
Abstract
Kinases continue to be one of the most important targets in today’s drug discovery efforts. Following the identification of lead compounds through screening efforts, it is important to profile these leads against other kinases within that family, as well as from other families, to ascertain potential off-target effects. Because many kinase assays require the use of different substrates, optimization time and costs during profiling can be prohibitive. Here we demonstrate the versatility of a luminescent ADP accumulation assay, where one set of reagents can be used for a wide variety of kinases with differing Km app for ATP and substrates. Assay sensitivity allows for the use of low enzyme concentrations and small percent ATP conversion levels while still maintaining high signal:background ratios. We have used a simple, inexpensive automated pipetting system to automate the entire process from enzyme optimization through generation of compound IC50 values. Agreement with literature values proves this combination of chemistry and instrumentation provides a simple, yet robust solution for automated kinase profiling.
INTRODUCTION
Kinases are one of the most diverse enzyme families being studied today. Because of the large number of genetic mutations inherent within this family, and their role in cell signaling, kinases have been implicated in a large number of diseases, including cancer and diabetes. This has also caused kinase-targeted drug discovery efforts to continue to grow in importance among today’s pharmaceutical companies. Inhibitors are designed to either act at the ATP-binding site (ATP competitive), preventing binding of ATP by the kinase, or at another domain (allosteric), causing a conformational change in the kinase1 leading to altered enzyme activity. Of recent particular interest has been the search for inhibitors of tyrosine kinases. Gleevec™ (imatinib; Novartis Pharmaceuticals, Basel, Switzerland), an inhibitor of Bcr–Abl fusion protein,1 and Tarceva™ (erlotinib; Genentech Inc., South San Francisco, CA), an inhibitor of EGFR,1 are just 2 examples of successful drug discovery campaigns to identify inhibitors for this kinase family. However other protein kinases, as well as lipid and sugar kinases, are also gaining interest as possible therapeutic targets.
As new lead compounds are identified for their primary target through screening efforts, it is becoming increasingly important for these compounds to be profiled against other kinases to determine any possible off-target effects for these compounds. This is primarily due to the nonspecificity of ATP competitive inhibitors. These compounds, as explained above, compete with ATP to bind at the ATP-binding site of the kinase. Due to the conserved nature of the ATP-binding domain among protein kinases, selectivity of the inhibitors has to be verified against other kinases. This is necessary in order to assess that they do not inhibit other nontarget kinases, resulting in undesirable adverse effects of the potential drug candidate. Therefore, profiling efforts that include testing multiple concentrations of lead compounds in an IC50 format against kinases within the same target class, as well as from other kinase families, are necessary in order to determine the selectivity of the compounds against the target of interest.
While the importance of kinase profiling is generally well recognized, caution must be exercised in generating accurate profiling data. For example, the inability to use the same assay components when testing multiple kinases may result in erroneous conclusions. Many of the commercially available kinase assays have to use multiple substrates when being run with a variety of kinases.2 Some require separate substrates for serine/threonine, tyrosine, or lipid kinases, while others require additional substrates within these groups.3 , 4 It can also be difficult to develop an assay that uses the same detection reagents for kinases having a broad range of ATP (µM to mM). Further complicating this situation is the fact that assay-specific peptide substrates are frequently used as surrogates of native kinase substrates. This can then raise the question of the relevance of information generated to the physiological environment. Together, these points cause optimization time and costs to increase, as each assay with each new substrate, or new concentrations of detection reagent, need to be optimized before being brought online for profiling.
Another important consideration in setting up and generating accurate profiling data is the sensitivity of the assay. The ability to retain a good dynamic range, while still being able to use a concentration of enzyme that maintains initial rate velocity, can be difficult to achieve without having a highly sensitive assay. As a general rule, 10% substrate conversion is considered safely within this initial rate. This means that the amount of enzyme used to achieve 10% conversion is usually quite small, especially when working with kinases that have low ATP K m app values. Thus, assay sensitivity is critical in these situations in order to detect low substrate conversion while maintaining suitable signal:background ratios.
Coupled with the desire to find the appropriate assay is the need for instrumentation that can accurately titrate and dispense compounds, as well as deliver assay components to the desired plate. Primary screening instrumentation, while being excellent for delivering assay components when testing a single target, is not suited for the complications that kinase profiling can present. Because multiple kinase/substrate mixes, and ATP concentrations need to be dispensed to each assay plate, it is critical to have an instrument that can accurately aspirate and dispense multiple reagents at a time in low volumes.
