized substrates required but also money for special ordering of such substrates. In many kinase reactions such as novel kinases where the substrate is not known, this feature of ADP-Glo makes it very attractive to monitor either the kinase activity that is mediated by hydrolysis of ATP, or by monitoring autophosphorylation of the enzymes. These features are not offered by nonradioactive-based assays that are dependent on substrate labeling. Also, at many times, labeling of large size substrates makes it difficult if not impractical to develop an assay that provides high dynamic range while maintaining robustness. This feature makes it feasible to use native substrates for the tested enzymes without taking a shortcut by using artificially generated peptides. In addition, the current nonradioactive assays that are based on modified product detection use antibodies to the phosphorylated product that precludes the use of any substrate that is already phosphorylated, a situation that arises when using enzymes such as glycogen synthase kinase 3 (GSK3) and casein kinase 1 (CK-1). These enzymes require substrates previously phosphorylated by other kinases for the addition of phosphate to new phosphorylation sites.
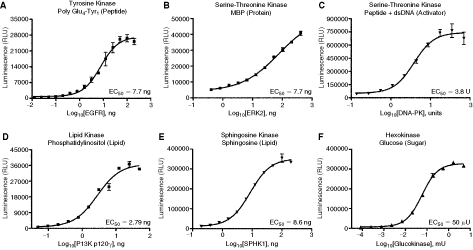
Universality of the ADP-Glo™ kinase assay. Titration of the enzymes was performed in 5 µL using ADP-Glo assay as described in Materials and Methods section. (
Determination of Kinetic Parameters of Kinases
Because ADP-Glo can be used with a wide range of ATP and substrate concentrations, it is possible to determine kinetic parameters for any enzyme. Thus higher concentration of ATP for high ATP requiring kinases and high substrate concentrations, for example proteins, peptides, lipids, sugars, can be easily used with ADP-Glo assay. The results shown in Figure 4 are representative of data generated with the catalytic subunit of cAMP-dependent protein kinase (PKA) at broad range of ATP concentrations to determine the K m for ATP and also using a broad range of peptide substrate concentrations to determine K m value for kemptide substrate. These results show that K m of 24.7 µM for ATP and a K m of 15.3 µM for kemptide substrate for PKA are in the range of the values reported in the literature.9 –11
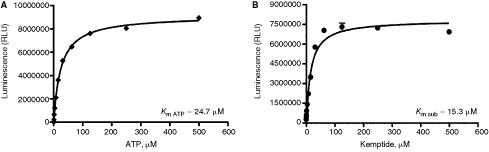
Determination of K
m values for peptide substrate and ATP. To determine the K
m for ATP and substrate in protein kinase catalytic subunit (PKA) assay, the reactions were performed in 25 µL and were incubated at room temperature for 20 min. (
Inhibitor selectivity of kinases was also tested using ADP-Glo and the catalytic subunit of PKA as a representative example. As shown in Figure 5, H-89, a known potent ATP competitive inhibitor of PKA inhibited the enzyme activity with an IC50 of 68 nM at 10 µM ATP. This value concurs with the values reported in the literature.12 Testing staurosporine, a promiscuous ATP competitive inhibitor of many kinases, on inhibition of PKA showed an IC50 of 13.76 nM at 10 µM ATP.13 When we tested the peptide inhibitor of PKA (PKI) that is known to be a non-ATP competitive inhibitor of the enzyme, we obtained an IC50 of 7.34 nM that is similar to what is reported in the literature.14 It is noteworthy that U0126, a known inhibitor of MEK/MAPK pathway15 but is not known to inhibit PKA, did not inhibit PKA till we used micromolar concentrations of the inhibitor (IC50 of 8.45 µM). Thus, ADP-Glo can distinguish between known inhibitors of kinases and discriminate against those that do not inhibit the enzyme.
