
Abstract
Neuroimaging and neuropathological studies implicate the hippocampus in schizophrenia pathogenesis [1]. Magnetic resonance imaging studies found decreased hippocampus volume in schizophrenia, which precedes onset of illness [2, 3]. A greater volume reduction in the anterior hippocampus (AH) compared to the posterior hippocampus (PH) has also been reported [4, 5]. A review of the neuropathological findings in the hippocampus of schizophrenia brains suggests subtle morphological alterations such as size, organization and shape of the neurons [6]. Molecular findings of decreased levels of synaptic proteins [7, 8] and dendritic indices [9] are consistent with findings of subtle morphological alterations suggesting a disturbance of connectivity within and between the hippocampus and other brain regions. Furthermore decreased levels of signalling proteins such as glutamate [10, 11], decreased γ-aminobutyric acid (GABA) interneurons [12] and serotonin and muscarinic receptor density [13, 14], decreased glucose metabolism [15], which has been associated with severity of positive symptoms [15–17] have also been reported. Finally, a large-scale microarray study has found the hippocampus to be one of the regions with the highest number of altered gene transcripts in schizophrenia [18] and an earlier proteomics study found 16 proteins altered in expression [19]. Because proteins are essential for cellular function, it is important to assess the changes in the proteome of the schizophrenia hippocampus in order to characterize in detail the molecular pathways responsible for abnormalities of this region in schizophrenia. Additionally, because the AH and PH are known to carry out different functions and also connect to different regions of the brain [5], it would be important to investigate the protein expression profiles in these two regions separately because this may provide a more detailed picture of the role of the hippocampus in schizophrenia. We hypothesize that the protein profiles of the AH are different to the PH based on the differences in the function and connectivity of each of these regions in the brain and we expect to find a larger number of alterations in the proteome of the AH compared to the PH based on the degree of pathology that has been reported for the AH in schizophrenia.
