
Abstract
Protein–protein interactions are increasingly of interest as targets in small-molecule drug discovery. The interaction between the Ca2+- and phospholipid-binding protein Annexin A2 and its binding partner S100A10 has been implicated in angiogenesis and cancer metastasis. Here, we present a methodology to screen for inhibitors of this protein interaction. We developed a Cy5-labeled S100A10 tracer and showed by circular dichroism spectroscopy that the secondary structure is indistinguishable from that of non-labeled S100A10. This tracer was used to develop a binding assay based upon fluorescence resonance energy transfer to a Cy3-labeled Annexin A2 peptide ligand. The binding parameters matched those for unlabeled components as observed by equilibrium dialysis, which we determined separately, as well as those determined by isothermal titration calorimetry. Binding of labeled and unlabeled peptide was specific and mutually competitive. We used this assay for screening a small compound library derived by computational interrogation of the S100A10-binding pocket. Hits were obtained with IC50 values in range of the IC50 of the cognate Annexin A2 peptide ligand. Hits were subjected to an exact parallel assay measuring an unrelated protein–protein interaction (antigen–antibody). In this way, we identified genuine hits that inhibited the interaction between S100A10 and Annexin A2 but do not affect the fluorescence readout. These compounds are potentially of interest as candidates for further analysis and medical chemistry exploration. The simple assay format described here can be employed in early-stage exploration of other protein–protein interaction targets.
INTRODUCTION
Protein–protein interactions are widely regarded as pivotal to cell regulation and increasingly of interest as targets in small-molecule drug discovery.1 –3 Here, we present a methodology to screen for inhibitors of the known interaction between the Ca2+- and phospholipid-binding protein Annexin A2 (also known as p36) and its accessory protein S100A10 (also known as p11). Annexin A2 is a member of the wider Annexin multigene family, which has been implicated in many intra- and extracellular processes.4 Annexin A2 exists as heterotetramer with members of the S100 family of Ca2+-binding proteins, in particular with the S100A10 protein.5 Two S100A10 molecules form a dimeric structure that yields 2 binding pockets, each of which accommodates the 14 amino acid N-terminal region of Annexin A2.6 It appears that this Annexin N-terminus is sufficient to mediate binding with the S100A10 dimer since Annexin A2(1–14) as an isolated peptide (referred to as AA2(1–14) hereafter) can dissociate a preformed complex7 and deletion of residues 1–14 from Annexin A2 results in a complete loss of the interaction.8 The AA2(1–14)-binding site on S100A10 is well characterized, both by mutagenesis and crystallography. It involves a comparatively deep concave binding pocket,9 , 10 which is within reach of what are commonly considered drug-like molecules.11 Using the AA2(1–14) peptide as competitor, the in vivo relevance of the interaction between Annexin A2 and S100A10 has been probed. AA2(1–14) inhibited neoangiogenesis into Matrigel plugs in a mouse model system and it also inhibited metastasis of prostate cancer cells into the bone marrow in mice.12 , 13 These studies suggest that the interaction between S100A10 and Annexin A2 may be important in angiogenesis and cancer metastasis.
Small-molecule blockers of protein–protein interactions have been successfully identified using a variety of screening techniques. Sandwich ELISAs have been applied widely, for example for the measurement of the p53-mdm2,14 the VLA4-VCAM,15 and the B7.1-CD2816 protein–protein interaction. Fluorescence polarization has also been used to measure the binding of a BH3 peptide to Bcl-XL17 and a p53 peptide to mdm2.14 Various FRET-based assay methods have been described to discover small-molecule inhibitors of protein–protein interactions, usually relying on tagging the target proteins. Thus a time-resolved FRET assay has been used to discover small-molecule blockers of the interaction between the GST-tagged estrogen-related receptor-〈 and its co-activator18 and genetically engineered fluorescent protein tags have been applied to identify small-molecule blockers of the interaction between protein kinase C-iota and its effector PAR6.19
Although the interaction between S100A10 and Annexin A2 is well established, no small-molecule blockers of this interaction have been reported. In order to make the interaction between S100A10 and Annexin A2 accessible for screening of small-molecule libraries, we developed a method that involves the labeling of the S100A10 protein using a Cy5 fluorophore and assessing the protein interaction by measuring FRET from a Cy3 donor engineered onto the C-terminus of AA2(1–14). We reasoned that direct labeling circumvents the use of a large tag on the relatively small S100A10 protein thus avoiding potential steric interference with its ability to form a productive dimer necessary for accommodating Annexin A2. Direct chemical labeling has been successful to measure the interaction between various other protein pairs by FRET.20 –22 A key part of the current method is the use of a parallel “counter screen” based upon monitoring an antibody–antigen interaction using the same fluorescence readout, which facilitates the identification of genuine hit molecules. The simple assay format can in principle be applied to other protein–protein interactions.
