
Abstract
The pathology of bipolar disorder (BPD) remains an enigma. The absence of animal models for the disorder has meant that the use of human postmortem brain tissue is still a central approach to understanding its biology. At the molecular level, this approach is beginning to suggest mechanisms that underlie the pathology of BPD that involve three key areas of brain function: neurotransmitters and their receptors; G-proteins; and signal transduction pathways. Current findings in these three areas will be reviewed with the aim of understanding the central pathology of BPD.
Difficulties in using postmortem CNS tissue
As with any other approach, the use of postmortem central nervous system (CNS) tissue to study the neurobiology of BPD has limitations and confounding factors. Because of difficulties in collecting postmortem CNS many studies in BPD are completed using tissue from a small number of subjects. However, the numbers studied do not usually differ greatly from the size of cohorts in functional neuroimaging studies. In addition, where tissue is collected ad hoc after death, a great deal of care needs to be taken to ensure an accurate diagnosis according to current criteria is attained. Significantly, as with diagnosis of the living, the use of structured instruments for diagnosis after death has been shown to take much of the variability from the diagnostic process [1]. In BPD, phase of illness at death becomes a significant factor. For example, while the tissue collection at the Mental Health Research Institute is from subjects with type 1 BPD with psychotic symptoms close to death [2], tissue from the Stanley Foundation collection consists of tissue from subjects with different types of BPD that are not in a particular phase of their illness [3]. However, as long as accurate diagnoses have been completed, it should be possible to determine whether a change in a particular marker in postmortem CNS from subjects with BPD is associated with a particular type of the disorder or phase of illness.
Another confounding factor when studying BPD using postmortem CNS is that most BPD subjects from whom tissue is collected would have received drug treatment during their lifetime. Clearly, in BPD this could involve treatment with mood stabilizers and antipsychotic drugs [4], [5]. There are approaches to nullify the impact of drug treatment as a significant confounding factor when using postmortem CNS. For example, comparing results from tissue from subjects with BPD who were receiving antipsychotic drugs at death with those from tissue from subjects with schizophrenia receiving similar drugs is becoming increasingly normal practice. In this situation, if a result is diagnostically specific this lessens the likelihood that a simple drug effect has been identified. In addition, showing that the protein or messenger RNA of interest is not altered in the CNS of rats which have been treated with mood stabilizers or ‘antipsychotics’ can give additional comfort that results observed in postmortem CNS from subjects with BPD are not simply a result of such drug treatments. Finally, undertaking a stringent review of the drug history of each donor and, wherever possible, attaining a plasma drug level at death can also help to assess the likely impact of drug treatment on protein and messenger RNA levels.
Given the difficulties in collecting postmortem CNS and the possible confounding factors that could influence outcomes from the study of such material the continuation of research in this field could be questioned. However, it must also be acknowledged that there are no good animal or cellular models of BPD, that both functional and structural neuroimaging have limitations with resolution and other problems surrounding their use in identifying changes in the BPD CNS and it is not clear if peripheral or genetic studies will provide information on the neurobiology of BPD. Thus, the study of postmortem CNS remains an important component of the broad research approach designed to identify causes of BPD and this area of research is likely to expand with the ability to use postmortem CNS with high-throughput screening technologies such as gene arrays [6] and 2D electrophoresis [7] that allows levels of thousands of messenger RNA and proteins to be measured at one time.
