
Abstract
Chronic hypoxia-induced pulmonary hypertension (PH) is characterized by increased pressure and resistance in the pulmonary vasculature and hypertrophy of the right ventricle (RV). The transcription factors, nuclear factor activated T-cells (NFAT), and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB/p65) contribute to RV hypertrophy (RVH). Because peroxisome proliferator-activated receptor gamma (PPARγ) activation attenuates hypoxia-induced PH and RVH, we hypothesized that PPARγ inhibits activation of RV hypertrophic transcriptional signaling mechanisms. C57BL/6J mice were exposed to normoxia (21% O2) or hypoxia (10% O2) for 21 days. During the final 10 days of exposure, selected mice were treated with the PPARγ ligand, pioglitazone. RV systolic pressure (RVSP) and RVH were measured, and NFATc2 and NF-kB/p65 protein levels were measured in RV and LV nuclear and cytosolic fractions. Cardiomyocyte hypertrophy was assessed with wheatgerm agglutinin staining. NFAT activation was also examined with luciferase reporter mice and analysis of protein levels of selected transcriptional targets. Chronic-hypoxia increased: (1) RVH, RVSP, and RV cardiomyocyte hypertrophy; (2) NFATc2 and NF-κB activation in RV nuclear homogenates; (3) RV and LV NFAT luciferase activity; and (4) RV protein levels of brain natriuretic peptide (BNP) and β-myosin heavy chain (β-MyHC). Treatment with pioglitazone attenuated hypoxia-induced increases in both RV and LV NFAT luciferase activity. Chronic hypoxia caused sustained RV NFATc2 and NF-κB activation. Pioglitazone attenuated PH, RVH, cardiomyocyte hypertrophy, and activation of RV hypertrophic signaling and also attenuated LV NFAT activation. PPARγ favorably modulates signaling derangements in the heart as well as in the pulmonary vascular wall.
Keywords
Pulmonary hypertension (PH) comprises a heterogeneous group of disorders associated with significant morbidity and mortality despite recent therapeutic advances. Previous work from our lab and others has shown that chronic hypoxia-associated PH promotes changes in pulmonary artery smooth muscle and endothelial cells.1–3 These changes in the pulmonary artery lead to increased pulmonary vascular pressures and resistance that promote changes in the right ventricle. Right ventricular (RV) hypertrophy (RVH) and ultimately RV failure is the main determinant of mortality in patients with PH. 4 Despite the significant role that RVH plays in PH-associated morbidity and mortality, current therapies for PH have not been shown to have direct effects on the right ventricle contractile apparatus. Thus, further understanding and therapeutic manipulation of RV responses to elevations in pulmonary vascular pressures may have important implications for survival. 5
Cardiomyocytes are terminally differentiated cells that lose their ability to proliferate soon after birth. Cardiac hypertrophy is associated with changes in muscle phenotype characterized by the expression of fetal-type genes such as α-actin and brain natriuretic peptide (BNP). 6 Hypertrophy in the ventricle is initiated by stimuli including wall stress, pressure overload, and hypoxia. To adapt to changes in cardiac workload, cardiomyocytes undergo hypertrophy defined as an increase in cell size and protein synthesis.7,8 Although growing interest has stimulated mechanistic studies focused on RVH, many presumed mechanisms of RVH have focused on the pressure overload associated with increased pulmonary vascular resistance rather than direct mechanisms of transcriptional regulation in the RV myocardium. Two well established transcription factors known to be involved in cardiomyocyte hypertrophy are NFAT and NF-κB, both members of the Rel transcription factor family that play critical roles in activating gene expression in hypertrophic cell signaling.9,10 Chronic hypoxia exposure activates both NFAT and NF-κB in pulmonary arteries and pulmonary vascular wall cells in experimental models.2,11,12 Further, pharmacological or knockdown approaches to inhibit these transcription factors attenuated pulmonary vascular remodeling and RVH.9,11,13,14 These observations suggest that strategies to attenuate activation of these hypertrophic transcriptional pathways might attenuate both pulmonary vascular remodeling and RVH.
Peroxisome proliferator-activated receptors (PPARs) are ligand-activated transcription factors belonging to the nuclear hormone receptor superfamily.15–17 These receptors are differentially expressed in various tissues and play important roles in the regulation of diverse cellular processes including metabolism, proliferation, and inflammation. For example, PPARγ is expressed at low levels in the heart where it regulates cardiomyocyte lipid and glucose homeostasis. 18 Work from our group and others has demonstrated that reductions in PPARγ expression are associated with PH,2,3,19–25 whereas activation of PPARγ with thiazolidinedione ligands, e.g. rosiglitazone or pioglitazone, attenuates PH.1,3,19,26–29 In addition to reducing right ventricular systolic pressure (RVSP) in experimental models of PH, PPARγ activation also attenuated RVH. However, the impact of PPARγ ligands on RV hypertrophic transcriptional pathways during chronic hypoxia-induced PH pathogenesis has not been defined. 30 Based on evidence that PPARγ activation can mediate transrepression of other transcriptional pathways, 31 we hypothesized that PPARγ would attenuate activation of NFAT and NF-κB in the RV, reduce expression of their downstream targets, and attenuate RVH and RVSP in a hypoxia-induced mouse model of PH.
Methods
Mouse model of chronic hypoxia-induced PH and RVH
A well-established mouse model of hypoxia-induced PH was employed to examine RVH in vivo. Male C57BL/6J mice aged 8–12 weeks were obtained from Jackson Labs (Bar Harbor, ME, USA) and exposed to normoxia (21% O2) or hypoxia (10% O2) for 3 weeks as we have previously reported. 3 During the final 10 days of exposure to normoxia or hypoxia, each animal was treated with pioglitazone (PIO) (10 mg/kg/d) or with an equivalent volume of vehicle (methylcellulose) by oral gavage. All animals had access to standard mouse chow and water ad libitum and were maintained on a 12:12-h light-dark cycle. All animal protocols employed were reviewed and approved by the Institutional Animal Care and Use Committee of the Atlanta Veterans Affairs Medical Center.