MATERIALS AND METHODS
Materials
Labeled AA2(1–14)-Cy3 was synthesized by Peptide Protein Research (Wickham, UK) with the Cy3 label linked to a cysteine residue introduced at the C-terminal end of the peptide. Labeling kits for Cy5 conjugation were obtained from GE Healthcare/Amersham (Buckinghamshire, UK). All molecular biology reagents and protein reagents were obtained from Promega, New England Biolabs, Qiagen, or Merck/Novagen. Chemicals were obtained from Sigma or Fisher. The small-molecule library was obtained from Asinex (Rijswijk, the Netherlands).
Preparation of Recombinant Human S100A10
S100A10 was amplified from a human placenta cDNA library (Biochain Institute, Inc., Hayward, CA) and cloned into pET14b (Merck-Novagen). For purification, a 1 L culture of Escherichia coli {Rosetta (DE3)pLacI} harboring pET14b-S100A10 was grown at 37°C to OD600 = 0.6; isopropyl-β-
Equilibrium Dialysis
For equilibrium dialysis of AA2(1–14)-Cy3 and S100A10, a DispoEquilibrium Dialyzer with a 10 kDa molecular weight cutoff (Harvard Apparatus, Holliston, MA) was used. Two micromolar S100A10 were placed in one cell of the dialyzer and 0.102 to 6.5 µM AA2(1–14)-Cy3 in the opposite cell. The dialyzers were sealed in foil and gently shaken at 20°C until equilibrium had been established. Samples were then withdrawn from each cell and the amount of AA2(1–14)-Cy3 in the cells was quantified against a standard curve of known AA2(1–14)-Cy3 concentrations.
Protein Labeling and Storage of Tracers
S100A10 (0.66 mL, concentration of 10 mg/mL) in buffer C was incubated with Cy5-monofunctional reactive dye (GE Healthcare) at 20°C for 30 min after which the buffer volume was adjusted to 2.5 mL. Incubation was continued for another 30 min and free dye was removed by gel filtration in buffer C using a 5-mL PD-10 column (GE Healthcare). The labeling was very reproducible and typically the stoichiometric ratio of Cy5 to S100A10 protein was 0.3 mol/mol. Labeled protein was stored at 4°C in buffer C.
AA2(1–14)-Cy3 was obtained as powder in 1 mg batches and kept at 4°C. When required, batches were reconstituted in buffer C to a concentration of 1 mg/mL and stored in aliquots at -20°C until needed. For the purpose of the assay, batches were matched onto each other based on the amount of peptide.
For counter screens, donkey anti-goat IgG (Sigma) was labeled with Cy5-monofunctional reactive dye (GE Healthcare) as described for S100A10 labeling. Cy3-conjugated rabbit anti-goat IgG was obtained from Sigma. Labeled protein was stored at 4°C in buffer C.
Circular Dichroism (CD) Spectroscopy
CD spectra from 190 to 250 nm were acquired on a π*-180 Applied Photophysics spectrophotometer at room temperature. Identical concentrations of Cy5-labeled S100A10 and non-labeled S100A10 in buffer C were diluted in water to give a final concentration of 10 µM. Control spectra of diluted buffer C were acquired separately and used to correct the signal obtained for the protein. The raw ellipticity value (θ) was converted into mean residue ellipticity ([θ]) by the formula:
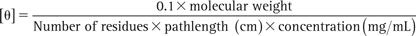
For the labeled protein, the molecular weight of the label was taken into account.