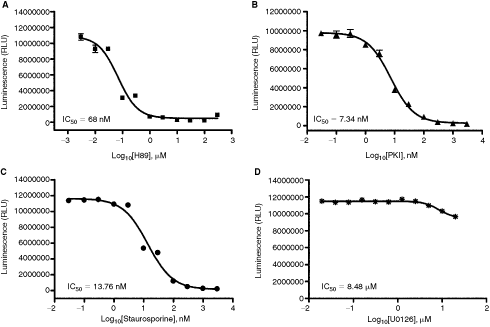
Determination of inhibitors selectivity using ADP-Glo™ assay. protein kinase catalytic subunit (PKA) kinase assays were performed in 25 µL in the presence of serial dilution of a PKA-specific ATP competitive inhibitor H-89 (
Because the assay is tolerant to a very high concentrations of ATP (up to 1 mM), it is suited to differentiate between the mechanism of inhibition by ATP competitive and noncompetitive inhibitors of kinases. To test this we used H-89, a known ATP competitive inhibitor of PKA and a peptide inhibitor (PKI) that is known to be an ATP-non-competitive inhibitor of the enzyme. Testing the inhibitor concentrations at 3 ATP concentrations ranging from 10 µM to 1 mM, we show that increasing ATP concentrations from 10 µM to 100 µM and to 1 mM increased the IC50 value for H-89 from 70 nM to 296 nM and to 2,694 nM, respectively, confirming the ATP competitive nature of this inhibitor (Fig. 6A–6C). In contrast, increasing ATP concentrations from 10 µM to 100 µM and to 1 mM showed similar IC50 values of 5, 4.8, and 7 nM, respectively, confirming that PKI is an ATP noncompetitive inhibitor of PKA. Thus, because of the flexibility of ADP-Glo in using any concentration of ATP up to 1 mM we were able to distinguish between ATP competitive and noncompetitive inhibitors of PKA.
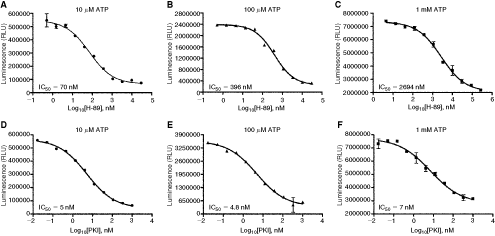
Determination of the mode of action (MOA) of protein kinase catalytic subunit (PKA) inhibitors. PKA kinase assays were performed in 25 µL in the presence of serial dilution of a PKA-specific inhibitors H-89 or PKI in the presence of 10 µM ATP (
Comparison With Other ADP Detection–Based Assays
Few other assays are commercially available that monitor ADP production using fluorescent detection. These assays gained recognition among scientists who are working on kinases and ATPases and interested in a universal platform so it will be convenient to monitor the activity of these enzymes without much effort on designing the substrate and also for enzymes that are not classical protein kinases such as lipids, and sugar kinases. One of these assays that is an anti-ADP antibody-based and has been used by several investigators is the Transcreener™ ADP assay from Bellbrook Labs (Madison, WI), which is also called Adapta by Invitrogen or HTRF Transcreener ADP assay by CisBio (Bagnols-Ceze, France). The Adapta assay is based on time-resolved fluorescence resonance energy transfer (TR-FRET) formats where ADP is conjugated to a fluorescent compound (ADP Tracer) and anti-ADP-antibody is linked to another fluorescent compound. The generation of ADP during enzyme reaction displaces the ADP tracer from the antibody and thus results in decrease in TR-FRET and hence it is a negative response assay (for detail see ref.16 –18). Because of the popularity of this assay, it was of interest to compare results obtained with ADP-Glo that is bioluminescent vs. Adapta that is a fluorescent-based assay. When we tested the tyrosine kinase activity of EGFR, we got a maximum-fold increase in signal of 3.6 using Adapta for the amount of enzyme required for half completion of the enzyme reaction while ADP-Glo gave a fold increase in signal of 29 for the same amount of enzyme using the same reaction conditions. In addition, the conversion of ATP to ADP is only linear in the first 10% substrate conversion with a maximum of 3- to 4-fold change in signal while that of ADP-Glo is linear till all ATP has been consumed with an increase in signal that is proportional to the percentage of ATP conversion. Thus ADP-Glo in the example given in Figure 7 gave an increase in signal of ∼55-fold for 30% conversion while that of Adapta gave only a maximum of 5. Thus the dynamic range of ADP-Glo is significantly higher than that of Adapta by at least a factor 10. In addition, the linear relationship between ATP conversion and RLU in ADP-Glo assay and its positive response makes it ideal for carrying out kinase assays at high sensitivity using low amount of enzymes and shorter enzyme reaction time. Table 4 shows a subset of over 100 enzymes tested with ADP-Glo where a signal-to-background ratio of 5 was generally produced by a low enzyme amount (SB5). This is important for determining IC50 for highly potent inhibitors using low amount of enzyme. As shown in Figure 8, it is difficult when using Adapta assay to determine IC50 for the EGFR inhibitor PD158780 using low amounts of EGFR (1.77 and 3.75 ng) while that was easily determined (IC50 of 0.93 nM and 0.77 nM) using ADP-Glo assay and the same amount of enzyme. Even increasing the amount of enzyme up to 50 ng, it was hardly possible to get a change in the activity profile and no accurate IC50 determination could be assessed using Adapta. Thus, due to the low dynamic range and the low SB, it is difficult to get an IC50 values accurately for potent inhibitors with Adapta unless large amount of enzyme is used.
