
Abstract
The term psychosis has been refined over the last 40 years from a loose definition of functional impairment due to gross psychiatric illness [1] to a more restrictive definition emphasizing delusions and hallucinations, or disorganized thought and behaviour. Although these symptoms are the hallmark of schizophrenia, they can also be present in a range of medical and neurological conditions which have long been described as ‘organic psychosis’ [2]. However, the definition of organic as ‘identifiable structural brain disease or toxic-metabolic brain dysfunction’ is problematic as the ‘non-organic’ psychoses also have demonstrable brain pathology, and the alternative classification of primary and secondary schizophrenia has been suggested to more appropriately capture the aetiologies of psychosis [3]. Furthermore, a broad definition of the term organic psychosis is often additionally used in the literature and clinical medical practice to refer to the presence of psychotic symptoms in dementia or delirium. A more restrictive definition excluding these is preferred for its diagnostic specificity and for its potential to shed light on the aetiopathological mechanisms of psychosis. Secondary presentations may account for approximately 3% of newly presenting schizophrenia [4], and the distinction is significant as many of the aetiologies are treatable. Although secondary schizophrenias may at times be discernible from primary schizophrenic illness on clinical grounds [5],[6], they are often indistinguishable [7] and we suggest that in this latter circumstance the term ‘schizophrenia-like psychosis’ (SLP) is used to denote these presentations that present with psychotic symptoms characteristic of schizophrenia and to exclude psychosis in the context of delirium and frank dementia, where clouding of consciousness or gross cognitive impairment co-present with psychotic symptoms. For the purposes of this review, we limit our discussion to reports of patients presenting with SLP without gross cognitive impairment or clouding of consciousness.
In a landmark review, Davison and Bagley reported a number of statistical associations between SLP and central nervous system (CNS) disorders [8], most notably epilepsy and head injury, but also infections, Wilson's disease, Huntington's disease, cobalamin deficiency and neoplastic disease [8]. Many of these associations have been strongly replicated since that time [4], leading to a greater understanding of the biology of schizophrenia by emphasizing the role of cortical and subcortical structures. Current neuroanatomical models of schizophrenia propose that there is aberrant integration of functionally interdependent circuits, particularly involving frontal and other association cortical regions and their interconnections and connections to limbic and other subcortical structures [9], and schizophrenia is now widely seen at a functional level as a ‘disconnectivity syndrome’. The putative anatomical substrates for this disconnectivity have been theorized as being at the level of the synapse [10],[11], within frontosubcortical [12] or frontotemporal [13],[14] circuitry, a cerebello-corticosubcortical loop [15] or interhemispheric callosal connections [16].
With the advent of newimaging and genetic methodologies, the role of white matter pathology in schizophrenia as a potential substrate for this disconnectivity has begun to emerge [17],[18]. Given that schizophrenia is a disorder of young adult-onset [19] that appears to affect frontotemporal [19], frontosubcortical [12] and callosal structures [20], it could be expected that CNS disorders affecting white matter tracts connecting these regions may result in psychosis [21]. This review will explore this hypothesis for dysmyelinating and demyelinating diseases, neoplastic and inflammatory diseases, callosal abnormalities and other developmental disorders, and outline mechanisms by which these diseases and their pattern of lesions may lead to psychosis.
White matter structure and development
Axons and their myelin sheaths make up close to half the adult brain's volume [22],[23], with left-greater-thanright asymmetry [24]. Myelination, which begins during the middle trimester of pregnancy [25], is 90% complete by 24 months of age [26] and then proceeds slowly until the sixth decade of life [27],[28]. The brainstem and cerebellum myelinate first, then diencephalon, posterior cortical and finally anterior cortical regions [29]. The last tracts tomyelinate are commissural and associative fibres [27]. White matter density and organization increases as this process ensues [30]. During adolescence and the first three decades of adulthood, grey matter volume loss occurs [31], while white matter volume expands, producing an overall constancy in brain volume across early to mid-adulthood [23],[27],[28],[32] with a negative correlation between grey to white matter ratio and age during this period [23],[32]. After this time, the loss of white matter as the brain ages exceeds that of grey matter due to progressive desaturation of myelin lipids [33], and age-related ‘cortical atrophy’ may relate more to loss of white than grey matter [29]. Given that chronic psychotic disorders such as schizophrenia present in adolescence or early adulthood, if disruption to white matter development is a substrate for psychosis in CNS diseases then it may be that disruptions to the structures myelinating during this period – such as frontotemporal connections or commissural tracts [28] – are responsible for the development of psychotic symptoms. Some key frontotemporal white matter bundles, such as the arcuate and uncinate fasciculi, have been implicated in the pathogenesis of schizophrenia [34],[35], as has the brain's largest commissural bundle, the corpus callosum (CC) [36],[37]. It could then be expected that disorders preferentially affecting these regions and structures are likely to present with SLP, and the nature of the psychotic phenomena may depend on brain maturational stage.