In the present study 2-D gel electrophoresis (2DGE) and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS) were used to investigate protein expression differences in the AH and PH in schizophrenia subjects compared to matched controls in an unbiased manner. Distinct protein expression profiles of the schizophrenia brains are expected to consist of a mixture of proteins that may be altered as a result of causative pathogenesis of the disease, the effects of neuroleptic medication and secondary changes induced during the course of this long-lasting illness. The aims of this investigation were twofold: to clarify the extent of involvement of the AH and PH in schizophrenia pathogenesis at the level of protein expression; and to identify schizophrenia-related molecular mechanisms in the AH and PH that may explain some of the observed subtle neuropathology and functional impairment in these regions.
Methods
Human brain tissue samples
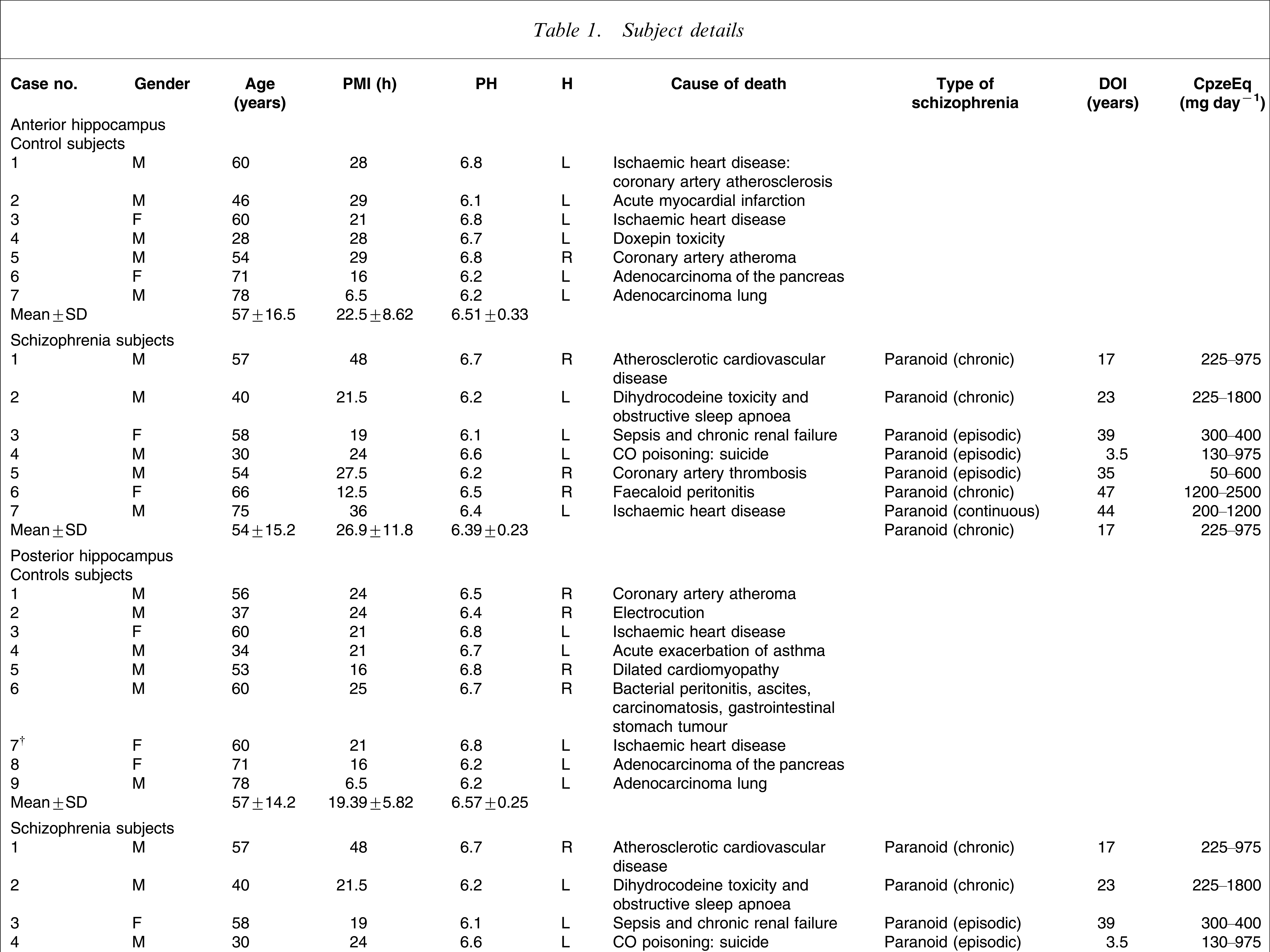
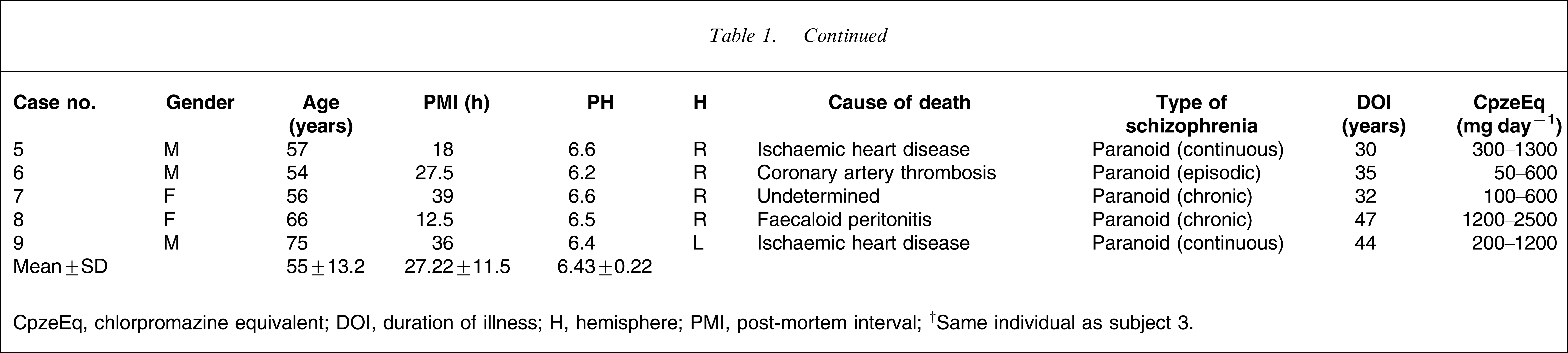
Fresh frozen post-mortem brain tissues from the AH and PH were provided by the NSW Tissue Resource Centre (University of Sydney, Sydney, NSW, Australia) from seven schizophrenia and seven control subjects, and nine schizophrenia and nine control subjects, respectively. AH was defined as where the amygdala ends and the ventral lateral thalamic nucleus appears. PH was defined as the region spanning the pulvinar thalami. Tissue was sectioned in the coronal plane and included all cornu ammonis regions and dentate gyrus. Subtle variations were expected in the proportion of neuronal populations sampled for each region from each case due to slight variations in the coronal blocks from which the hippocampus was sampled and from the sampling itself. The impact of these differences on protein analysis was minimized using biological replicates and a selection criterion that excluded from further analysis protein spots present in <30% of the cases examined. All schizophrenia subjects fulfilled DSM-IV criteria [20]. Subjects were determined to be free of significant neuropathology, and controls had no history of major psychiatric, neurological illnesses or drug abuse. All subjects were of Caucasian origin and were matched for gender, age, hemisphere, post-mortem interval (PMI) and brain pH, where possible. No significant difference was present between the schizophrenia and control groups in age, PMI or pH (p > 0.05). Information regarding schizophrenia subtype, duration of illness (DOI), neuroleptic dose and cause of death was provided for each subject (Table 1). Patients were treated with a range of typical neuroleptics except schizophrenia subject 4, who was treated with both typical and atypical neuroleptics. An estimated total amount of