Here we evaluate the utility of a luminescent ADP accumulation assay, where one set of reagents can be used for a wide variety of kinases with differing ATP requirements, to address all the points mentioned above. Because of the high sensitivity (high signal:background ratio), and the large dynamic range of the assay, we were able to detect small percent conversions of ATP to ADP using low amounts of enzyme. The fact that it is also a universal, luminescent assay means that it can be used with any kinase:substrate combination, and is less susceptible to fluorescent compound interference.5 Furthermore, because the assay uses a genetically engineered luciferase (Ultra-Glo™Luciferase6), which is more resistant to chemical interference than native luciferase, generation of false hits is minimized. The assay has a sensitivity that is comparable to radiometric methods without the need for disposing radioactive waste. The assay is performed in 2 steps. After the kinase reaction is completed, an equal volume of ADP-Glo™ reagent is added to the well. This reagent serves 2 purposes: to terminate the kinase reaction and to deplete the remaining ATP. In the second step, Kinase Detection Reagent is added that simultaneously converts ADP generated in the kinase reaction to ATP, and then allows this de novo ATP to be measured through the use of a luciferase/luciferin reaction.5
Here we demonstrate the performance of this ADP detection technology in combination with simple instrumentation in kinase characterization, assessment of various kinase inhibitors, and finally kinase profiling.
MATERIALS AND METHODS
Materials
SRC, Active (Catalog # S19-18G), AKT2, Active (Catalog # A17-10H), PI3K (p110α/p85α), Active (Catalog # P27-18H), ROCK1, Active (Catalog # R10-11G), and ZAP70, Active (Catalog # Z02-10G) were purchased from SignalChem (Richmond, BC Canada). CHUK (IKK alpha; Catalog # PV4310) was purchased from Invitrogen Corp. (Madison, WI). cAMP-dependent protein kinase catalytic subunit (PKA; Catalog # V516A), protein kinase C (PKC; Catalog # V526A), and DNA-dependent protein kinase (DNA-PK; Catalog # V581A) were obtained from Promega Corporation (Madison, WI).
Poly(Glu, Tyr) sodium salt (Catalog # P0275) was purchased from Sigma-Aldrich (St. Louis, MO). IKK peptide substrate (Catalog # P226-0001) and crosstide peptide substrate (Catalog # P149-0001) were purchased from BIOMOL International (Plymouth Meeting, PA). S6K Substrate (Catalog # S05-58) was purchased from SignalChem (Richmond, BC Canada). Kemptide (PKA) peptide substrate (Catalog # V5601), neurogranin (28–43) (PKC) peptide substrate (Catalog # V5611), DNA-dependent protein kinase peptide substrate (Catalog # V5671), and ADP-Glo kinase assay kit components (Catalog # V9101) were obtained from Promega Corporation (Madison, WI).
PP2 (Catalog # AG 1879) was purchased from EMD (Gibbstown, NJ). Staurosporine (Catalog # S4400), Akt 1/2 kinase inhibitor (Catalog # A6730), and dimethyl sulfoxide (Catalog # D2650) were purchased from Sigma-Aldrich (St. Louis, MO). Wortmannin (Catalog # W-2990) was purchased from LC Laboratories (Woburn, MA). Y27632 (Catalog # EI299-0001) was purchased from BIOMOL International (Plymouth Meeting, PA).
Liquid handling. A Precision™ XS Microplate Sample Processor from BioTek Instruments, Inc. (Winooski, VT) was used to perform all ATP, enzyme, and compound titrations, as well as to dispense all kinase and ADP-Glo reaction components to the wells of the assay plates.
Luminescent signal detection. All assay plates were read using a Synergy MX Monochromator-based Multi-Mode Microplate Reader from BioTek Instruments, Inc. (Winooski, VT). The luminescence detection method was chosen, and a 1 s integration time was used. Automatic sensitivity adjustment was used to detect the well on the plate containing the highest luminescent signal. All wells were appropriately adjusted to that well.
Methods
Buffers and substrates. Enzyme reactions for ROCK1, IKK alpha, PI3 kinase, and PKA were performed using Reaction Buffer A (40 mM Tris, pH 7.5, 20 mM MgCl2, and 0.1 mg/mL BSA). AKT2 reactions were performed using Reaction Buffer A plus 1 mM DTT, while Src and ZAP70 reactions were performed using Reaction Buffer A plus 2 mM MnCl2, 0.1 mM NaVO3, and 2 mM DTT. PKC reactions were performed using PKC Activation Buffer (0.32 mg/mL phosphatidylserine, 0.032 mg/mL diacylglycerol, 20 mM Tris–HCl, pH 7.5, 10 mM MgCl2), and PKC Co-activation Buffer (0.25 mM EGTA, 0.4 mM CaCl2, 0.1 mg/mL BSA). DNA-PK reactions were performed using DNA-PK Reaction Buffer (50 mM HEPES (KOH, pH 7.5), 100 mM KCl, 10 mM MgCl2, 0.2 mM EGTA, 0.1 mM EDTA, 1 mM DTT), in the presence of 10 µg calf thymus DNA for activation.