Fluorescence Assay
Fluorescence assays were carried out in Nunc Black nontreated 384-well plates at 20°C in 50 µL buffer C. All incubations were performed in quadruplicate. Compounds, peptides, and buffer controls were added to the wells in a 10 µL volume in 5% DMSO. For concentration curves, dilutions were prepared in 5% DMSO and 10 µL of each dilution was added to the wells. AA2(1–14)-Cy3 and S100A10-Cy5 were preincubated for 5 min at 20°C and 40 µL of the preformed complex was then added to the wells and mixed for 10 s to yield a final DMSO concentration of 1%. After 5 min incubation at 20°C, readings were taken on a Molecular Devices FlexStation by excitation at 525 nm and emission at 695 nm unless otherwise indicated. Each screening plate contained as controls the individual labeled proteins, as well as the 2 protein partners with and without 12.8 µM of the non-labeled AA2(1–14) competitor (all in quadruplicate). Fluorescence resonance energy transfer (FRET) was calculated by subtracting the sum of the fluorescence emission of S100A10-Cy5 and AA2(1–14)-Cy3 individually from that measured for the co-incubated partners. The non-labeled AA2(1–14) peptide was used as control for inhibition, allowing positive identification of inactive compounds. For IC50 determination, data were analyzed using GraphPad Prism.
Compound Selection
Candidate inhibitors were selected by computational interrogation of a small-molecule 3D database23 using the known crystal structure of the S100A10–AA2(1–14) complex (PDB entry code 1BT6) as a guide. Selected compounds were evaluated using the virtual screening program GOLD24 and hits showing high fitness scores and plausible binding modes were selected and purchased. Compounds were supplied as powder in 5 µmol quantities. Compounds were solubilized in DMSO to yield a 5 mM stock solution that was kept frozen. A 50 µM working solution in 5% DMSO was made from the stock solution and used to pipette into the wells. A detailed explanation of the computational chemistry approaches and compound structures will be reported elsewhere.
Protein Analysis
Purity was assessed by SDS-PAGE with Coomassie blue staining. Protein concentrations were measured by determining absorbance at 280 nm. The extinction coefficient for each protein, based on primary structure, was determined using the program ProtParam from the ExPASy Proteomics Server (http://www.expasy.org/). Absorbance was converted to concentration using the formula: concentration (M) = absorbance · (path length (cm) · extinction coefficient (M−1 cm−1))−1.
Mass Spectrometry
Proteins were digested with sequencing grade porcine Trypsin (Promega) and the resulting digest was purified by filtration through a 5-kDa cutoff spin column. Extracted peptides were run on a Walters QTOF2 hybrid quadrupole mass spectrometer via an autosampler comprising of an analytical capillary column and nanospray device. Data-dependent switching was incorporated so that whenever a peptide with an associated charge of 2+ or 3+ was detected above a preset threshold signal, the mass spectrometer would automatically switch to ms–ms mode to generate fragmentation data from the detected peptide.
RESULTS
Recombinant S100A10 and Equilibrium Dialysis
S100A10 was produced in E. coli and protein extraction under native conditions was found to be effective, yielding around 20 mg of soluble protein from a typical 1 L culture. For all subsequent experiments, the N-terminal His-tag was removed and the resulting sample was subjected to gel filtration and concentration steps to yield a pure protein fraction. Purity was estimated to be 99%, based on SDS-PAGE and identity of the protein was confirmed by mass spectrometry (data not shown).