Neurotransmitters and their receptors
As in other areas of postmortem CNS research into the neurobiology of psychiatric illnesses [8], [9], there has been a focus on markers of various neurotransmitter systems and whether they are affected by the pathology of BPD. The major components of these systems that have been studied in postmortem CNS research are the neurotransmitters themselves plus their transporters and receptors (Fig. 1). The monoamine neurotransmitter transporters are proteins on the presynaptic neurones which act to take up the neurotransmitter that has been secreted by that neurone and thus act as signal modulators [10]. More recently, other neurotransmitter transporters, such as the glutamate transporter, have been demonstrated on non-neuronal cells [11] and it is suggested these transporters may have diverse roles such as an involvement with neuroprotection. Neurotransmitter receptors can be present on both the pre- and postsynaptic neurone [12], with presynaptic receptors having a number of functions, including regulation of neurotransmitter production and release [13]. Importantly, most studies thus far completed using postmortem CNS do not use techniques that allow the pre- and postsynaptic distribution of receptors to be differentiated and generally look at global effects on receptor densities.
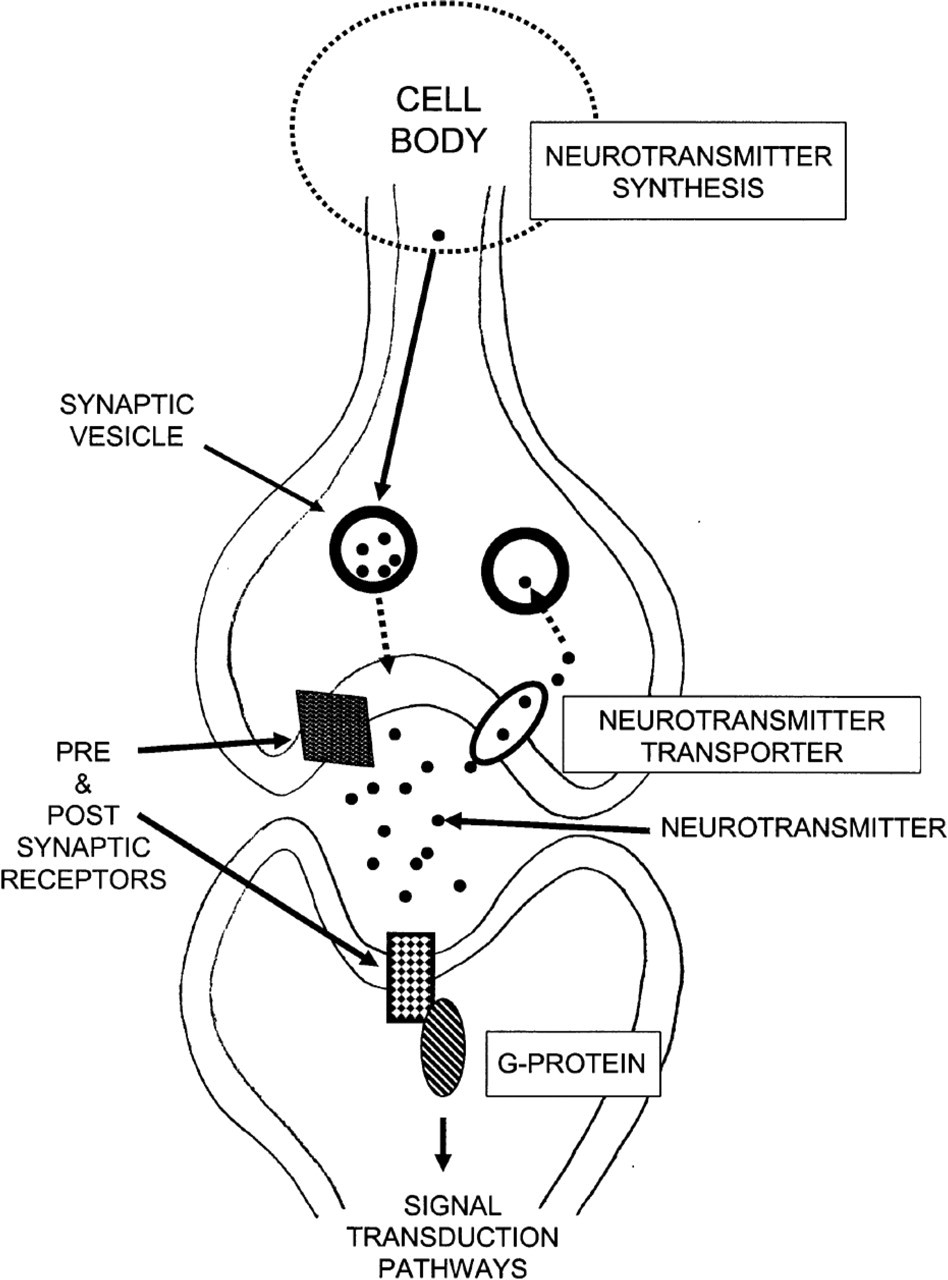
Simplified schematic diagram showing some of the processes involved in neurotransmitter release at the monoamine synapse. The schema shows neurotransmitter transporters located on a presynaptic neurone with neurotransmitter receptors localized on the pre- and postsynaptic neurones. The neurotransmitter is shown contained in synaptic vesicles and the fusion of these vesicles with the presynaptic neuronal membrane results in the neurotransmitter being discharged into the synaptic cleft. Once in the synaptic cleft the neurotransmitter can activate both pre- and postsynaptic receptors, can be taken up back into the presynaptic neurone by the neurotransmitter transporter or can be degraded and excreted.
Given the central role of the neurotransmitters in CNS function, it was logical for early studies to focus on these chemicals in the CNS from subjects with BPD. An extensive study using tissue obtained postmortem from BPD subjects failed to show any changes in the levels of serotonin (5HT), noradrenaline (NA) or dopamine (DA) in the hippocampus, caudate nucleus, putamen, mediodorsal thalamus or in the frontal, parietal, occipital or temporal cortices [14]. However, in the same study, a number of changes in the ratio of neurotransmitters and their breakdown products, an indirect measure of neurotransmitter turnover, were reported. These data supported the hypothesis that there were decreases in 5HT turnover in the frontal, parietal and temporal cortices from subjects with BPD. In addition, there appeared to be decreased DA turnover in the parietal and occipital cortex with increased NA turnover in the thalamus, frontal, temporal and occipital cortices. Unfortunately, this study provided no clear picture as to the cause of these complex changes in neurotransmitter turnover in BPD, however, such changes would have profound effects on CNS functioning that could well result in the symptoms associated with BPD.