Saturating substrate concentrations were used with each enzyme reaction for all experiments. The concentrations used were: Src and ZAP70:0.13 mg/mL poly(Glu, Tyr); ROCK1:152 µM S6K; AKT2:50 µM crosstide; PI3 kinase alpha: 66.67 µg/mL phosphatidylinositol/16.67 µg/mL phosphoserine; IKK alpha: 200 µM IKK peptide; PKC:100 µM neurogranin peptide; PKA:50 µM kemptide; DNA-PK: 2 mg/mL DNA-PK peptide.
Enzymes and incubation conditions. Enzyme concentrations for Src, ZAP70, ROCK1, AKT2, and PI3 kinase alpha were kept at a nonrate limiting concentration of 10 ng/rxn during the ATP K m app determination. For the first step of the 10× signal:background (SB10) determination, enzymes were titrated 1:2, beginning at a top concentration of 130 ng/rxn for Src, AKT2, and PI3 kinase alpha, and 200 ng/rxn for ZAP70 and ROCK1. For the second step of the SB10 determination, the following concentrations were individually prepared to confirm the actual SB10 value. Src: 0.25, 0.5, 1.0, 2.0, and 4.0 ng/rxn; ZAP70: 5.0, 7.5, 10.0, 12.5, 15.0, and 25.0 ng/rxn; ROCK1: 10.0, 15.0, 20.0, 25.0, 30.0, and 35.0 ng/rxn; AKT2: 2.0, 4.0, 6.0, and 8.0 ng/rxn; PI3 kinase alpha: 0.5, 1.0, 2.0, 3.0, 4.0, and 5.0 ng/rxn. Enzyme concentrations for profiling dose–response curves were as follows: Src: 0.68 ng/rxn; ZAP70: 11.24 ng/rxn; ROCK1: 11.5 ng/rxn; AKT2: 4.12 ng/rxn; PI3 kinase alpha: 3.43 ng/rxn; IKK alpha: 90 ng/rxn; PKC: 15 ng/rxn; PKA: 0.05 U/rxn; DNA-PK: 1.0 U/rxn.
All kinase reaction incubation times were set at 60 min in order to standardize the conditions for profiling. The exceptions were ZAP70 and ROCK1, and PKA and PKC, due to the low and high activity levels, respectively, of the enzymes. Enzyme reaction incubation times were therefore increased to 90 min for ZAP70 and 120 min for ROCK1, and decreased to 10 min for PKA and 30 min for PKC.
ATP solutions. As recommended by the ADP-Glo supplier, the Ultra Pure ATP included in the kit was used in each reaction. ATP concentrations used in Src, ZAP70, ROCK1, AKT2, and PI3 kinase alpha enzyme reactions were based upon the determined K m,app value (see Results and Discussion section). These values were determined by running an ATP titration with each of the enzymes mentioned above, where ATP was titrated 1:2.5, beginning at a concentration of 1,000 µM. These values were then used in each subsequent experiment. The ATP concentrations used for the additional kinases included in the profiling validation were based upon existing literature sources. The concentrations were: IKK alpha (42 µM),7 PKC (100 µM),8 PKA (25 µM),9 and DNA-PK (100 µM).10
ATP/ADP conversion curves. ATP/ADP conversion curves were prepared for each enzyme included in the enzyme titration and confirmation experiments. ATP and ADP were created at 100 and 1,000 µM, containing 1× Reaction Buffer A. The 100 µM ATP and ADP were then combined following the manufacturer’s protocol, to create the various ADP and ATP percent levels represented in the curve (100/0, 80/20, 60/40, 40/60, 20/80, 10/90, 5/95, 4/96, 3/97, 2/98, 1/99, and 0/100, respectively). Therefore point #1 of the curve will contain 100 µM ADP/0 µM ATP, point #2 of the curve will contain 80 µM ADP/20 µM ATP, and so on. The 100 µM curve was then diluted to 1.32 and 4.62 µM, which represented the correct ATP concentration for ZAP70 and ROCK1, respectively, using enzyme diluent containing buffer and substrate. The 1,000 µM ATP and ADP were then combined using the method described above to create the same conversion curve. After the combination procedure was complete, the 1,000 µM curve was diluted to 13.9, 225.9, and 26.41 µM, which represented the correct ATP concentration for Src, AKT2, and PI3 kinase alpha, respectively, following the same dilution procedure previously described.
Kinase inhibitors. For the selective inhibitor IC50 and Z′-Factor determinations, known inhibitors were identified for each of the 5 optimized kinase reactions. PP2 (Src), Y27632 (ROCK1), and AKT 1/2 kinase inhibitor (AKT2) were titrated 1:4, beginning at a starting concentration of 10,000 nM. Staurosporine (ZAP70) and Wortmannin (PI3 kinase alpha) were titrated 1:3, with starting concentrations of 50 and 25 nM, respectively. For the Z′-Factor calculation, 24 replicates of all compounds were performed at 0 nM for the positive control and 10,000 nM for the negative control.