Dysmyelinating disease
Disorders interrupting myelination in adolescence/adulthood
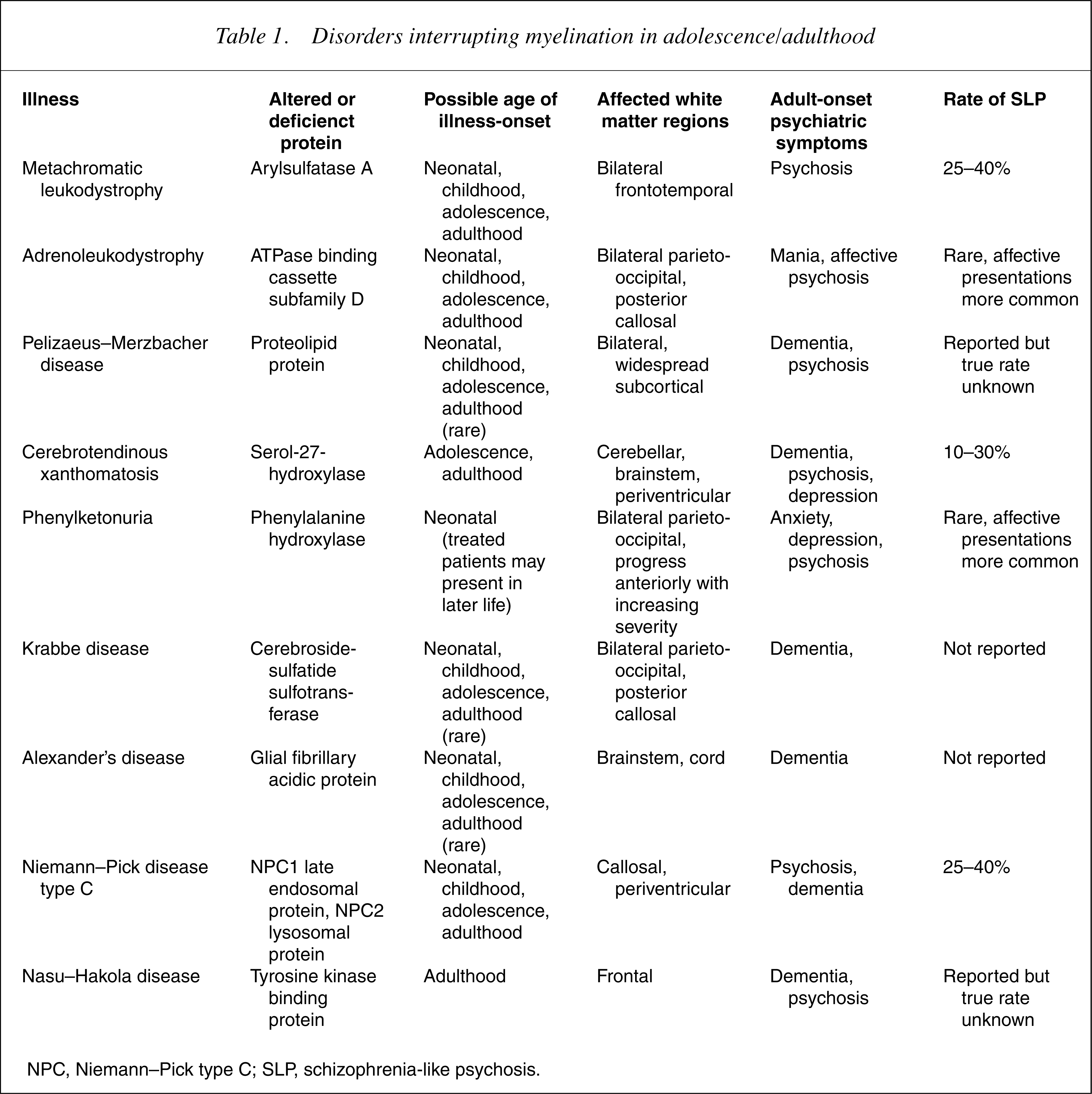
NPC, Niemann–Pick type C; SLP, schizophrenia-like psychosis.