If there were changes in neurotransmitter turnover that were affecting prevailing levels of neurotransmitters in the brains of subjects with BPD, it would be predicted such changes would be reflected in changes in key B. DEAN 137 markers associated with such neurotransmitter systems, such as neurotransmitter receptors and transporters. Unlike schizophrenia [15], there has been little study of these important components of the neurotransmitter pathways in BPD. In the case of 5HT, changes in the densities of the 5HT2A and 5HT1A receptors have not been detected in the frontal cortex [2] or hippocampus [16] from subjects with the disorder. The density of the 5HT1D and 5HT1F receptors have also been shown not to be altered in that region of the CNS from subjects with BPD [16].
A change in the affinity of [3H] citalopram binding to the serotonin transporter (SERT) has been reported in the hippocampus [16], but not frontal cortex [2], of subjects with BPD. Similarly, a change in the affinity of [3H] paroxetine binding to SERT in the hippocampus, but not frontal cortex or caudate-putamen, has been reported in schizophrenia [17], [18]. These data would be consistent with a conformational change in SERT being present in the hippocampus of subjects with BPD and schizophrenia [18]. It has been argued that the conformational change in SERT in the hippocampus of subjects with schizophrenia would result in a prevailing hyperserotonergic state in that brain region, which in turn could induce some of the symptoms of the illness [19]. If that were the case, and as a similar change has been observed in BPD and schizophrenia, it could be argued that the conformational change in SERT would be associated with a common symptom across the two disorders. Such a symptom would be psychoses. However, one would also expect that an increased level of 5HT would be associated with an increased turnover of the neurotransmitter in the hippocampus of BPD subjects, a finding not supported by the study of hippocampal 5HT turnover [14]. Further evidence involving SERT in the pathology of BPD comes from a serial analysis of gene expression in the frontal cortex of BPD patients that, despite examining levels of mRNA from several hundred genes at one time, reported only mRNA for SERT to be altered in the tissue from BPD subjects [20]. Thus, current data would suggest that changed 5HT neurotransmission are important in BPD but further data is required to determine the exact nature of that involvement of 5HT in the pathology of the disorder.