During the universal kinase inhibitor experiment, Staurosporine was prepared at multiple starting concentrations (ZAP70 and ROCK1: 50 nM, Src and AKT2: 500 nM, and PI3 kinase alpha: 10,000 nM). The compound was then titrated 1:3 for all kinase reactions. Finally for profiling, Wortmannin was titrated 1:5 beginning at a starting concentration of 10,000 nM for all 9 kinases included. All reactions containing compound had a final DMSO concentration of 1%. During the reaction optimization experiments, this same DMSO concentration was added to each well to simulate the presence of compound in the reaction.
AUTOMATED WORKFLOW
Assay Setup
Labcon 200 µL Robotic Tips: Non-sterile, low retention (BioTek Catalog # 98254) were used for diluent addition and serial titrations, and delivery of ADP-Glo reagents to the assay plate. Labcon 50 µL Robotic Tips: Non-sterile, low retention (BioTek Catalog # 98250) were used to transfer compounds, DMSO solutions, enzyme/substrate mixes, and ATP to the assay plate. Titrations were carried out in 96-well format using 96-well Greiner U-bottom plates (Promega Catalog # A9161). Enzyme reaction components were also placed into this type of plate for the automated methods. ADP-Glo components were placed into Greiner MASTERBLOCK®, 1 mL, U-bottom, solid, natural, non-sterile 96-well plates (Greiner Bio-One Catalog # 780 201) when running the automated methods. Assays were run in 384-well format using Corning® 384-Well Low Flange White Flat Bottom Polystyrene Not Treated Microplates (Catalog # 3572) from Corning Life Sciences (Lowell, MA). Table 1 describes the general protocol followed for kinase profiling. The 12-point compound titrations, including diluent only point, were completed prior to the transfer of enzyme reaction components to the assay plate. DMSO or inhibitors, kinase/substrate mixes, and ATP were dispensed at 5 µL volumes, making the kinase reaction a total of 15 µL. After the appropriate room temperature (RT) reaction incubation time, 15 µL of ADP-Glo reagent was then added to each well. The plate was once again incubated at RT for 40 min. Finally, 30 µL of Kinase Detection Reagent was added to each well. Through this delivery method, a 1:1:2 ratio of kinase reaction:ADP-Glo reagent:Kinase Detection Reagent is created within the well. Assay components were pre-mixed 5 times before delivery, to ensure reagent consistency within the well. The exception was Kinase Detection Reagent, where a one-time mix was only necessary to pre-wet the tips. All assay plates were mixed on an orbital shaker for 30 s at ∼1,000 rpm following the enzyme reaction component, ADP-Glo, and Kinase Detection Reagent additions. This was done to increase reaction consistency among replicates.
ATP Titration
100 µL of 3× concentrated ATP solution was dispensed to column 1 of a 96-well Greiner U-bottom plate (Promega Catalog # A9161). 60 µL of dilution buffer, containing 1× Reaction Buffer was then dispensed to columns 2–12. 40 µL of ATP was then transferred from column 1 to column 2 and mixed 10 times using a volume of 80 µL. The process was repeated to create a serial 1:2.5 titration from column 1 to 11. 5 µL of the titration was then transferred in duplicate to the assay plate. This was followed by 5 µL additions of 3% DMSO in 1× Reaction Buffer (DMSO solution). The remaining kinase reaction components, ADP-Glo, and Kinase Detection Reagents were then added to the assay plate as previously described in Table 1.
Enzyme Titration
80 µL of 3× concentrated plus enzyme mixes were dispensed to the first column of a 96-well Greiner U-bottom plate. 40 µL of minus enzyme mixes were then dispensed to columns 2–12. 40 µL of enzyme was then serially titrated 1:2 from column 1 to 12. Enzyme plus diluent was mixed 10 times using a volume of 60 µL after each dispense into a new column across the titration plate. 5 µL of the titration was then transferred in duplicate to the assay plate, followed by duplicate 5 µL transfers to separate wells of the 3× concentrated ATP/ADP conversion curve samples. A no enzyme control was also transferred in duplicate to the plate from the Greiner U-bottom plate containing the reaction components. All plus and minus enzyme mixes, as well as the no enzyme control, contained buffer and substrate. 5 µL of DMSO solution and 3× ATP were then added to the enzyme reaction wells, and 5 µL of DMSO solution and 1× Reaction Buffer were added to the conversion curve wells. Subsequent incubations and ADP-Glo and Kinase Detection Reagent additions were as previously described.