medication administered to each patient was determined by multiplying the DOI (days) with the median dose of medication (mg day−1 chlorpromazine equivalent units, because the medication information provided by the NSW Tissue Resource Centre (TRC, University of Sydney, NSW, Australia) was the lifetime range from the lowest to the highest prescribed dose). Approval from the Central Sydney Area Health Service (Protocol Number X03-00285) was obtained for the use of human post-mortem brain tissue in the present study.
Subject details
CpzeEq, chlorpromazine equivalent; DOI, duration of illness; H, hemisphere; PMI, post-mortem interval; †Same individual as subject 3.
Two-dimensional gel electrophoresis
2DGE analysis was performed as described in detail in Sivagnanasundaram et al. [21]. Briefly, proteins were extracted and protein concentration determined by the Bradford method using a complex human brain protein mixture as a standard. A total of 350 µg of each protein sample was profiled in duplicate in the first dimension on precast immobilized 11 cm pH 4–7 gradient strips and in the second dimension on precast 8–16% Tris-HCl sodium dodecylsulfide–polyacrylamide gel electrophoresis. The protein profile of each gel was visualized by colloidal Coomassie Blue staining [22].
Image acquisition and analysis
Gels were scanned using Phoretix Powerscan software (Nonlinear Dynamics, Newcastle-upon-Tyne, UK) with a UMAX flatbed scanner (light path: transmissive, brightness: 40.55, contrast: 80.12, 400 dots per inch). Intensity calibration was performed prior to gel scanning using a 25×125 mm AGFA strip (Ridgefield, NJ, USA). The resulting gel images were analysed using Phoretix 2D Expression software (Nonlinear Dynamics), which allows spot detection, spot matching between different gels, and spot quantification. Spots detected automatically by the software were checked manually to ensure consistency across all gels. Mode of non-spot background subtraction was applied, in order to remove background intensity present in the gels, which would otherwise artificially raise spot volume. Total spot volume normalization was then applied, which corrects for the differences in protein loading concentration between gels, by dividing the volume of each spot by the total volume of all spots in the gel and multiplying the resulting figure by 100. A reference gel was created consisting of all protein spots detected on all subgels within the schizophrenia and control groups and all protein spots detected on the subgels were matched to the reference gel.