Following exposure to normoxia or hypoxia and treatment ± PIO, RVSP were measured as previously reported. 32 Briefly, RV systolic pressure (RVSP) was measured in mice lightly anesthetized with isoflurane. A 0.8-F microtip pressure transducer (Millar Instruments, Houston, TX, USA) was inserted into the surgically exposed right jugular vein and advanced into the RV. RVSP was continuously monitored for 10 min and data were analyzed using a Powerlab system (AD Instruments, Denver, CO, USA). Hearts were removed and the RV free wall was dissected from the left ventricle and septum (LV+S). Ratios of the weight of the RV to the LV+S were calculated as an index of RVH. 32 In selected studies, the RV and LV+S tissues were minced and homogenized in ice-cold buffer (200 mM Sucrose, 20 mM Tris, 2 mM ethylene glycol tetraacetic acid, 0.3 mM phenylmethylsulfonyl fluoride plus protease and phosphatase inhibitors) then centrifuged at 1000 × g for 10 min at 4℃. The resulting supernatant was centrifuged again at 10,000 × g for 30 min at 4℃ to separate the cytosolic and nuclear fractions. The nuclear pellet was washed in ice-cold buffer three times and re-suspended in radioimmunoprecipitation assay buffer containing phosphatase and protease inhibitors for 30 min at 4℃ and then centrifuged at 21,000 × g for 30 min at 4℃ to obtain the nuclear fraction.
Western blot analysis
Immediately after sacrifice, homogenates of the RV and LV were prepared and their protein concentrations determined. After cellular fractionation, proteins (20 µg per lane) were subjected to SDS-PAGE (4–12% gradient gels) (Invitrogen, Carlsbad, CA, USA) followed by electroblotting of proteins onto nitrocellulose membranes. After appropriate blocking (5% non-fat dried milk for NFATc2 and 3% bovine serum albumin for NF-κB), the blots were probed with primary antibodies (1:1000) specific to NFATc2 and NF-κB/p65 (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Total RV homogenates (20 µg per lane) were also subjected to SDS-PAGE then probed with primary antibodies to PPARγ, BNP (1:1000, Abcam, Cambridge, MA, USA), β-MyHC (1:4000, Abcam), phospho-NFATc2, or NFATc2 (1:1000, Santa Cruz Biotechnology) in 5% powdered non-fat dry milk or 3% bovine serum albumin, respectively, on a rocking platform overnight at 4℃. After washing, PPARγ and NFATc2 membranes were incubated with horseradish peroxidase-conjugated secondary antibody (Jackson Immuno Research Labs, West Grove, PA, USA). BNP and β-MyHC membranes were incubated with anti-rabbit antibody. Proteins were normalized to the β-actin, histone, or fibrillarin content of the same sample. Levels of protein in the cytosolic fraction were normalized to α-tubulin. Total protein was normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) or β-actin in the same sample. For all blots, immunodetection was performed using a LI-COR Odyssey infrared fluorescence imaging system (LI-COR Biosciences, Lincoln, NE, USA). Quantitative analysis of blots was performed with the use of Scion Image software (Scion based on NIH image).
Analysis of cardiomyocyte hypertrophy
Changes in the size of mouse cardiomyocytes were measured by fluorescence staining of ventricular sections from each experimental group. Hearts were perfusion-fixed with 4% paraformaldehyde and embedded in Tissue-Tek. Fluorescence-tagged wheat germ agglutinin (WGA, Sigma-Aldrich, St. Louis, MO, USA) was employed to measure the cardiomyocyte cross-sectional area as previously reported. 9 Briefly, myocyte cross-sectional areas were visualized using a membrane staining fluorescein isothiocyanate-conjugated WGA (10 µg/ml) for 2 h at room temperature. Images of WGA-stained myocytes were captured digitally and the cross-sectional areas were calculated using NIH Image J software. Three sections randomly selected from each of four animals in each experimental group were examined and the cross-sectional areas were calculated for at least 50 myocytes per section.
RNA isolation, reverse transcription, and quantitative PCR
Total RNA was isolated from the RV using the RNeasy Mini Kit (Quiagen, Valencia, CA, USA) and quantitated using NanoDrop spectrophotometry (Thermo Scientific, Wilmington, DE, USA). cDNA was prepared using the iScript cDNA Synthesis Kit (Bio-Rad Laboratories, Hercules, CA, USA) and reverse transcribed from 1 µg of total RNA. Real-time PCR was then performed to characterize the expression of target genes with primers based on human RNA sequences using Applied Biosystems® 7500 fast Real-Time PCR System (Thermo Fisher Scientific, Waltham, MA, USA). Primers for amplification of BNP and GAPDH were as follows: BNP: 5′-GCTCCTGCTCTT CTTGCATC-3′ and 5′-AGGGATGTCTGCTCCACCT-3′; GADPH: 5′-ACAAGCTTCCC GTTCTCCAG-3′ and 5′-GGTCACCAGGCTGCTTTTAAC-3′. All samples were analyzed in duplicate. The relative abundance of target mRNA in each sample was calculated using the ΔΔCt method. 33
NFAT-luciferase reporter mice
To confirm NFAT activation, NFAT-Luc transgenic reporter mice aged 8–12 weeks (generously provided by Dr. Jennifer Gooch) were utilized. This NFAT-Luc construct was designed using nine copies of an NFAT binding site from the IL-4 promoter (5’-TGGAAAATT-3’) positioned 5’ to a minimal promoter from the alpha-myosin heavy chain gene (–164 to +16) and inserted upstream of the luciferase reporter in pGL-3 Basic (Promega Corporation, Madison, WI, USA) to create NFAT-luc. This NFAT-luciferase transgene was injected into newly fertilized oocytes to generate phenotypically normal transgenic mice (FVBN background). 34 Briefly, these reporter mice were exposed to 3 weeks of hypoxia or normoxia ± treatment with pioglitazone. RV and LV tissues were isolated, homogenized in lysis buffer (1 µL/µg), and particulate matter was separated by centrifugation at 12,000 × g for 1 min. Luciferase assay reagent (100 µL) was added to 20 µL of supernatant and luminescence was measured for 10 s using an OptoComp luminometer (MGM Instruments, Hamden, CT, USA) as previously reported. 35 As an internal control for background luminescence, luminescence values were obtained from littermate mice that did not express the construct.