Enzyme Confirmation
50 µL enzyme/substrate mixes were created using various concentrations of each kinase. 5 µL of DMSO solution was added to the assay plate, in duplicate. 5 µL of the enzyme/substrate mixes or ATP/ADP conversion curve were then added in duplicate to the wells containing the DMSO solution. Finally, 5 µL of ATP was added to the enzyme wells, while 5 µL of 1× Reaction Buffer A was added to the standard curve wells. Subsequent incubation and ADP-Glo assay component additions were as previously described.
Selective Inhibitor IC50 and Z′-Factor Validation
The inhibitor titrations were prepared in the following manner. 100 µL of 3× concentrated PP2, AKT1/2 kinase inhibitor, and Y27632, and 90 µL of Staurosporine and Wortmannin were dispensed to column 1 of a 96-well Greiner U-bottom plate. Each inhibitor contained 3% DMSO. 75 µL of DMSO solution was then dispensed to columns 2–12 for the PP2, AKT1/2 kinase inhibitor, and Y27632 titrations, followed by 60 µL of DMSO solution being dispensed into columns 2–12 for the staurosporine and wortmannin titrations. 25 µL of the compounds was transferred from column 1 to 2 for the initial 3 compounds whereas 30 µL of compound was transferred for the remaining 2 compounds, in order to generate a 1:4 and 1:3 dilution through the plate, respectively. Compound plus DMSO solution was mixed 10 times using a volume of 75 µL for all compounds. 5 µL of the titrations were then transferred in duplicate to the assay plate. The process followed to test all 5 optimized kinase reactions with Staurosporine was the same as that used to test Staurosporine with ZAP70 alone in the previous experiment.
For the Z′-Factor determination, twenty-four 5 µL replicates of 0 or 30 µM inhibitor, in DMSO solution, were added to the assay plate. Subsequent enzyme reaction and ADP-Glo assay component additions, along with incubation times for both experiments, were as previously described.
Wortmannin Kinase Profiling
100 µL of 3× concentrated wortmannin, in 3% DMSO, was dispensed to the first column of a 96-well Greiner U-bottom plate. 80 µL of DMSO solution was then added to columns 2–12. 20 µL of Wortmannin was then transferred from column 1 to 2. The compound with diluent was then mixed 10 times using a volume of 75 µL. The process was repeated to create serial 1:5 titrations from columns 1–11. The 5 µL of the titration was then transferred in duplicate to the assay plate, followed by 5 µL of enzyme/substrate mix, and 5 µL of ATP. The 5 optimized reactions were incubated for the times mentioned above, while the IKK alpha, PKA, PKC, and DNA-PK enzyme incubations were incubated for 120, 10, 30, and 60 min, respectively, according to the manufacturer’s recommendation. All other incubations and ADP-Glo assay component additions were as previously described.
Data Analysis
ADP concentrations, % ATP conversion, and signal:background (SB) values were calculated as follows. ATP/ADP conversion curves were plotted on an X–Y graph, with [µM ADP] on the X-axis and relative light units (RLUs) on the Y-axis. Linear regression was then performed to determine the equation of the curve. [µM ADP] values were then computed for the enzyme titration or enzyme confirmation concentrations by comparing the luminescent values at each point with the luminescent values of the conversion curve and interpolating the [µM ADP] value for that enzyme concentration. The computed [µM ADP] values were then divided by the maximum concentration of ADP that could be created by a completed reaction, which is equivalent to the initial [ATP] in the well. This number was multiplied by 100 to generate the % ATP conversion value. Finally, the SB value for each enzyme concentration was computed by dividing the average RLU value for that concentration by the average RLU value for the no enzyme control.
The % enzyme activity was calculated first by subtracting the luminescent values for wells containing all reaction components, plus maximum inhibitor, from the values for each reaction well. The luminescent values for the 0 µM inhibitor point were then averaged. Each row luminescent value was then divided by this value. The results were multiplied 100 times to determine the % enzyme activity in that reaction well.
Prism, version 5.01, from GraphPad Software (La Jolla, CA), was used to calculate all ATP K m, enzyme EC50, [µM ADP], and compound IC50 values.
RESULTS AND DISCUSSION
In order to ensure proper pharmacology when performing kinase profiling experiments on lead compounds, it is imperative that each kinase assay has been previously optimized. These optimization experiments include ATP titration, enzyme titration, and enzyme amount confirmation. Validation of the quality of assay performance can be achieved by determining Z′-factor scores, as well as performing kinetic studies using known kinase inhibitors.
The ATP titration is performed in order that an appropriate ATP concentration is chosen to ensure proper reaction kinetics. This concentration should be sufficient to create an appropriate dynamic range in the assay, while not being too great so as to adversely affect the pharmacology results seen with the enzyme. The enzyme titration and confirmation are included to once again ensure that a proper dynamic range is maintained during profiling applications. Proper compound inhibition curves and IC50 values can be generated when using an enzyme concentration that creates a sufficient signal:background ratio, while still being in the linear range of the enzyme reaction. Once the optimization process is complete, it is then necessary to validate each enzyme reaction, through the use of Z′-factor and known inhibitor IC50 studies. Only a robust assay that provides accurate inhibitory results with predetermined compound/kinase pairs can be trusted to deliver correct kinase profiling data as part of lead compound toxicity studies.