Statistical analysis
Average gels were created for each group, to assist comparison and reduce variations within subgels of a group. Averaging parameter was set to 70%, that is, a protein spot had to be present in at least 70% of the subgels within a group to be represented in the average gel for subsequent statistical analysis. Protein spots showing statistically significant difference in expression between schizophrenia and control average gels, generated with an averaging parameter set at 70%, were assessed initially for significant difference using t-test and then verified using two-level nested ANOVA (http://udel.edu/∼mcdonald/statnested.html). Nested ANOVA analysis is suitable for equal and unequal sample sizes and takes into consideration technical versus biological replicates [23]. Significant outliers (p < 0.05) of protein spot intensity for each sample within the groups were assessed using Grubbs’ test (http://www.graphpad.com/quickcalcs/Grubbs1.cfm) and removed prior to nested ANOVA analysis. Pearson's correlation analyses were performed to examine whether protein expression levels differed as a function of either, age, PMI, DOI, brain pH or medication. Values were calculated using the average normalized volumes from each subject for each statistically significant protein.
Mass spectrometry and identification of protein spots
The differentially expressed protein spots were excised for identification by mass spectrometry, using spot picker tips (1.5 mm tips for PDN 1.5 spot picker; The Gel Company, San Francisco, CA, USA) and subjected to in-gel trypsin digestion (Porcine trypsin; Promega, Sydney, NSW, Ausralia). The digested peptides were desalted and concentrated using C18 Perfect Pure Tips (Eppendorf, Sydney, NSW, Ausralia) as described in Clark et al. [24]. The mass spectrometry was performed using a QSTAR XL mass spectrometer with an oMALDI ion source (Applied Biosystems, Scoresby, Victoria, Australia). The monoisotopic peptide mass data were generated manually or automatically using the oMALDI Xpert software (version 2.0; threshold was set to 10, exclusion window of 3 amu and maximum of 50 peaks were generated; Applied Biosystems) and was used to perform searches of the mammalian SWISS-PROT, NCBI and TrEMBL databases using the programs Aldente (www.expasy.ch) and MASCOT (version 2.0, www.matrixscience.com). For MASCOT, identification parameters were set to allow one possible missed tryptic cleavage per peptide, peptide mass accuracy within ±0.15 Da and variable oxidation of methionine checked. For MASCOT a minimum of four matched peptides and a MOWSE probability score of >64, which corresponds to p < 0.05, was required for confidence in identification. For Aldente, peptide mass accuracy was allowed within ±0.2 Da. If the programs returned more than one protein match, the best match was chosen based on the observed isoelectric point (pI) and molecular weight values of the protein, the number of matching peptide masses, the percentage sequence coverage and the agreement between matches returned by the two programs.
Results
Proteins isolated from the AH and PH from each schizophrenia and control sample was assessed for differential expression in duplicate. In total the AH and PH reference gels consisted of 979 and 1042 protein spots, respectively, representing protein spots identified in both schizophrenia and control gels. Among these, 919 and 936 protein spots were identified in both schizophrenia and control samples in the AH and PH, respectively. Of these, the control samples showed greater variability in the AH when all 919 protein spots were assessed on standard deviation (SDcontrols=0.93 compared to SDschizophrenia=0.33), and the schizophrenia samples showed greater variability in the PH when all 936 protein spots were assessed (SDschizophrenia=0.095 compared to SDcontrol=0.052). A total of 518 and 490 protein spots were assessed for differential expression in the AH and PH, respectively, using an averaging parameter of 70%. Significant changes in the relative abundance of 43 and 16 protein spots were found between the control and schizophrenia groups in the AH and PH, respectively, using two-level nested ANOVA, taking into consideration the technical (in-duplicate) and biological (schizophrenia and control samples) replicates (p < 0.05; Tables 2 and 3; Figure 1).