Statistical analysis
For all experiments, statistical analysis was performed using one-way analysis of variance (ANOVA) followed by a Tukey’s post-hoc analysis to detect differences among experimental groups. The level of statistical significance was set at an alpha value of P ≤ 0.05. Statistical analyses were carried out using GraphPad Prism, Version 5.0 software (LaJolla, CA, USA)
Results
Elevations in RVSP and RVH are hallmark features of PH. Previous studies from our lab have shown that treatment with the PPARγ ligand, rosiglitazone, attenuates hypoxic increases in RVSP and RVH in the mouse model.
3
As illustrated in Fig. 1, the current study extends previous observations by confirming that hypoxia increased both RVSP and RVH in mice, and these hypoxic increases in RVSP and RVH were similarly attenuated by treatment with an alternative thiazolidinedione PPARγ ligand, pioglitazone (Fig. 1a and b). While the magnitude of pioglitazone-induced attenuation of hypoxia-mediated PH and RVH was small, pioglitazone normalized hypoxia-induced increases in the cardiomyocyte cross-sectional area (Fig. 1c and d).
Pioglitazone attenuates hypoxia-induced increases in RVSP, RVH, and cardiomyocyte surface area. C57BL/6J mice were exposed to normoxia (NOR, 21% O2) or hypoxia (HYP, 10% O2) for 3 weeks. During the last 10 days of exposure, mice were treated with pioglitazone (PIO) or vehicle (VEH). (a) Each point represents the RV systolic pressure from 13–15 animals in mmHg with mean RVSP ± SEM presented in superimposed lines. *P < 0.05 vs. NOR and +P < 0.05 vs. HYP. (b) Ratios of the weights of the RV to the LV + septum from 15–17 animals are presented as individual points with mean RV: LV+S ± SEM presented as superimposed lines. *P < 0.05 vs. NOR and +P < 0.05 vs. HYP. To demonstrate hypertrophy on the cellular level, sections of the RV were labeled with fluorescence-tagged wheat germ agglutinin, images were captured digitally, and cross-sectional area was measured using Image J. (c) Each bar represents the mean cardiomyocyte surface area ± SEM from 50 cells from at least three 7 µm sections per animal (n = 4). *P < 0.05 vs. NOR and +P < 0.05 vs. HYP. (d) Representative images are shown. Magnification, ×40.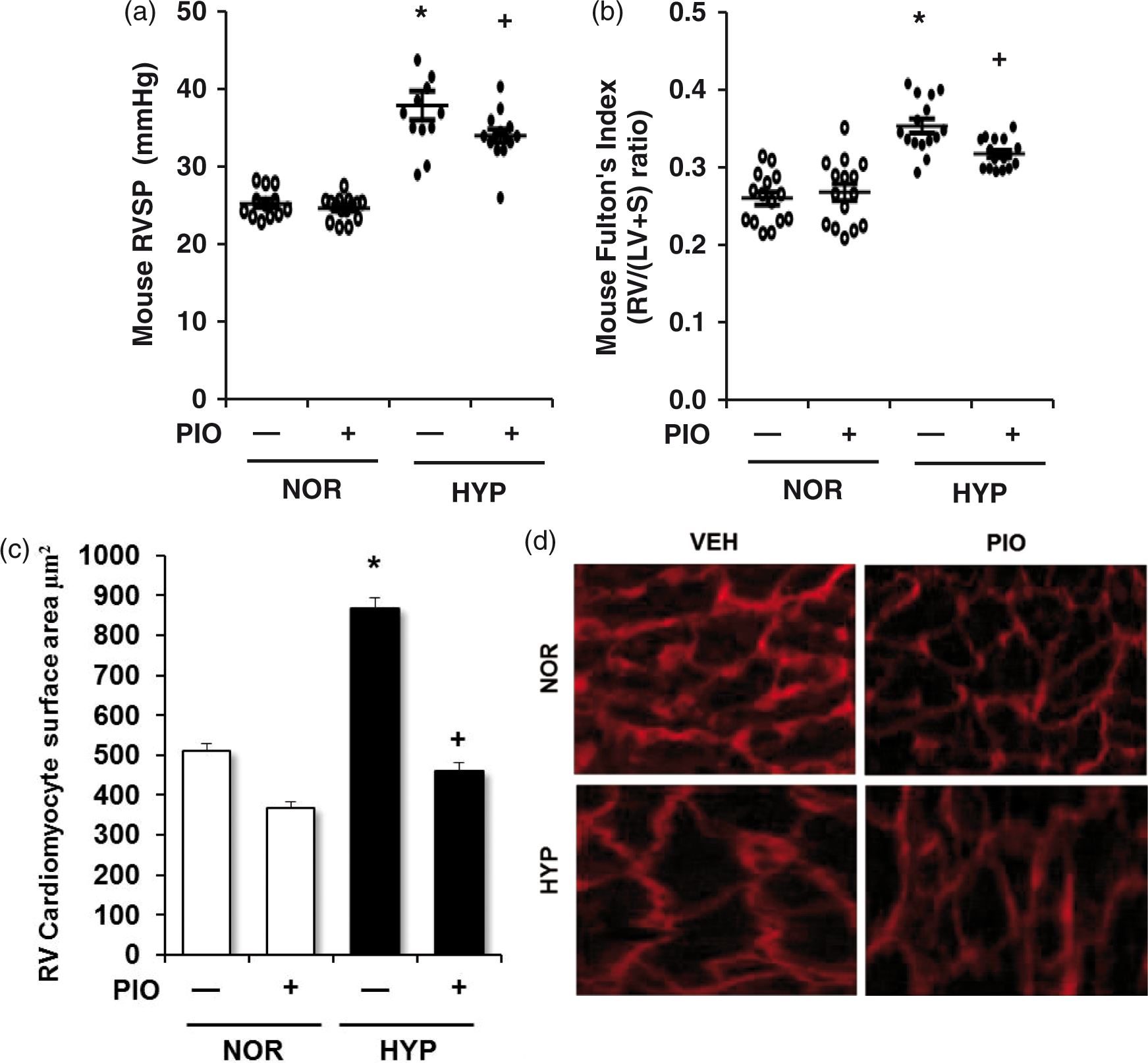
To further examine the impact of pioglitazone treatment on hypertrophic transcriptional signaling pathways in the right ventricle, NF-κB and NFAT activation were examined by determining their nuclear translocation in fractions prepared from RV homogenates and subjected to western blotting. Compared to normoxic exposure, chronic hypoxia caused a roughly sixfold increase in nuclear NF-κB/p65 levels and a concomitant reduction in cytosolic NF-κB/p65 (Fig. 2a and c). In contrast, compared to normoxia exposure, hypoxia increased NFAT nuclear translocation (Fig. 2b) but alterations in cytosolic NFAT were not detected (Fig. 2d). Treatment with pioglitazone attenuated hypoxia-induced increases in nuclear and decreases in cytosolic NF-κB/p65 and attenuated hypoxic NFAT nuclear translocation.