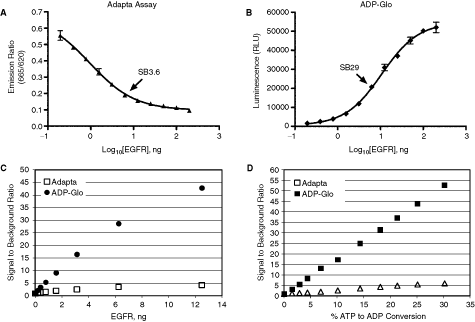
Comparison of the sensitivity of ADP-Glo™ and Adapta assay. For both assays, titration of EGFR Kinase was performed in 8 µL using tyrosine kinase buffer with 0.4 µg/µL poly(Glu4, Tyr1) substrate, 20 µM ATP, and incubated at room temperature for 60 min. (
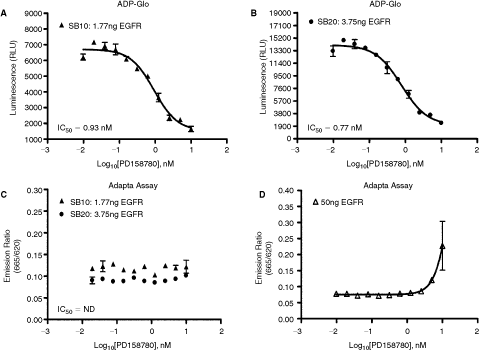
Comparison of ADP-Glo™ and Adapta assays in generating inhibitor dose response using small amount of enzyme. A dose response of the PD158780 inhibitor in EGFR kinase reaction was performed in 8 µL using tyrosine kinase buffer with 0.4 µg/µL poly(Glu4, Tyr1) substrate, 20 µM ATP, serial dilution of the inhibitor, and incubated at room temperature for 60 min. The effect of the inhibitor on enzyme activity was detected with ADP-Glo (
aSB5 value corresponds to amount of enzyme to give a SB ratio of 5.
DISCUSSION
Radiometric assays have been considered the “gold standard” and the most reliable assay for monitoring the activity of protein kinases19 and later on for screening chemical and natural product libraries in search for potential new kinase-based therapeutics. However, due to health and environmental issues regarding radioactivity and disposal of radioactive compounds, alternative technologies were developed. In addition, adapting radiometric assays into a homogeneous assay formats for high-throughput screening was fraught with difficulties. Thus, several approaches have been developed to address these issues (reviewed in ref.6 , 7). Most of the assays that were developed are based on fluorescent technologies using either fluorescence polarization (FP),20 , 21 fluorescence enhancement readout,22 , 23 or TR-FRET.24 Each of these assays has limitations such as sensitivity, high percentage of false hits, the need for customization of substrates for each enzyme, and cost. Some of these assays are universal but lack the ability to be used at a broad range of either the substrate or ATP18 or require large amounts of enzyme to generate meaningful data. Others have to be optimized for each enzyme and thus a novel strategy for developing a kinase assay that meets the various requirements was needed. The ADP-Glo described here meets these requirements. In comparison with Adapta (anti-ADP antibody-based assay), the assay has a broader range of ATP (up to 1 mM) while Adapta is limited to a concentration of up to 100 µM ATP due to cross reactivity of the anti-ADP tracer with ATP precluding its usefulness for high ATP K m enzymes. This is important since the potency of inhibitors that are ATP competitive is dependent on cellular concentration of ATP that is normally present in millimolar concentration. Thus, for the inhibitor to be effective in vivo, it has to be tested at a minimum of 100 µM and better if tested at 1–2 mM ATP. In addition, the narrow range with Adapta (maximum of 4- to 5-fold) makes it less desirable for high-throughput screeners due to the need for high amount of enzymes18 and makes it less useful for screening high-potency inhibitors. Thus, the narrow dynamic range, limitations on ATP concentrations, low signal to background, and the large amount of enzyme needed makes this assay less suitable for HTS.