There have been only limited studies of other neurotransmitter receptors in BPD. Radioligand binding studies have reported no change in the density of the ionotrophic glutamate receptors (N-methyl-D-aspartate [NMDA], D,L-α-amino-3-hydroxy-5-methyl-4-isoxazole [AMPA] and kainite receptors) in the frontal cortex from BPD subjects [2]. Similarly, the density of AMPA receptor has been reported not to be altered in the striatum [21]; however, the same study did show a decrease in mRNA encoding the gluR1 subunit of the AMPA receptor. The significance of a change in mRNA encoding a subunit of the receptor when the levels of that receptor appear not to be altered are not easily interpreted; however, it would be predicted that such a change would have no functional significance.
γ-aminobutyric acid (GABA) is the most abundant neurotransmitter in the human cortex and it has been suggested to have a role in the pathology of BPD [22]. Importantly, the GABAA receptor has a number of discrete binding sites, one of which is the binding site for the benzodiazepines (Fig. 2) [23]. This is significant because these drugs are used in the control of mood [24] and therefore changes to the site at which benzodiazepine binds in the CNS of BPD subjects could be associated with the mood swings that are characteristic of the disorder [25]. Thus, the presence of an increased number of benzodiazepine binding sites in the frontal cortex of subjects with BPD [2] could be an important finding implicating the GABAA receptor in the pathology of BPD. This finding is made more intriguing in that, in the same individuals, the number of GABA binding sites on the GABAA receptor (Fig. 2) measured using [3H] muscimol binding was not altered. Importantly, expression