Pioglitazone attenuates chronic hypoxia-induced NF-κB and NFAT nuclear translocation in the right ventricle. Nuclear and cytosolic fractions were prepared from the RV homogenates of mice treated as described in Figure 1. (a) Nuclear NF-κB/p65 levels were normalized to histone levels in the same sample. Each bar represents mean ± SEM nuclear NF-κB/p65: histone levels expressed as fold-change vs. control (n = 4 animals/group). (b) Nuclear NFATc2 levels were normalized to histone levels in the same sample. Each bar represents mean ± SEM nuclear NFATc2:histone levels expressed as fold-change vs. control (n = 4 animals/group). (c) Cytosolic NF-κB/p65 levels were normalized to α-tubulin levels in the same sample. Each bar represents mean ± SEM cytosolic NF-κB/p65:α-tubulin levels expressed as fold-change vs. control (n = 4 animals/group). (d) Cytosolic NFATc2 levels were normalized to α-tubulin levels in the same sample. Each bar represents mean ± SEM cytosolic NFATc2: α-tubulin (n = 4 animals/group). In all graphs *P < 0.05 vs. NOR and +P < 0.05 vs. HYP. Representative immunoblots are presented above each bar graph.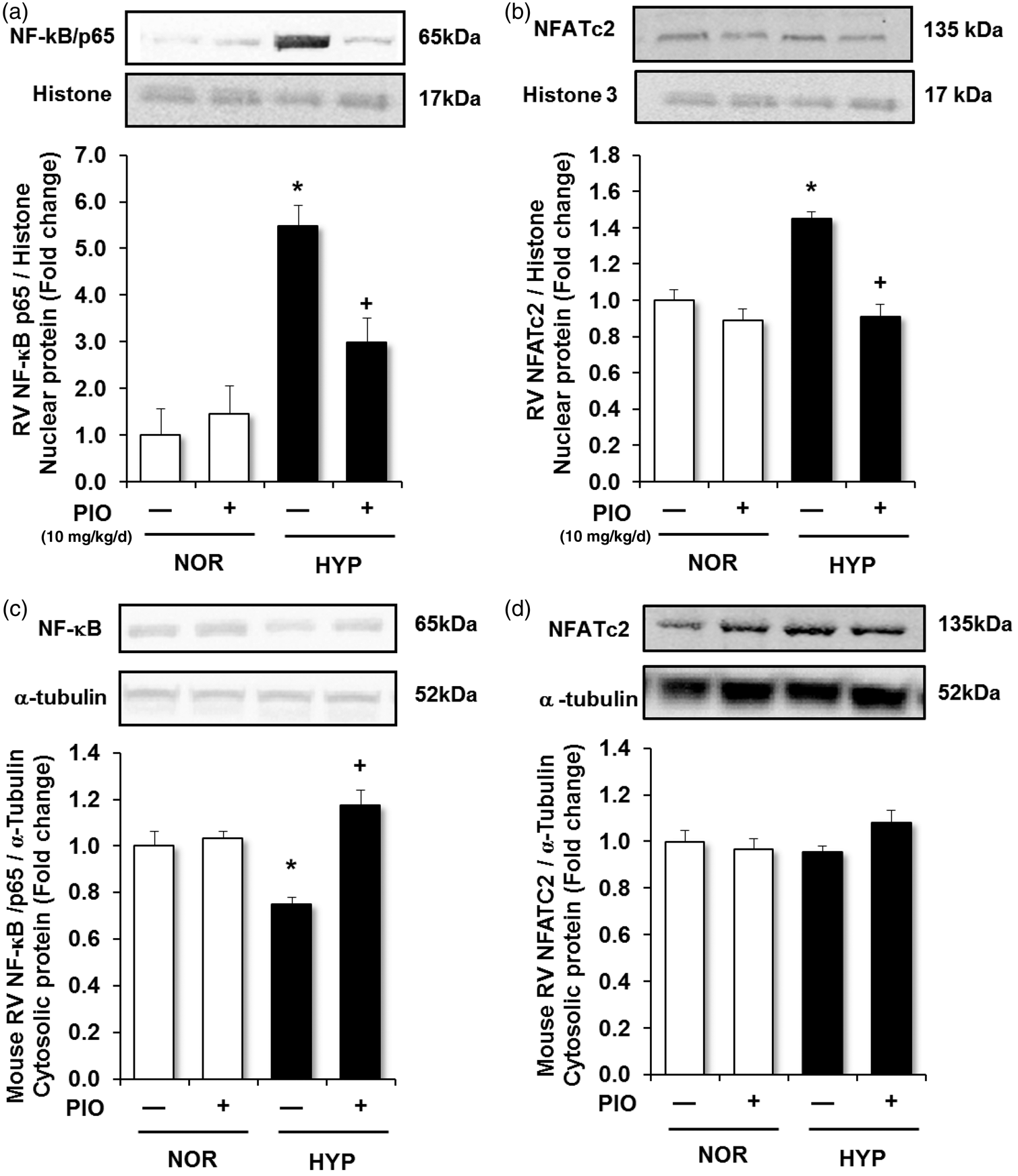