In order to determine the parameters of AA2(1–14) binding to recombinant S100A10 (rec-S100A10), we performed an equilibrium dialysis experiment using AA2(1–14) that was chemically synthesized to contain a Cy3 fluorophore at the C-terminus. AA2(1–14)-Cy3 was incubated with rec-S100A10 until equilibrium was established. The concentration of free peptide was determined against a standard curve measuring the fluorescence of known concentrations of peptide (Fig. 1A). Under equilibrium conditions, the binding isotherm showed clear saturation, the curve fitting well to a one-site binding model. The equilibrium dissociation constant determined in this way was 1.3 ± 0.3 µM (K d ± standard error of the fit) (Fig. 1B). This value of the equilibrium dissociation constant is very close to that recently measured by isothermal titration calorimetry (ITC) at room temperature using a non-labeled AA2(1–14) peptide.25
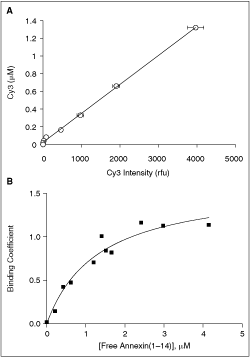
Equilibrium dialysis of AA2(1–14)-Cy3 and S100A10. (
S100A10 Labeling
Inspection of the crystal structure of the complex between AA2(1–14) and S100A10 (PDB entry code: 1BT6) revealed that the C-terminus of the AA2(1–14) peptide (where the Cy3 fluorophore is attached) is within 50Å of 11 free amino groups on S100A10 (Table 1). This suggests that it should be possible to measure the interaction by FRET, with a donor fluorophore engineered onto the C-terminus of AA2(1–14) and an acceptor linked to one of these amino groups of S100A10. We chose Cy3 as donor fluorophore and synthesized a AA2(1–14)-Cy3 peptide and Cy5 as acceptor fluorophore, which was linked to S100A10 using standard NHS-ester protein coupling chemistry. In order to evaluate the effect of Cy5 labeling on the structure of S100A10 protein, CD spectroscopy was performed. As shown in Figure 2, S100A10 is characterized by extensive helical conformation, compatible with observations made on this protein using X-ray crystallography.6 Cy5 labeling of S100A10 did not affect the CD spectrum, indicating that labeling had no implications for the secondary structure of S100A10 (Fig. 2). Thus it was concluded that S100A10 labeled in this way by Cy5 provides a bona fide tracer to use in resonance energy transfer assays.
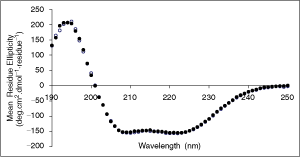
Circular dichroism spectroscopy. CD spectra of Cy5-labeled S100A10 (filled circles) and unlabeled S100A10 (open circles). The spectra were obtained at a protein concentration of 10 µM in diluted buffer C at 20°C. Data are expressed as mean residue ellipticity values.
Chain A, S100A10; Chain C, first AA2(1–14) peptide in the structure; Chain D, second AA2(1–14) peptide in the structure.
As mentioned, 11 free amino groups on S100A10 are potentially exposed to the labeling agent. In order to define the site of labeling more precisely, non-labeled and Cy5-labeled S100A10 were trypsinized and the tryptic digests was analyzed by time-of-flight positive ion electrospray mass spectrometry. The deconvoluted single charge ES+ spectrum of the non-labeled protein allowed ready identification of peptide species with a mass of 1,208 and 2,494, corresponding to tryptic peptides EFPGFLENQK and PSQMEHAMETMMFTFHK, respectively. The overall spectrum of the Cy5-labeled protein was qualitatively and quantitatively identical to that of the non-labeled protein, except for a reduction in the peak at 2,494 (data not shown). This reduction is most likely caused by Cy5 labeling of the lysine at position 17 interfering with the tryptic cleavage process. Thus the lysine at position 17 is a likely site of Cy5 linkage.
FRET-Binding Assay
Figure 3A shows emission wavelength scans (530–750 nm) of incubations of individual and of co-incubated fluorescent-binding partners using an excitation wavelength of 488 nm. Excitation of S100A10-Cy5 alone resulted in marginal emission fluorescence across the wavelength spectrum, comparable to the fluorescence observed in buffer only. Excitation of AA2(1–14)-Cy3 at 488 nm resulted in an expected emission peak at around 570 nm, which tailed off at longer emission wavelengths. Excitation of AA2(1–14)-Cy3 in the presence of Cy5-S100A10 resulted in a peak of fluorescence emission from Cy5, which the spectral overlay in Figure 3A shows not to be due to bleed-through fluorescence from excitation of the Cy3 fluorophore alone. It was concluded that this peak at 670 nm is due to energy transfer between the fluorophores. This suggests that the 2 proteins are sufficiently close to allow measurement of the interaction between the 2 partners by this method.