The traditional approach of TR-FRET using anti-phosphorylated peptide approach18 , 25 has shown promise with anti-phosphorylated tyrosine containing substrates but due to the limitations on substrate concentrations used in the assay (maximum of 1 µM) makes it less desirable for enzymes that require high substrate concentrations. Also, the assay uses mostly peptide substrates and thus it is difficult to use with proteins, lipids, sugars, or any other substrates, thus precluding its use for lipid kinases, sphingosine kinases, sugar kinases, and so on. Also, this technology requires optimization of enzyme reactions with each kinase and is difficult to apply to many serine/threonine kinases particularly when the substrate is not known. These drawbacks also apply to other technologies that are dependent on fluorescently labeled peptide substrates such as the Z-lyte26 and any other fluorogenic assay.22 The latter assays also lack the universality feature that makes them limited in their scope. Previously, we have developed Kinase-Glo® that is based on the bioluminescent detection of ATP depletion during a kinase reaction. The light output from this assay is inversely proportional to the kinase activity. This assay is simple to use, can detect up to 500 µM ATP, is universal, and was validated with numerous kinases.27 –29 However, Kinase-Glo performance can be limited when assaying low-activity kinases that result in low ATP depletion at high initial ATP concentration. This is due to the low dynamic range generated at this level of ATP conversion. Thus, there is no technology that is currently available that meets the universality requirement, high sensitivity, robustness, and broad range of substrate concentrations and that matches those features of the radiometric assay. ADP-Glo technology meets all the requirements that are critical for HTS such as being homogeneous, large-scale formatting, evasion of high cost of radioactive waste disposal, and minimal false hits.
Since ADP-Glo assay monitors the ADP released in the kinase reaction, it requires the use of purified kinases that are free of ATPases or other enzymes that produce ADP. However, the activity of these contaminating enzymes can be easily detected using a kinase inhibitor in a control reaction. Also, the ATP used in the kinase reaction should be of high purity in order to minimize the background. Because the assay is a 2-step enzymatic reactions, we wanted to ensure that screening of compounds is not influenced by the effect of the compounds on assay performance. To address this issue, we have tested the effect of a library of pharmacologically active compounds (LOPAC) that contains 1,280 compounds at 10 µM using ADP-Glo on the first step (ATP depletion and kinase termination) and the second step (conversion of ADP to ATP and generating light using luciferase/luciferin reaction), and also on the overall performance of the assay. The results show that only 2 compounds have inhibited the assay by 50% that is translated to <0.2%. The use in ADP-Glo Assay of Ultra-Glo™ luciferase that is resistant to interference from common native luciferase inhibitors is another reason for the low false hits encountered by us and by others using Kinase-Glo®.30 In addition, screening 1,300 compounds containing kinase-focused scaffolds against CLK 4 using ADP-Glo showed that the false hits rate is also minimal.31 We and others have shown that the percentage of compound interference with ADP-Glo is very minimal but it is always important to perform control experiments in order to pinpoint those false hits. For example, to test hits for the possibility of chemical interference with enzymatic depletion of ATP or generation of the luminescent signal, mock reactions containing a mixture of ADP and ATP without the kinase could be subjected to the appropriate concentration of test compound or vehicle control (eg, 1% DMSO). A true kinase inhibitor should not alter luminescence by >20% compared to vehicle-only control. Alternatively, compound interference can be also checked using another luciferase-based assay, such as Kinase-Glo, as a counter-screen assay for the identified hits.
In conclusion, the results presented here validate the use of this assay not only in early kinase assay development but also for screening a large set of chemical libraries as well as profiling various kinases. Thus, the fact that this assay has important features like Z′ value of 0.7 or higher, high signal to background at low substrate conversion, universality, high sensitivity, ease of use, and no fluorescence interference resulting in extremely low false hits makes it ideal for any phase of kinase assay-related projects in drug discovery.
AUTHOR DISCLOSURE STATEMENT
H.Z., M.Z., K.H., and S.A.G. are employees of Promega Corporation. S.A.G. is also an employee of the University of Wisconsin School of Medicine and Public Health.