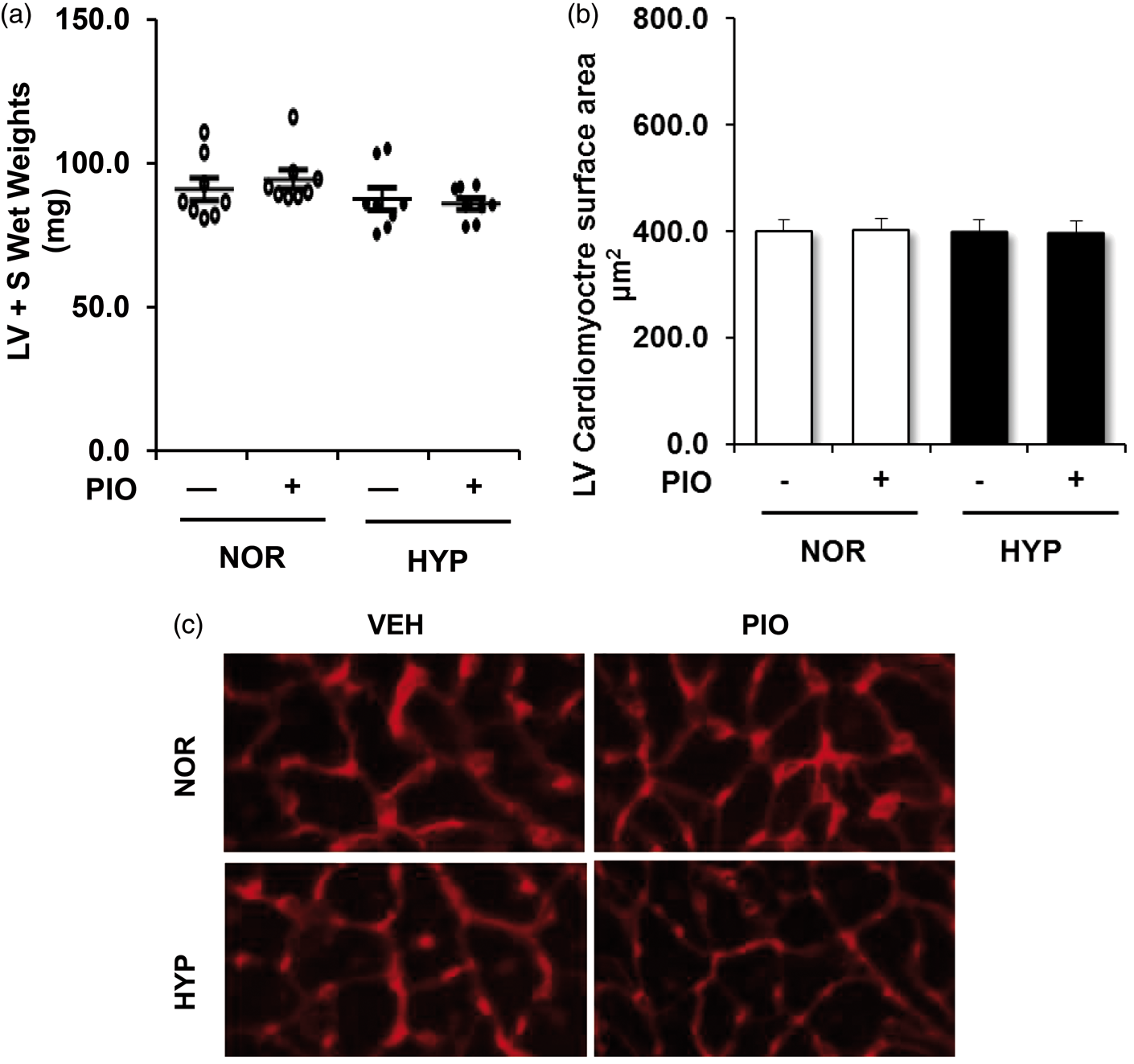
To further confirm hypoxia-induced alterations in NFAT activity in the right ventricle, we employed transgenic NFAT luciferase reporter mice. Chronic hypoxia stimulated a roughly fourfold increase in NFAT activity in the RV that was attenuated by pioglitazone treatment (Fig. 3a). Surprisingly, chronic hypoxia also stimulated a roughly twofold increase in NFAT activity in the LV that was also attenuated by pioglitazone treatment (Fig. 3b).Because hypoxia exposure activated NFAT activity in LV homogenates (Fig. 3), we sought to more carefully analyze the impact of pioglitazone treatment on LV hypertrophic signaling pathways. However, in contrast to observations in the RV, hypoxia failed to stimulate activation and nuclear translocation of either NF-κB/p65 (Fig. 3c) or NFAT (Fig. 3d). Chronic hypoxia also failed to increase LV weight (Fig. 4a) or LV cardiomyocyte surface area (Fig. 4b and c).