studies have shown that changing the levels of the γ2 subunit of the GABAA receptor will change the density of benzodiazepine binding on the GABAA receptor without the density of GABA binding sites [26]. Thus, together with the data from the study of postmortem brain tissue, it can be argued that there is an increase in the levels of GABAA receptor containing the γ2 subunits in the frontal cortex of BPD. The consequence of such a change in the makeup of GABAA receptors still awaits elucidation.
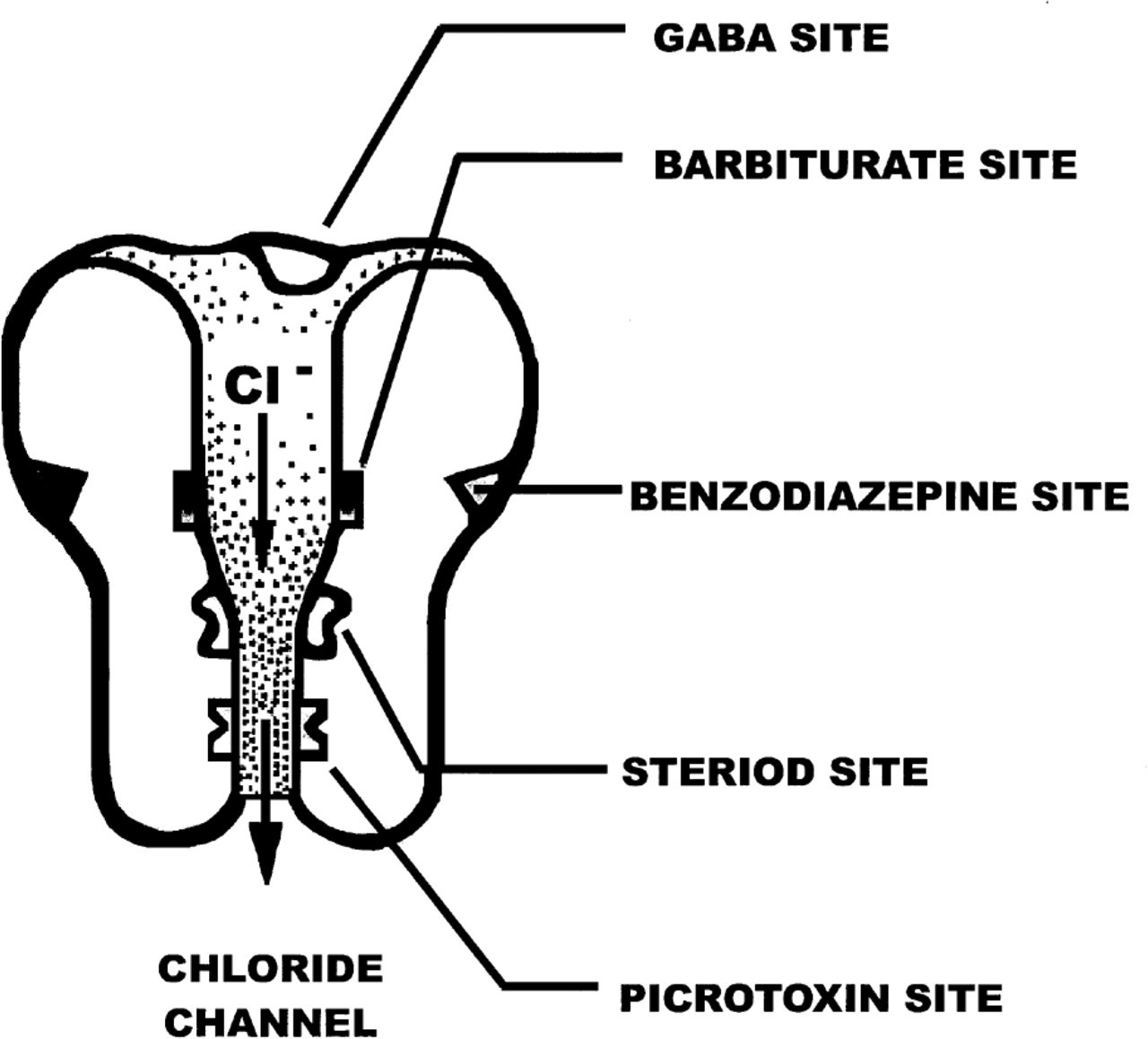
Schematic diagram showing the various binding sites on the GABAA receptor.
G-proteins
It is significant that lithium and antidepressants, both of which are used in the treatment of BPD [25], affect the function of G-proteins [27], [28]. Importantly, the ability of many neurotransmitter receptors to appropriately modulate intracellular pathways following activation is dependent on their ability to complex with G-proteins [29] (Fig. 1).
There is direct evidence to suggest there may be abnormalities in the interactions between neurotransmitter receptors and G-proteins in BPD. Data to support this hypothesis comes from the demonstration that there is a decrease in 5HT, isoproterinol and carbachol stimulated guanosine 5′-o-(3-[35S])thiophosphate binding in cortical tissue from subjects with BPD [30]. These data suggest that there are abnormalities in the way 5HT, adrenalin (isoproterenol), acetylcholine (carbachol) and possibly other receptors are interacting with G-proteins in the CNS from BPD subjects. Moreover, these data raise the possibility that abnormal neurotransmission could be present even if, as is the case for certain 5HT receptors, there is no change in receptor density in specific CNS regions.