ATP K m app Determination
Assay sensitivity to weak inhibitors is improved by using a concentration of ATP that is at K m app for the enzyme. The same point is also true for working with ATP competitive inhibitors. Using concentrations that are either above or below the K m app value can cause compounds to appear more or less potent, respectively, than what they actually are. This is an important consideration for profiling applications. Table 2 shows the K m app values determined by our protocol as well as the literature values for various kinases. Each was generated by plotting relative luminescence values vs. ATP concentration. The Michaelis-Menten equation was then used to generate the K m app value. By comparing the 2 data sets, it can be seen that the ATP K m app values for all kinases tested closely agree with the literature values, except for Src, where a 6-fold difference was noticed. This difference can be accounted for by the difference in the ability of the kinase to phosphorylate the substrates that were used in the kinase reaction. These results confirm that the automated ADP-Glo Assay is capable of generating appropriate ATP K m app values that agree with existing literature. It is also apparent that the ADP accumulation assay can accommodate a wide range of K m app spanning >2 orders of magnitude.
aReference listed for each literature ATP K m app value.
bIndicates value measured in the presence of manganese.
cLiterature ROCK1 substrate not reported.
Enzyme SB10 Determination
Once ATP K m app values are established, appropriate enzyme concentrations need to be determined. This is typically a balance of minimizing enzyme consumption (ie, cost) and obtaining sufficient signal relative to background (SB) for adequate performance in profiling experiments. It is also important to be certain that the enzyme concentration chosen results in a low % conversion of ATP to ADP to maintain proper kinetics for the enzyme reaction. A concentration that gives high conversion of ATP to ADP would right shift IC50 values of compounds tested with that assay. Often in profiling settings, enzyme concentrations are chosen that would yield luminescent values ∼10 times over the background values (SB10). The first part of the SB10 determination process involved generating enzyme titrations for each of the 5 included kinases. Figure 1 shows the results that were generated with each. [µM ADP], % ATP conversion, and SB levels were then determined using the methods previously explained. The ATP/ADP conversion curves were used not only to compute the [µM ADP] values, but also to verify that the range of luminescent signal produced from each well was within the linear range of the microplate reader. The 100% ADP/0% ATP point on the curve will generate the highest luminescent signal possible for that reaction, due to the fact that all of the ADP was detected. Therefore, since the detection instrument is able to automatically adjust its gain according to this value, all other values will also be correctly quantified. Following this initial quantitation, the data were reorganized into a format where signal:background ratios could be compared to conversion levels of ATP to ADP. This was done in order to see that the % ATP conversion values would be around the SB10 level. Figure 2 demonstrates this comparison. By viewing the results, it is apparent that concentrations of enzyme generating an SB value of ∼10 also result in low conversion levels of ATP to ADP; generally resulting in a % ATP conversion level around 10% or below. Even low activity enzymes, such as ZAP70,16 yield a relatively low % ATP conversion level at SB10. Because of these findings, it was confirmed that by using the concentration of enzyme yielding an SB10 value for each kinase reaction, acceptable data would be generated. Therefore, this amount of enzyme would be used for all validation experiments, as well as kinase profiling.
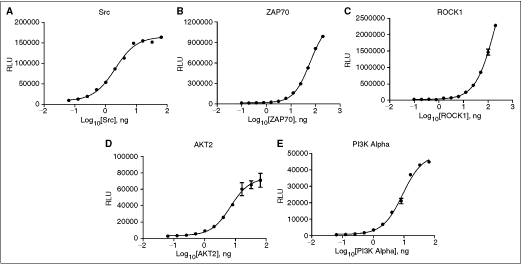
Enzyme titration. 13-point titrations were created for each kinase using the previously determined ATP K
m app values, as well as saturating substrate concentrations. All kinases titrations used a 1:2 dilution scheme. (
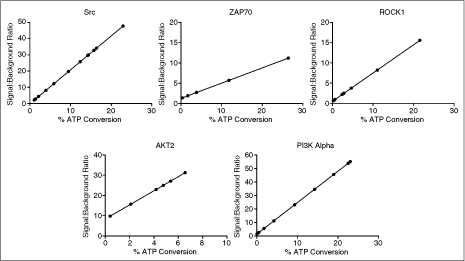
Signal:background ratio vs. % ATP/ADP conversion comparison. Signal:background ratio and % ATP/ADP conversion values were computed from the enzyme titration data and ATP/ADP conversion curve using the methods previously explained in the Materials and Methods Data Analysis section. Signal:background ratios (Y-axis) are shown for 0%–30% ATP/ADP conversion (X-axis), except for AKT2, where the highest % ATP/ADP conversion value attained was ∼7%. Data points represent the average of n = 2 values.