A number of other disorders where the development of white matter may be disrupted in adolescence or early adulthood warrant mention. Phenylketonuria (PKU) impairs the myelin-forming capacity of oligodendrocytes if not treated with a phenylalanine restricted diet [62],[63], although adult PKU sufferers who have been appropriately treated may show subtle white matter changes in parieto-occipital regions on magnetic resonance imaging (MRI) [62]. Psychosis has been reported [64–66], although is comparatively rare compared to anxiety and depression [67], perhaps due to the relative sparing of frontal white matter. Nasu–Hakola disease, or membranous lipodystrophy, results in frontal myelin loss in addition to cancellous bone fractures [68] and usually presents as a mid-life frontal dementia, although cognitive change may be preceded by SLP [69],[70], potentially due to disrupted frontotemporal white matter. The adult-onset form of Niemann–Pick type C (NPC) disease, a lipid storage disorder that affects white matter early but later also affects grey matter [71],[72], shows white matter disruption in the CC [73] and periventricular white matter [74] and presents initially as psychosis in up to 40% of cases [74–78], a rate comparable with MLD.
The regional distribution of white matter disruption in the disorders most strongly associated with psychosis, MLD and NPC, suggests that interruptions to myelination in frontotemporal, callosal and periventricular zones are crucial to the development of SLP in disorders where adolescent/early-adult myelination is disrupted – although the true strength of this association is difficult to define quantitatively from the available data. As can be seen in Table 1, disorders with a posterior predominance rarely present with psychosis, suggesting the importance of connections between, and within anterior regions of, the cerebral hemispheres in the origins of psychosis.
Demyelinating disorders
Compared to dysmyelinating disease, demyelinating disorders – where normally formed myelin is lost – appear to present with a much lower rate of psychosis. The most common of these is multiple sclerosis (MS), where small, variably distributed lesions to previously normal white matter are the hallmark of the disease [79]. Neuropsychiatric presentations are most commonly affective [48], and presentations of SLP occur in up to 5% of cases [80],[81]. Appearance of psychosis in MS appears to relate to lesion site rather than load [82], with frontal and temporal periventricular lesions most strongly associated with psychosis [81],[83–87], particularly where these lesions are very large, such as in Schilder's disease [88]. Progressive multifocal leukoencephalopathy, a rare post-viral demyelinating condition resulting in multifocal demyelination of subcortical white matter [89] has only very rarely been associated with psychosis [90]. The paucity of psychosis in demyelinating disease is likely related to the maturity of CNS structures that are affected, focal rather than diffuse white matter involvement (causing psychosis perhaps only when critical circuits are disrupted to a marked degree), and regional lesional variability; when SLP does present, frontotemporal white matter disruptions again appear to be the norm.
White matter tumours
Common gliomas (astrocytomas, oligodendrogliomas and ependymomas) arise principally from cells that predominate in white matter such as glia, and disrupt function through local deviation of and dissemination along adjacent axons [91]. The neoplasms that affect white matter structures, such as the developmental disorders, appear to produce psychosis when they impinge on anterior rather than posterior structures [92], as do those that affect the callosum or periventricular white matter [93]. Up to 20% of oligodendrogliomas, which commonly localize anteriorly, present with psychosis [94], and the rare infiltrative gliomatosis cerebri presents predominantly with mental state changes [95], including psychosis [96], although it is not regionally specific. Psychotic symptoms may arise from interruption of crucial networks linking frontal to subcortical or other cortical regions rather than perturbation of normal developmental processes, as these neoplasms can produce these mental state changes across the lifespan.