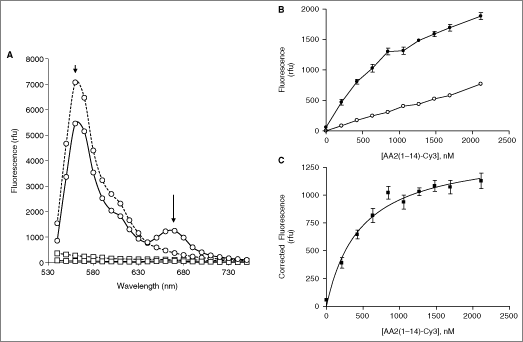
S100A10–Annexin A2-binding assay. (
For routine measurements (which did not employ wavelength scanning), the excitation wavelength was adjusted from 488 to 525 nm. This allowed a reliable FRET signal to be measured with minimal bleed-through. The incubation time was set at 5 min at which time point the signal was stable. Titration of AA2(1–14)-Cy3 against 407 nM S100A10 showed clear concentration dependence (Fig. 3B), the net signal above background (no S100A10) showing saturation with the curve fitting well to a one-site binding model (Fig. 3C). The apparent K d under these conditions was 890 ± 200 nM (5 independent determinations, n = 4 each), which compares well with that measured by equilibrium dialysis (Fig. 1) and ITC.25 In order to verify competition with binding of the Cy3-labeled peptide, we employed non-labeled AA2(1–14). As shown in Figure 4, the binding could be competed by non-labeled peptide in a manner compatible with a one-site competition binding model (Fig. 4). At 407 nM S100A10-Cy5 and 1.33 µM AA2(1–14)-Cy3, the pIC50 was 5.63 ± 0.05 (n = 8; SE). At 65 nM S100A10-Cy5 and 680 nM AA2(1–14)-Cy3, the pIC50 was 5.89 ± 0.04 (n = 4; SE).
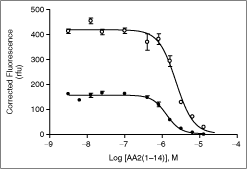
Inhibition of AA2(1–14)-Cy3 and S100A10-Cy5 interaction by non-labeled AA2(1–14). Different concentrations of non-labeled AA2(1–14) were incubated with either 1.33 µM AA2(1–14)-Cy3 and 407 nM S100A10-Cy5 (condition 1; open symbols; (n = 8 ± SE) or 680 nM AA2(1–14)-Cy3) and 65 nM S100A10-Cy5 (condition 2; filled symbols; n = 4 ± SE). Fluorescence intensity was measured and the background was subtracted as described in the Materials and Methods section. Data were fit using the sigmoidal dose–response (variable slope) curve fit in GraphPad Prism. The subtracted background fluorescence was 92 ± 6 rfu (condition 1; n = 4 ± SE) and 44 ± 2 (condition 2; n = 4 ± SE).
Compound Screening and Primary Plate Analysis
The conditions that afforded a larger absolute rfu count in Figure 4 were chosen for screening. We used the assay to screen several small-molecule libraries to identify blockers of the interaction between S100A10 and Annexin A2 and report an example of the screening of one such small-molecule library of 227 compounds. Z′-factor analysis of the screening plates showed an average Z′-factor of 0.62 ± 0.03 (mean ± SE; 22 plates), suggesting sufficient assay reliability for screening purposes. Each primary screening plate contained compounds at a concentration of 10 µM (n = 4), as well as positive and negative controls as described in the Materials and Methods section (Fig. 5). Plates were analyzed in 2 ways based upon (i) the percentage inhibition compared to control or (ii) the percentage inhibition compared to the average signal of the compounds on the plate (ie, excluding all the controls). Both measures exhibited a good overall correlation. There was little difference in absolute values of these 2 measures.