In order to verify the SB values generated for the various kinase concentrations during the enzyme titration, an enzyme confirmation was performed. This was done due to the small variances in concentration that can happen across a serial titration. The goal of the experiment was to identify the exact kinase concentration that would yield an SB10 value. Kinase concentrations were chosen that yielded signal:background levels between SB5 and SB20. Individual enzyme/substrate mixes were then prepared containing the specific concentration of kinase in the assay. Figure 3 shows how the data was then organized to show the SB value in relation to the concentration of enzyme in the reaction. Linear regression formulas from each graph were used to determine the enzyme concentration yielding an SB10 value. The ATP/ADP conversion data also determined from this enzyme confirmation, and listed in each graph of Figure 3, confirms the low % ATP conversion levels generated by this kinase concentration. These values illustrate the sensitivity of the assay. Thus, low ng/rxn amounts of enzyme can be used in each reaction, while still generating SB levels suitable for profiling.
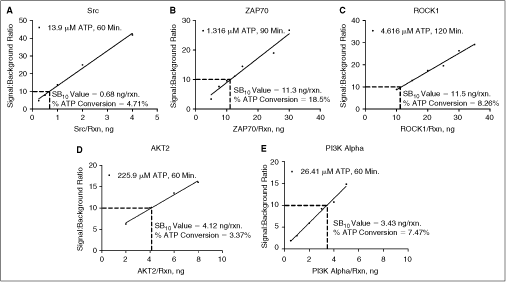
Enzyme SB10 determination. Specific kinase concentrations were identified from the kinase titration that yielded signal:background values between SB5 and SB20. These concentrations were then individually prepared to determine the SB10 value. (
Selective Inhibitor Mode of Action Determination
Before proceeding with kinase profiling, it is necessary to evaluate the optimized kinase reactions, as well as the instrumentation and methods being used to perform each assay. Comparing IC50 values from compound titrations of known inhibitors with existing literature values is a common practice used to accomplish this goal. These values are influenced not only by the assay condition such as enzyme and substrate concentrations, pH, temperature, and so on, but also by the preciseness of compound titration and reagent delivery. Determining Z′-factor17 as a statistical parameter has also been commonly considered an indicator of robustness of assay performance. It incorporates not only assay and background signals, but also the variability in each. For these reasons, each of the parameters listed has been widely adopted as measures of assay robustness in drug discovery applications.
For the IC50 determination, a single inhibitor was identified for each of the 5 optimized kinase reactions. Each inhibitor was titrated and added to a reaction using the K m app and SB10 concentrations of ATP and the corresponding kinase, respectively. Figure 4 shows the inhibition curves that were generated, along with the determined IC50 values. Literature values for these enzyme/inhibitor combinations are as follows: Src/PP2 5 nM,18 ZAP70/Staurosporine 55.8 nM,19 ROCK1/Y27632 140 nM,20 AKT2/AKT 1/2 kinase inhibitor 210 nM,21 and PI3 kinase alpha/Wortmannin 7 nM.22 It is apparent that the IC50 values generated by the automated assay are in agreement with those reported in the literature values.
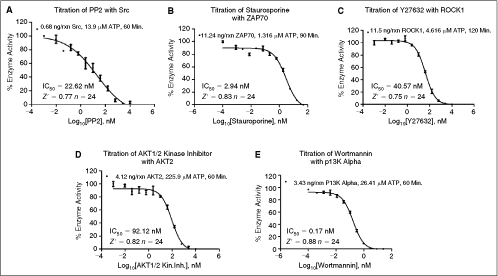
Selective inhibitor assay validation. IC50 determination: 12-point titrations were created for each individual known inhibitor, using the SB10 kinase concentration and determined ATP K
m app value. (
For determining the Z′-factor, the same inhibitors of each kinase were used at 0 and 10 µM concentrations for the positive and negative controls, respectively. Z′ factors generated using 24 replicates ranged from 0.75 to 0.88 (see Fig. 4). This confirms that each of the optimized kinase reactions is robust, using the concentrations of enzyme, ATP, and substrate, as well as the instrumentation described.
Universal Kinase Inhibitor Validation
In order to critically evaluate the performance of the automated ADP-Glo assay, we carried out additional studies. Staurosporine was chosen due to its nonselective inhibition of kinases as it binds to ATP-binding sites that are largely conserved through the kinome.23 Thus it is a useful tool to evaluate the validity of the assay pharmacology for kinase assays. Figure 5 shows the inhibition curves for each of the kinases, as well as the individual IC50 values. Results were in agreement with existing literature values, as IC50 values for Src, ZAP70, ROCK1, and AKT2 were all below 15 nM.24 The IC50 value for PI3 kinase alpha, 2 µM, also closely paralleled what has been reported in the literature where staurosporine is less potent against the PI3 kinase family.25 PI3 kinase alpha, being a lipid kinase, reacts differently with Staurosporine and will possess IC50 values generally orders of magnitude higher26 than for kinases with nonlipid substrates. The results from this study, combined with those from the selective inhibitor mode of action study, confirm that each assay using ADP-Glo technology should yield accurate results when used in a kinase profiling setting.