Inflammatory diseases
The inflammatory diseases associated with psychosis appear to exert their effect through a combination of vascular lesions affecting white and grey matter structures, and autoantibodies to axonal and other tissue. The main disorder meeting each of these criteria is systemic lupus erythematosus (SLE), which affects the CNS in up to 70–80% of SLE sufferers [97]. Ischaemic lesions in grey and white matter occur [98] as well as axonal tract degenerationmediated by CNS autoantibodies and cytokine activation [99],[100]. Although mood disorders are more common, psychosis occurs in up to 5% of cases [97],[101–103]. Psychosis in SLE has been linked to titres of antiribosomal P antibody (ARPA) and the prothrombotic antiphospholipid antibody (APLA) in some [101],[104–110] but not all [111–113] studies, and anti-DNA antibodies have been associated with NMDA-mediated excitotoxicity in the hippocampus [114]. The demyelinating antiganglioside antibody (AGA) is also seen in up to 30% of SLE patients [115–117]. In the related disorder Sjogren's syndrome (SS), significantly fewer (25%) patients suffer CNS involvement [118–119]. Sjogren's syndrome lacks ARPAs and AGAs [120], and SS-characteristic autoantibodies are associated with neurologic rather than psychiatric disturbance [120]. Mood or anxiety disorder is present in up to half of the patients [121] but psychosis is rare [122]. This relative infrequency in SS compared to SLEmay be due to its differing autoantibody profile, suggestive of less direct impact on myelinated structures, and the later age of onset in SS, again reinforcing that the underlying maturity of CNS structures may be as important as lesion location in generating psychotic symptoms.
Velocardiofacial syndrome
Velocardiofacial syndrome (VCFS) is caused by microdeletions on chromosome 22q11 [123] and results in facial dysmorphology, cardiac anomalies and learning disabilities [124] with rates of SLP of up to 30% [125–128]. Neuroimaging findings often mirror the frontotemporal grey matter deficits seen in schizophrenia [129], but also reveal similar white matter abnormalities, including reduced total volume [130],[131], callosal abnormalities [132],[133], cavum septum pellucidum [133–135] and focal hyperintensities [136]. Disrupted white matter tracts between frontal and other association cortex reported in VCFS [133] are additionally consonant with findings in schizophrenia [137]. Of note, posterior WM regions are affected in VCFS children and frontal regions in adults [133], consistent with the rostrocaudal direction ofmyelination across the life cycle [29], but also consistent with the high prevalence of psychosis in disorders disrupting anterior white matter structures.
Abnormalities of the corpus callosum
If disruptions to specific white matter structures are posited to predispose to psychosis, then lesions to the largest tract, the CC, would be expected to show increased rates of the disorder. The CC has frequently been implicated in the pathogenesis of schizophrenia due to its role in interhemispheric connectivity [138], and alterations in size and shape have been consistently reported [139–142]. Two developmental malformations of the CC associated with psychosis are partial or complete agenesis of corpus callosum (ACC) and callosal lipoma [143], which commonly co-occur [144]. Partial or complete ACC is associated with a range of heritable and acquired diseases that disrupt normal white matter development [145], including mutations in genes controlling axonal growth [146], and intrauterine insult such as infection or toxin exposure [147]. When psychiatric disturbance presents in ACC sufferers, it is psychotic in nature in at least half of the patients [37] and undiagnosed ACC has been detected in schizophrenia populations at a significantly increased rate [148]. Andermann's and Apert's syndrome both cause ACC and have significantly higher rates of psychosis than healthy controls [149],[150]. Agenesis of corpus callosum also occurs in greater rates in VCFS [151], perhaps as a result of the deletion or altered regulation of genes involved in neural or axonal migration. Acquired adulthood callosal disorders such as Marchiafavi–Bignami disease, an acute myelinosis related to thiamine deficiency, are rarely associated with psychosis [152]. Dysgenetic but not acquired callosal disorders are strongly associated with psychosis, suggesting that developmental pathology of interhemispheric connectivity, or white matter as a whole, plays a role in psychotic symptoms.
Discussion
Because of the multiplicity of illnesses that are associated with psychosis, it follows that a range of different processes at the cellular or molecular level are the substrate for these. Considering the correlation between clinical syndromes and white matter ‘lesions’ requires acknowledgement of two issues: the difficulty in defining white matter as an anatomically ‘pure’ entity (given that myelinated structures exist in grey matter regions, and a range of cell types including neurons populate white matter regions); and that the neuron and axon are essentially part of a single cellular unit. Thus, diseases primarily affecting one compartment will necessarily also affect the other, making pure ‘white matter’ or ‘cortical’ diseases relatively rare in clinical practice.