Pioglitazone attenuates hypoxia-induced RV and LV NFAT luciferase reporter activity. NFAT-luciferase reporter mice were exposed to normoxia (NOR) or hypoxia (HYP) and treated ± pioglitazone (PIO) as described in Fig. 1. (a) Each bar represents mean ± SEM luciferase activity in RV homogenates expressed as fold-change vs. control. (b) Each bar represents mean ± SEM luciferase activity in LV homogenates expressed as fold-change vs. control (n = 6). (c) LV nuclear NF-κB/p65 levels were normalized to fibrillarin levels in the same sample. Each bar represents mean ± SEM nuclear NF-κB/p65:fibrillarin levels (n = 6). (d) Nuclear NFATc2 levels were normalized to fibrillarin levels in the same sample. Each bar represents mean ± SEM nuclear NFATc2: fibrillarin levels (n = 6). In all graphs *P < 0.05 vs. NOR and +P < 0.05 vs. HYP. Chronic hypoxia does not induce LVH or LV cardiomyocyte hypertrophy. C57BL/6J mice were exposed to normoxia (NOR) or hypoxia (HYP) and treated ± pioglitazone (PIO) as described in Fig. 1. (a) Each point represents the LV + S weight in mg from eight animals with the mean ± SEM values presented as superimposed lines. (b, c) LV sections were labeled with fluorescence-tagged wheat germ agglutinin, and LV cardiomyocyte cross-sectional area was measured. (b) Fifty cells from at least three sections per animal were analyzed (n = 4 mice/group). Each bar represents the mean ± SEM LV cardiomyocyte surface area in µm2. (c) Representative images are presented. Magnification, ×40.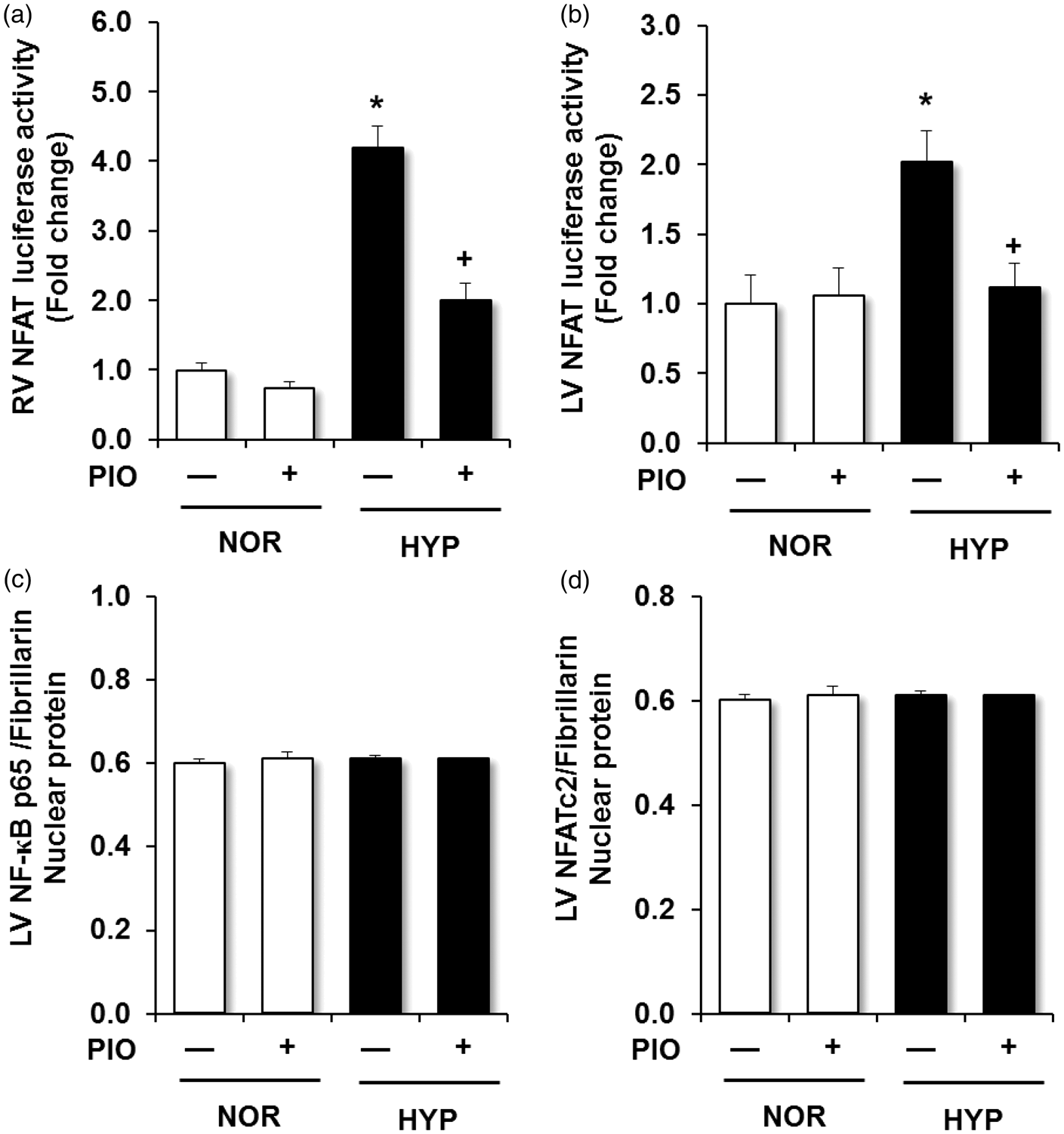
To further examine these transcriptional pathways activated in the RV during hypoxic PH pathogenesis, downstream targets previously reported to be regulated by NFAT such as BNP and β-MyHC were examined.
10
Consistent with hypoxic NFAT activation, levels of BNP and β-MyHC were increased following hypoxia exposure, and pioglitazone treatment attenuated hypoxic increases in BNP and β-MyHC (Fig. 5).
Pioglitazone attenuates hypoxia-induced increases in RV BNP and β-MyHC. C57BL/6J mice were exposed to normoxia (NOR) or hypoxia (HYP) and treated ± pioglitazone (PIO) as described in Fig. 1. RV homogenates were prepared and subjected to western blotting. (a) Each bar represents mean ± SEM BNP protein levels normalized to β-actin in the same sample and expressed as fold-change vs. control. (b) Each bar represents mean ± SEM β-MyHC protein levels normalized to β-actin in the same sample and expressed as fold-change vs. control (n = 5). *P < 0.05 vs. NOR and +P < 0.05 vs. HYP. Representative immunoblots are presented above each bar graph.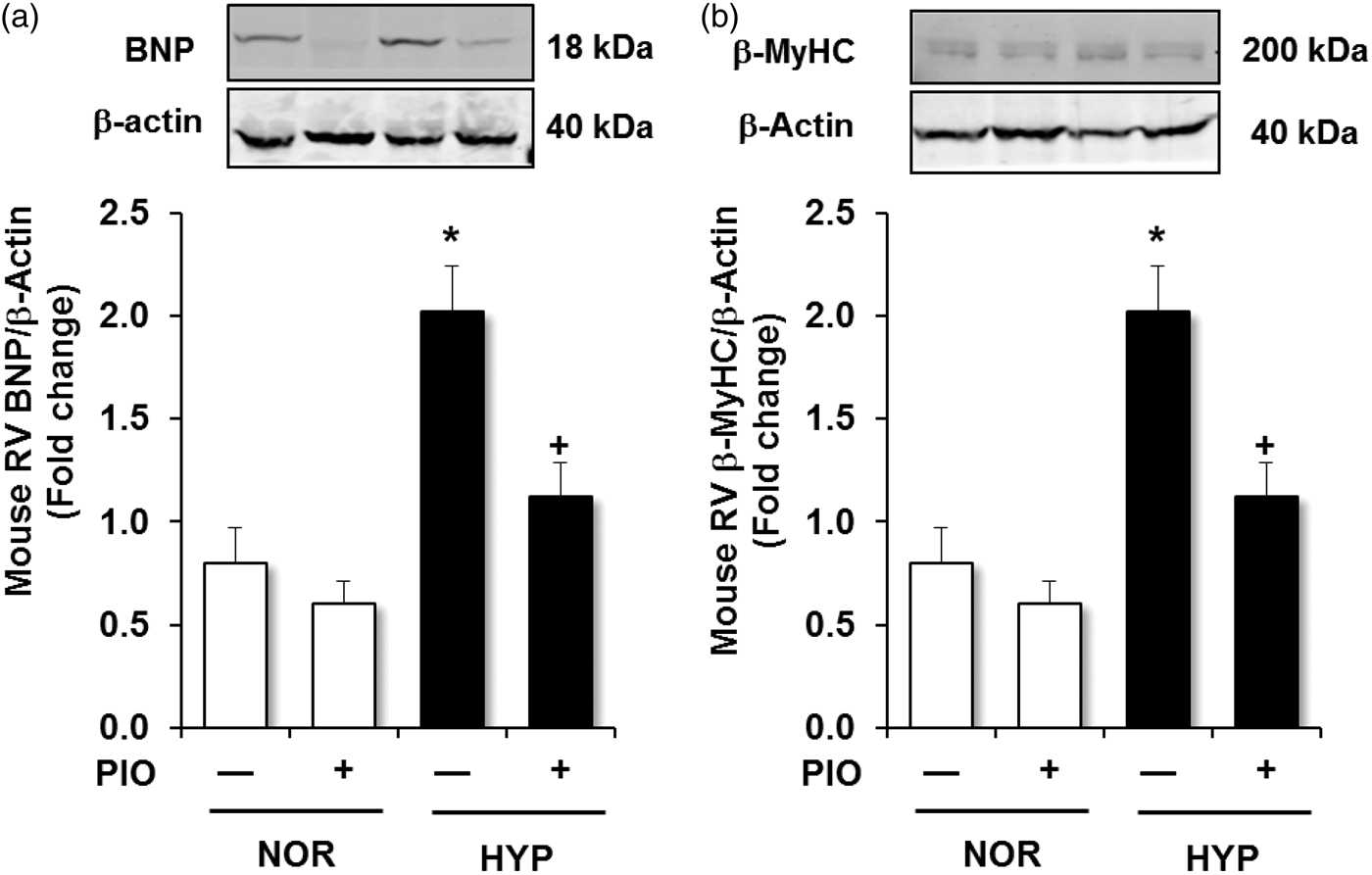
Discussion
The current study demonstrates that NFAT and NF-κB are activated in the RV myocardium following exposure to chronic hypoxia in vivo and that activation of these transcriptional pathways can be attenuated by therapeutically targeting PPARγ with the thiazolidinedione ligand, pioglitazone. Although previous studies have shown that PPARγ activation attenuates PH, RVH, and pulmonary vascular remodeling in several experimental models,1,3,19,26–28,36 this is the first report to describe PPARγ-mediated regulation of transcriptional pathways controlling RV cardiomyocyte hypertrophy. Daily oral administration of pioglitazone attenuates the activation and nuclear translocation of NFATc2 and NF-κB/p65 in the right ventricle. These transcriptional pathways coordinate a program of hypertrophic gene expression that includes reactivation of fetal ventricular gene expression profiles and enhanced expression of downstream targets such as BNP and β-MyHC.10,37,38 Coupled with previous evidence that PPARγ ligands attenuate PH and activation of proliferative pathways in the lung and pulmonary vasculature,39,40 these findings suggest that PPARγ activation may represent a novel therapeutic intervention that can attenuate pathobiological derangements in both pulmonary vascular and RV signaling.