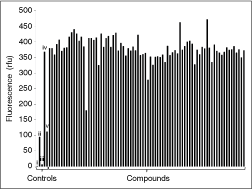
Small-molecule screening. An example of the results from one screening plate is shown. Each compound and each control was tested in quadruplicate and the bars represent the average. Ten micromolar compound was added to 1.33 µM AA2(1–14)-Cy3 and 407 nM S100A10-Cy5 after which fluorescence was determined as described in the Materials and Methods section. The following controls were included on each plate (i) buffer C, (ii) AA2(1–14)-Cy3 alone, (iii) S100A10-Cy5 alone, (iv) AA2(1–14)-Cy3 and S100A10-Cy5, and (v) AA2(1–14)-Cy3, S100A10-Cy5, and 12.8 µM non-labeled AA2(1–14). The average coefficient of variance (CV) of the compound samples on this screening plate was 7.2%.
Three cutoff criteria for compound activity were used. First, compounds that showed inhibitory activity that differed >20% from the average activity of all the compounds on the plate were chosen (Fig. 6A; Table 2). Second, compounds that showed inhibitory activity that exceeded 3 standard deviations from control value on the relevant plate were chosen (Fig. 6B; Table 2). And third, compounds of which at least one of the replicates showed an inhibition value differing >3 standard deviations from control were chosen (Fig. 6C; Table 2). The last measure was the least stringent and encompassed all compounds identified by the first 2 measures. Compounds fulfilling either of these cutoff criteria were reconfirmed in repeat assays. Compounds 1–6 showed a combined average inhibition of >20% from the non-compound control and were profiled further by IC50 determination (Table 2). Compounds 7–23 (largely consisting of compounds selected based upon the third cutoff criteria) showed <20% inhibition of the interaction and were not profiled further. The most potent compounds were compounds 1 and 2. Compounds 3–6 were less potent and fell into 2 groups based upon the characteristic of the free floating curve fit. For compounds 3 and 5, the value of the bottom fit was >50% of that of the top fit, indicating that these only partially inhibited the binding. Compounds 4 and 6 showed >50% inhibition of binding at infinite concentrations. Some compounds showed a high variability between and within assays (eg, compounds 2 and 3) that may be a reflection of their solubility.
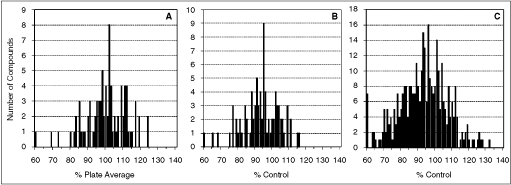
Frequency distribution of compound binding activity. For one screening plate, the compound activity spectrum was determined by calculating: (
A total of 227 compounds were screened at 10 µM final concentration and the average binding activity of the hits is shown as percentage of noncompound control (% cont; n = 4 ± SE) or as percentage of average binding activity of all compounds in the plate (% plate av; n = 4 ± SE). For the primary screen, only hits that fulfilled the cutoff criteria are shown: % plate av: average binding activity of the quadruplicate incubation differs by >20% of the average binding activity of all compounds in the plate; % cont, values underlined average binding activity of the quadruplicate incubation differs by >3 standard deviations (measured in the noncompound control) from the average noncompound control binding activity; % cont, not underlined binding activity of one or more compound-containing wells of the quadruplicate incubation differs by >3 standard deviations (measured in the noncompound control) from the average noncompound control binding activity. Repeat assays were performed at 10 µM concentration and a composite average was calculated (n = 8 ± SE). For compounds with average inhibition >20%, IC50 profiles were generated in the target assay and the counter assay. Midpoint pIC50 values are reported (n = 4 ± SE of the fit) and the bottom fit is shown as % of the top fit (n = 4 ± SE). NF = no fit.Fluorescence interference is a well-known phenomenon affecting the reliability of primary screens. Therefore, we performed a counter screen on those compounds selected above for IC50 profiling. The counter screen consisted of the same fluorescence system (Cy3–Cy5 FRET) but monitored a different protein interaction, namely the interaction between an antibody and an antigen. Compounds 1, 2, and 5 significantly inhibited the antigen–antibody interaction, and the inhibition curve overlapped with the curve obtained for the S100A10–AA2(1–14) interaction, suggesting that these hits may interfere with the fluorescence signal (Table 2; Fig. 7B); however, compounds 3, 4, and 6 only inhibited the interaction between S100A10 and AA2(1–14) and did not affect the interaction between antigen and antibody (Table 2; Fig. 7A). Thus these compounds may represent valuable starting points for further characterization and medicinal chemistry exploration.