Clinical symptomatology following white matter disruption can be understood as a consequence of disruptions to neural networks, distributed neural assemblies that subserve integrated cortical functions [153]. A range of networks have been defined, and each is associated with a broad neurobehavioural function [154]. Functional disruption of these networks can occur through a number of mechanisms. If the quality or speed of conduction across networks is reduced, then cortical regions that need to operate in a synchronous manner fall into asynchrony, leading to functional disintegration. Disruption to inhibitory systems leads to overactivity of downstream networks, or reduced activity in an area dependent on activation from another region [155]. This has been well shown with focal lesions [156–158], and in a diffuse process some networks may be more susceptible than others resulting in subtle global impairment of function with more significant dysfunction in a smaller number of key networks. This may be particularly the casewhere pathology affects important regions where there is a confluence of relay pathways [159].
At an anatomical level, developmental axonal pathology may cause alterations in neuronal structure and function. This will depend significantly on the nature of the white matter pathology itself; an abnormality in the capacity of oligodendrocytes to maintain myelination is likely to produce different results on neuronal populations than a dysfunction of axonal sprouting or guidance, or abnormal turnover of myelinated structures. For example, reduced input from neighbouring cells can result in neuronal cell loss, a phenomenon known as trans-synaptic degeneration (TSD) [160], which can occur following white matter dysfunction or insult and evolves slowly over many years [161]. In TSD, cells may atrophy, or undergo apoptosis in response to the decreased or absent afferent input [162],[163], or show reductions in synaptic density [164]. Although this has been best studied in lesion models, it is possible that an early static or neurodevelopmental process affectingwhite matter could result in slow changes to neuronal population number, size or dendritic arborization which could account for grey matter volumetric changes, such as those seen in schizophrenia [165].
The reasons why demyelinating illness such as MS appears to result in psychosis at a much lower rate than dysmyelinating disease are unclear, although it may be that the relatively discrete nature of the lesions, their rate of demyelination and the timing of their appearance do not disrupt connectivity in the psychotogenic way other diseases do [38]. Smaller focal lesions may allow for compensation in local or adjacent networks in a fashion that more diffuse disease does not, although this may depend on site, where lesions to relay areas through which a large amount of inter-regional information passes may have the greatest impact [159]. The timing of lesion onset inMS is decades after the appearance of symptoms in schizophrenia, and the relative maturity of affected networks also appears to play a crucial role. The association of discreteWMlesions and late-onset psychosis [56],[166] may appear to mirror the process in MS, although older individuals' cortical networks may be already compromised by age-related neuronal and synaptic loss [167], and the superimposition of WM lesions may work synergistically with these disrupted connections to produce psychosis. Alternatively, the myelin-degenerative theory of ageing suggests that age-related functional decline is secondary to gradual desaturation of myelin with resultant impaired transmission in heavily myelinated regions [168], particularly frontally [169] and thus any lesions that occur in association tracts leading to frontal regions in older individualsmay be sufficient to produce SLP. The instances of psychosis in MS could either result in psychotic symptoms as a result of a particular distribution of lesions, lesions ‘unmasking’ a vulnerability to psychosis that may not otherwise have become apparent without cerebral insult, or coincidence.
The key factors that appear to play a role in the genesis of SLP in white matter disorders relate to the underlying maturity of the CNS when lesion ‘load’ begins to significantly disrupt function, the location of lesions, and whether these lesions cause abnormal formation of, or destroy already normally formed, white matter structures. The developmental disorders listed in 19], such that although positive symptoms of psychosis may present with disruptions from adolescence through to later adulthood, more typical schizophrenia-like negative symptoms may require disruption to late adolescent/early adulthood maturational processes.
Conclusions
The increased rates of psychosis in some white matter diseases point to a possible role of white matter structures in the pathogenesis of schizophrenia, and white matter is an attractive candidate as an anatomical substrate for a disconnectivity syndrome. The differential effect of dysmyelinating versus demyelinating illnesses points to a role for diffuse white matter disease, and how the timing of insult appears to be crucial. In addition, regional pathologies – particularly those affecting the CC and frontotemporal zones – are associated with psychosis in away that suggests dysfunctional interhemispheric and corticocortical connectivity plays a role in the origins of psychotic symptoms. Psychiatric presentations in organic CNS illness – the ‘outliers’ of psychiatric presentation – continue to play a role in emphasizing the neurobiological underpinnings of mental illness, and in suggesting structures and substrates for further research with the aim of developing more targeted therapeutic interventions. Therapeutic modalities directly targeting white matter structures may prove useful novel therapies for sufferers of psychotic disorders such as schizophrenia.