As previously reported, using the current mouse model of hypoxia-induced PH, treatment with the PPARγ ligand, rosiglitazone, attenuated hypoxic increases in RVSP, RVH, and pulmonary vascular remodeling. 3 In the rat model of hypoxia-induced PH, in one study treatment with rosiglitazone attenuated hypoxia-induced RV hypertrophy and pulmonary vascular remodeling but not increases in RVSP, 1 whereas in others, treatment with rosiglitazone attenuated hypoxia-induced increases in RVSP and pulmonary vascular remodeling but not increases in RVH.19,36 Treatment with PPARγ ligands also attenuated experimental PH and RV hypertrophy: (1) in ApoE–/– mice caused by high fat diets; 26 (2) in rats caused by monocrotaline; 28 or (3) in rats treated with hypoxia or monocrotaline. 27 The differences in responses to PPARγ ligands among these models is most likely attributable to differences in dosing, route, or duration of treatment, the species studied (mouse versus rat), and stimulus used to provoke PH. The current report extends those studies to demonstrate that pioglitazone also attenuates hypoxia-induced increases in RVSP and RVH, albeit to a smaller degree than those reported for rosiglitazone. Both rosiglitazone and pioglitazone are synthetic thiazolidinedione PPARγ ligands employed clinically to enhance insulin sensitivity in patients with type 2 diabetes. Pioglitazone was selected for study in the current report because, unlike rosiglitazone, it has not been associated with adverse cardiovascular outcomes in diabetic patients. 15 And in post-marketing trials in millions of patients with type 2 diabetes, neither of these thiazolidinedione medications has been associated with reports of adverse pulmonary effects.
Pioglitazone treatment caused small but significant reductions in hypoxic increases in RVSP and RVH. However, compared to normoxic controls, these indices of PH remained elevated despite pioglitazone treatment (Fig. 1). In contrast, pioglitazone fully normalized hypoxic increases in RV cardiomyocyte size (Fig. 1), caused relatively greater reductions in hypoxic activation of hypertrophic transcriptional signaling in the RV (Fig. 2), and normalized the levels of selected protein targets of hypertrophic transcriptional signaling in the RV (Fig. 5). Pioglitazone treatment also attenuated activation of NFAT in the LV detected using the NFAT-luciferase mouse model (Fig. 3). Collectively these findings suggest that pioglitazone exerts direct effects on the ventricular myocardium during chronic hypoxia. Our studies do not address the nature of the persistent increases in RV mass in hypoxic animals following treatment with pioglitazone. We speculate that while hypoxia-induced cardiomyocyte hypertrophy may be particularly susceptible to reversal by PPARγ activation, other components of RVH, such as fibroblast or inflammatory cell infiltration, endothelial cell proliferation and increased capillary density, extracellular matrix deposition, and potentially influx and differentiation of progenitor cells, 5 may be less or unresponsive to PPARγ activation and thereby lead to persistent increases in RV mass. Additional studies will be required to more precisely characterize the relative contributions of these processes to hypoxia-induced RVH, the signaling pathways involved in activating these processes, and whether these elements might be susceptible to inhibition by more prolonged treatment with pioglitazone.