A change in receptor/G–protein interaction could be precipitated if there were changes in intracellular G-protein pools. The family of G-proteins is made up of three groups of proteins, termed α, β and γ subunits, and within each family the proteins have a high degree of homology [31]. Trimeric assemblies, made up of one member of each subunit family must come together and associate with G-protein linked receptors to facilitate signal transmission from the receptor to intracellular pathways. Because of the link between the actions of lithium and antidepressants with G-proteins there have been extensive studies on these proteins in postmortem tissue from subjects with BPD. These studies have shown changes in different G-proteins in different brain regions from subjects with BPD (Table 1) [30],[32–34]. Thus, there is clearly an opportunity for abnormalities in receptor/G–protein interactions to be involved in the pathology of BPD. As yet a clear mechanism implicating such an abnormality in BPD has yet to be demonstrated.
Changes in G-protein subunits reported in postmortem central nervous system tissue from subjects with bipolar disorder. Increases (⇑) or lack of change (⇔) in G-protein are shown by arrows with conflicting findings being indicated by the presence of multiple arrows
Signal transduction
Signal transduction pathways are the mechanisms by which extracellular stimuli can modulate the function of a cell [35] (Fig. 1). Modulation of these signal transduction pathways often occurs when a receptor interacts with specific G-proteins. Thus, changes in the interaction between receptors and G-proteins might be expected to cause downstream changes in signal transduction pathways. This hypothesis has led to the study of critical components of signal transduction pathways in postmortem tissue from BPD subjects. Decreased levels of inositol, an important component of the inositol phosphate signalling system and another target for lithium therapy [36], has been shown to be decreased in the frontal cortex but not the occipital cortex or cerebellum of subjects with BPD [37]. There is also a report of increases in cytosolic and membrane-associated protein kinase C α, γ, ζ and ε but not β or δ in frontal cortex from BPD subjects [38]. Finally, decreased [3H]cyclic AMP binding has been reported in the frontal, occipital and parietal cortices from these subjects [39]. Cyclic-AMP binding proteins are proteins that are affected by the product of adenylate cyclase stimulation, a key process in signal transduction pathways [40]. Thus, a decrease in these proteins in brain tissue obtained postmortem from BPD subjects could indicate a re-setting of signal transduction pathways as a result of decreased activation by G-protein coupled receptors. Unfortunately, like many areas in the study of the neurochemistry of BPD, these findings on signal transduction pathway markers need replication and therefore must remain tentative.
Neurotransmitter-related neuroimaging studies
Recently positron emission tomography (PET) has been used to study the molecular architecture of the brain of BPD subjects. A study the thalamus and midbrain in a group of subjects with either major depression (n = 7) or BPD (n = 6) showed an increased level of SERT in the thalamus [41]. These data add weight to the data from the study of postmortem tissue that abnormalities in SERT are involved in the pathology of the illness. In another study with PET on a mixed disorder group it was shown that there was a decrease in the 5HT1A receptor in the raphe and mesiotemporal cortex, particularly in the subjects with BPD [42]. Thus, both postmortem and neuroimaging studies would strongly suggest that changes in serotonergic systems of the CNS are important in the pathology of BPD.
While postmortem studies in BPD have yet to focus on the dopaminergic pathways of the brain a PET study has reported a decrease in DA D1 receptor in the frontal cortex, but not striatum, from subjects with BPD [43]. These data would suggest a study of dopaminergic markers in postmortem brain tissue is warranted.
Conclusion
Evidence from postmortem and neuroimaging studies strongly implicate changes in 5HT systems in the pathology of BPD. The fact that the studies have focused on multiple regions using multiple techniques means that further, more coordinated, research is required to fully define the relationship between changes in the serotonergic systems and BPD. There is also a considerable body of data that would suggest that there are abnormalities in receptor signalling in the brains of subjects with BPD. As with the changes in the serotonergic system, further research will be required to understand the contribution these changes make to the symptomatology and pathology of the illness. Finally, the body of data suggesting changes in SERT may be important in BPD could also indicate, as has been suggested in schizophrenia [15], that changes in presynaptic neuronal function might be an important area of research yet to be explored in BPD.