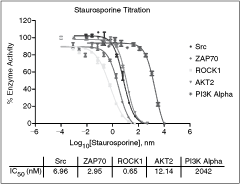
Staurosporine selectivity data. The 12-point titrations of Staurosporine were created at multiple starting concentrations using the SB10 kinase concentration and determined ATP K m app value. All staurosporine titrations used a 1:3 dilution scheme. The Staurosporine starting concentration was 500 nM for Src and AKT2, 50 nM for ZAP70 and ROCK1, and 10,000 nM for PI3 kinase alpha. The % enzyme activity was calculated as described in the Materials and Methods Data Analysis section, and plotted on the Y-axis, based on the log10[Staurosporine] (X-axis). IC50 values were computed using the nonlinear regression sigmoidal dose–response (variable slope) curve fit. Error bars represent standard error of n = 2 values.
Kinase Profiling
The 5 optimized and validated kinase assays were profiled, along with 4 additional kinases (IKK alpha, PKA, PKC, and DNA-PK). The 4 new kinase reactions were previously optimized using the manufacturer’s recommended concentrations and existing literature conditions. These kinases were included in the profiling panel in order to provide data from additional kinase families. Wortmannin was used as the model lead compound of interest. This compound, being a specific inhibitor for PI3 kinase, generates IC50 values typically in the low nanomolar range for this kinase. At higher concentrations, this compound has also shown inhibitory effects on DNA-PK.27 Results once again agreed with literature values, with IC50s for PI3 kinase alpha and DNA-PK being 0.07 and 6.85 nM, respectively (Fig. 6). Wortmannin showed little or no effect on the remaining protein kinases, with only partial or no inhibition curves being seen through the highest concentration of inhibitor tested.
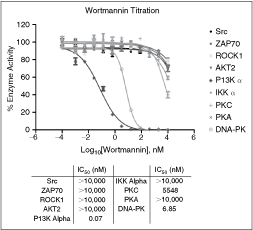
Wortmannin profiling data. The 12-point titrations of Wortmannin were created using a starting concentration of 10,000 nM. All titrations used a 1:5 dilution scheme. Kinase reaction conditions: Src: 0.68 ng/rxn, 13.9 µM ATP, 0.13 mg/mL poly(Glu, Tyr), 60 min. ZAP70: 11.24 ng/rxn, 1.32 µM ATP, 0.13 mg/mL poly(Glu, Tyr), 90 min. ROCK1: 11.5 ng/rxn, 4.62 µM ATP, 152 mM S6K substrate, 120 min. AKT2: 4.12 ng/rxn, 225.9 µM ATP, 50 µM crosstide peptide, 60 min. PI3 kinase alpha: 3.43 ng/rxn, 26.41 µM ATP, 66.67 µg/mL phosphatidylinositol/16.67 µg/mL phosphoserine, 60 min. IKK alpha: 90 ng/rxn, 42 µM ATP, 200 mM IKK peptide, 60 min. PKA: 0.05 U/rxn, 25 µM ATP, 50 µM kemptide, 10 min. PKC: 15 ng/rxn, 100 µM ATP, 100 mM neurogranin peptide, 30 min. DNA-PK: 1.0 U/rxn, 100 µM ATP, 2 mg/mL DNA-PK Peptide, 60 min. The % enzyme activity was calculated as described in the Materials and Methods Data Analysis section, and plotted on the Y-axis, based on the log10[Wortmannin] (X-axis). IC50 values were computed using the nonlinear regression sigmoidal dose–response (variable slope) curve fit. Error bars represent standard error of n = 2 values.
In conclusion, we have successfully validated the new ADP detection technology for use with a simple, inexpensive automated pipetting system. We automated the entire assay process in 384-well format from the optimization of 5 kinase reactions: Src, ZAP70, ROCK1, AKT2, and PI3 kinase alpha; and through generation of kinase profiling data for 9 enzymes. Concordance of the kinetic parameters generated by this assay with those reported in the literature validates the use of the assay chemistry with instrumentation used for automation and provides a simple yet robust solution for automated kinase profiling.
Footnotes
ACKNOWLEDGMENT
The authors would like to thank Sarah Shultz at Promega Corporation for her assistance in the Applications Lab.
AUTHOR DISCLOSURE STATEMENT
B.L. and P.B. are employees of BioTek Instruments, Inc. H.Z. and S.A.G. are employees of Promega Corporation.