Coupled with previous reports demonstrating that PPARγ effectively reduces alterations in the pulmonary vasculature during PH pathogenesis, the current findings suggest that therapeutic strategies, such as PPARγ activation that target pathogenic derangements in both pulmonary vascular and myocardial tissues, may be uniquely effective as novel therapeutic agents in PH. However, current concepts suggest that cardiomyocyte hypertrophy is initially adaptive, enabling the right ventricle to generate flow against the increased resistance in the pulmonary circulation. Isolated reductions in cardiomyocyte hypertrophy in the absence of reductions in pulmonary vascular resistance might therefore be maladaptive in PH pathogenesis. Because pioglitazone reduced both cardiomyocyte hypertrophy and pulmonary vascular remodeling, we speculate that these combined effects might be therapeutically beneficial; however, this hypothesis will require more rigorous testing in animal models of PH associated with more severe RV dysfunction and failure.
The current study employed a combination of approaches to examine NFAT and NF-κB activation in the heart. Translocation of these transcription factors from the cytosol to the nuclear compartment was analyzed with western blotting. Hypoxia stimulated a roughly fivefold increase in p65 nuclear translocation in the RV that was accompanied by reciprocal reductions in cytosolic p65 (Fig. 2). Hypoxia also caused a 40% increase in NFAT nuclear translocation detected by similar methods that was not associated with reciprocal reductions in cytosolic NFAT levels (Fig. 2). Therefore, to confirm myocardial NFAT activation, we employed NFAT-luciferase reporter mice. In this model, hypoxia activated not only RV NFAT activity, but also caused smaller increases in LV NFAT activity (Fig. 3). This level of LV NFAT activation was not detected by analysis of NFAT nuclear translocation (Fig. 3) nor was it associated with LVH or LV cardiomyocyte hypertrophy (Fig. 4). Hypoxia-induced upregulation of protein targets of NFAT provides additional evidence for RV NFAT activation (Fig. 5). These results suggest that the luciferase reporter mouse provides a more sensitive assay of NFAT activation in the heart. Taken together, these observations indicate that: (1) systemic as well as pulmonary molecular signaling mechanisms are activated during exposure to chronic hypoxia; and (2) treatment with pioglitazone effectively attenuates these pathways in both systemic and pulmonary compartments. While our studies do not directly examine the nature of the signals activated by hypoxia that cause LV NFAT activation, it is well established that chronic hypoxia increases circulating concentrations of vasoactive mediators and stimulates neurohormonal responses that can impact the LV. 5 Thus chronic hypoxia may be sufficient to activate signaling in the LV, but in the absence of LV pressure overload, insufficient to induce LV hypertrophy. Evidence that PPARγ plays an important role in regulation of systemic cardiovascular signaling and function 16 suggests that studies characterizing PPARγ-mediated regulation of hypoxic LV signaling will provide interesting avenues for future investigation.
Our study has several important limitations that merit further consideration. First, the hypoxia-induced PH model in the mouse fails to recapitulate many of the pathobiological derangements observed in patients with PH. 41 As a result, findings using this model will require confirmation in additional experimental models and/or in human tissues. Further, hypoxia-induced mouse models of PH do not develop RV failure. Thus, the exploration of PPARγ activation as a novel therapeutic approach in PH will require further testing in experimental models associated with more severe derangements in RV function. Second, this study employs systemic administration of the thiazolidinedione, pioglitazone, as a means to activate PPARγ. Although rosiglitazone and pioglitazone are both strong PPARγ agonists, drugs in this class can also exert biological effects through PPARγ-independent mechanisms. PPARγ activation stimulates the expression of selected genes by binding to PPAR response elements in their promoters. However, PPARγ activation also suppresses the activity of other pro-inflammatory transcription factors, including NF-κB and NFAT, through transrepression mechanisms. 31 Thus we speculate that pioglitazone activates PPARγ to directly inhibit NFAT and NF-κB although this inhibition could also be due to more upstream effects of pioglitazone and PPARγ inhibiting hypertrophic transcriptional signaling pathways through indirect mechanisms. Additional study will be required to define the precise molecular mechanisms involved in this inhibition. Finally, although rosiglitazone therapy has been associated with adverse cardiovascular outcomes in diabetic patients, pioglitazone therapy exerts beneficial effects on cardiovascular outcomes in these patients. 42 These findings emphasize that subtle alterations in the chemical structure of PPARγ ligands can induce unique biological effects. Exploiting the pharmacological potential of this receptor by designing ligands that maximize therapeutic and minimize adverse effects provides opportunities for developing new classes of therapeutic agents for PH.
In summary, the current report provides novel evidence that PPARγ regulates hypertrophic transcriptional pathways that are activated in the RV during chronic hypoxia. In concert with previous reports that PPARγ activation attenuates pathogenic pathways in the pulmonary vasculature, these results provide additional evidence that pharmacological PPARγ activation may provide a novel approach for reducing both pulmonary vascular and cardiac pathophysiology in PH.
Footnotes
Acknowledgments
The authors thank Ms. Jennifer Kleinhenz and Ms. Tamara Murphy for their excellent technical support. This work was presented in part in abstract form at the American Thoracic Society Meeting in 2015.
Conflict of interest
The author(s) declare that there is no conflict of interest.
Funding
This study was supported by funding from: Veterans Affairs Basic Laboratory Research and Development Merit Review Award (1I01BX001910 to CMH); NIH NHLBI R01 grant (HL102167 to CMH and RLS) and T32 training grant (HL076118 for KAC); and American Heart Association National Scientist Development Grant (13SDG14150004 to BYK).