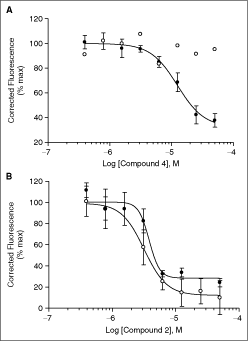
Selective inhibition of Annexin A2(1–14)–S100A10 protein interaction. A range of concentrations of compound 4 (
DISCUSSION
We present a screening assay that allowed us to identify candidate small-molecule blockers of the interaction between the protein S100A10 and Annexin A2. The assay consisted of a FRET-based system using Cy3 and Cy5 fluorophores to monitor the interaction between these proteins, as well as a counter screen that monitored a different protein interaction.
The screen involves the labeling of S100A10 with Cy5 fluorophore. It may be argued that the structure of S100A10 would be compromised by the labeling process. Whilst this formally remains a possibility, 2 pieces of evidence suggests that the structure of S100A10 is retained after labeling. First, CD spectroscopy showed that there is no loss of secondary structure after labeling of the protein. And second, the apparent K d of the interaction between Cy3-labeled Annexin A2 peptide and Cy5-labeled S100A10 is similar to that measured by equilibrium dialysis of Cy3-labeled Annexin A2 peptide against unlabeled S100A10. Furthermore, the apparent K d matches closely that reported from isothermal titration calorimetry experiments at room temperature.25
The binding as measured by the FRET signal can be competed by unlabeled ligand, indicating that the assay can be used to screen for inhibitory substances. We adopted a 384-well plate format for screening and showed that in this format the assay is sufficiently robust to support screening of a small-molecule library. Hit cutoff criteria employed here were based on deviation from the average binding activity of all compounds on the screening plate as well as deviation from separately assessed non-compound controls and in practice we observed little difference between these. In order to capture most hits, we preferred to use the 3 standard deviations from control as the primary hit cutoff criteria. There was no further merit in relaxing these criteria by including compounds of which at least one replicate met the cutoff value, since these were all excluded in the second screening round.
It has been frequently observed that compounds with good potency in primary screening assays turn out to be potentially artifactual with respect to the primary target. By using a counter screen based upon a different protein–protein interaction, we observed this to be the case for some primary hits since the most potent hits were inhibitory in the counter screen assay; however, moderately active hits (around 10 µM IC50) appeared selective and these could provide suitable starting points for further analysis. Confirmation that these hits can inhibit the interaction between the 2 native protein partners is required and whilst the counter screen presented here is useful, it itself monitors a specific interaction and it would be desirable to evaluate the hits in another protein interaction counter screen in order to discriminate further between specific and general protein disruptors.
Compounds that specifically inhibit the interaction between S100A10 and Annexin A2 would be useful as probes to analyze the function of this interaction in cells. So far, the interaction has largely been probed using the N-terminal Annexin A2 peptide as inhibitor; however, this peptide cannot penetrate the cell membrane and can only be used if access to the cell interior can be gained (eg, in microinjection or patch clamp experiments) or to monitor extracellular functions of this complex. Peptide competition experiments have suggested a role for the S100A10–Annexin A2 interaction in angiogenesis, prostate cancer cell metastasis, bone marrow cell homing (all likely due to an extracellular action), as well as ion channel activity, secretion, and endosomal fusion. Compounds identified here, or derivatives thereof, may be useful to further our understanding of the role of the interaction in these processes and to define other processes in which this interaction may play a role.
The assay method described in this manuscript requires medium level quantities of protein to be obtained and in addition it may not always be possible to use direct labeling as a technique to obtain a tracer. Nevertheless, the approach of direct labeling and counter screening could be useful for many protein–protein interactions that are currently described only at a cellular level and for which assay techniques are required to make the first step into the “drug discovery arena.”
Footnotes
ACKNOWLEDGMENT
The authors wish to thank Professor Mark Searle for help with the CD experiment.
AUTHOR DISCLOSURE STATEMENT
C.L., T.R.K.R., P.M.F., and L.V.D. are employees of the University of Nottingham.