2-D gel electrophoresis of the (a) anterior hippocampus and (b) posterior hippocampus proteome showing the protein spots differentially expressed in schizophrenia brains compared to controls and their corresponding spot numbers.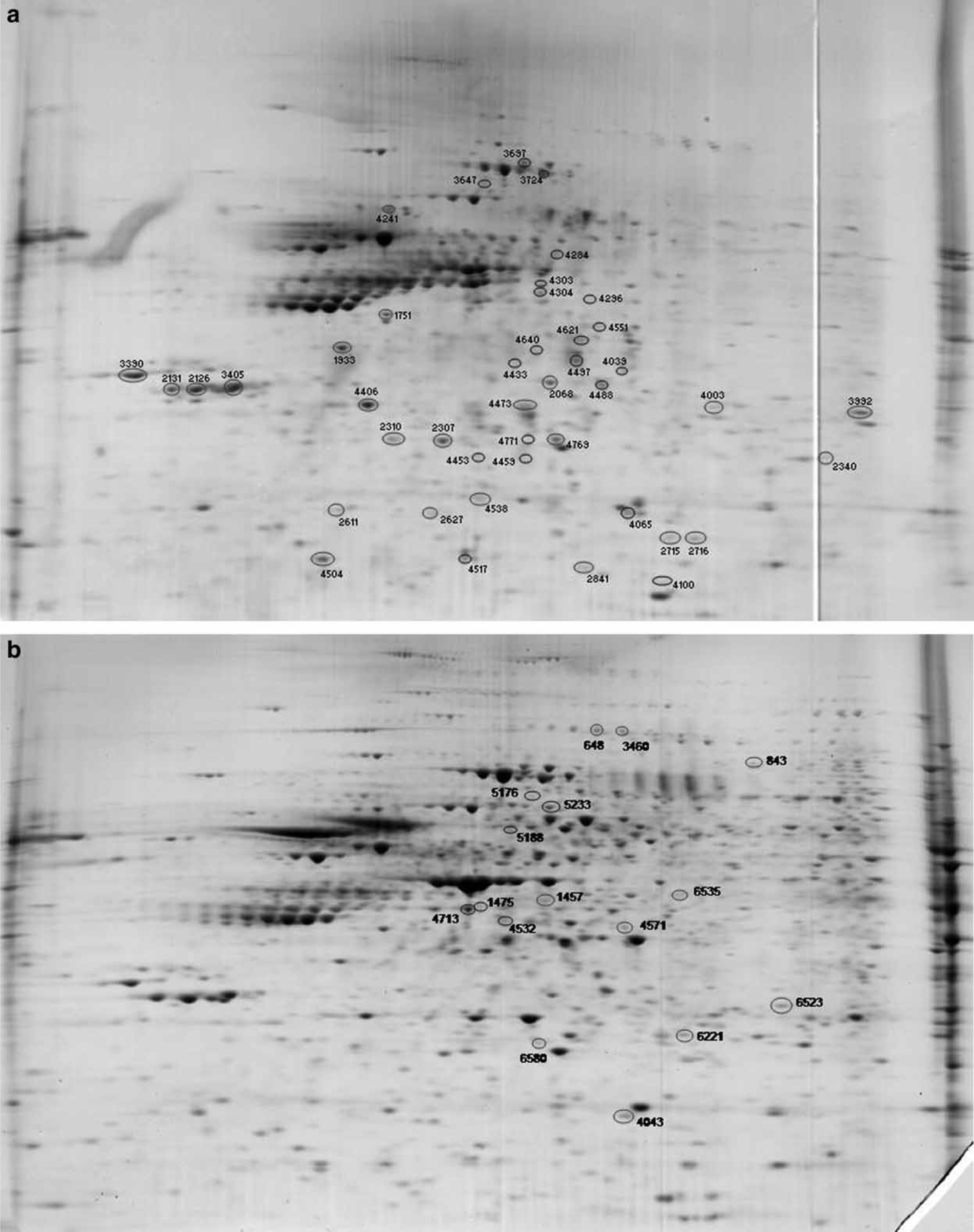
Proteins differentially expressed in the anterior hippocampus of schizophrenia brains
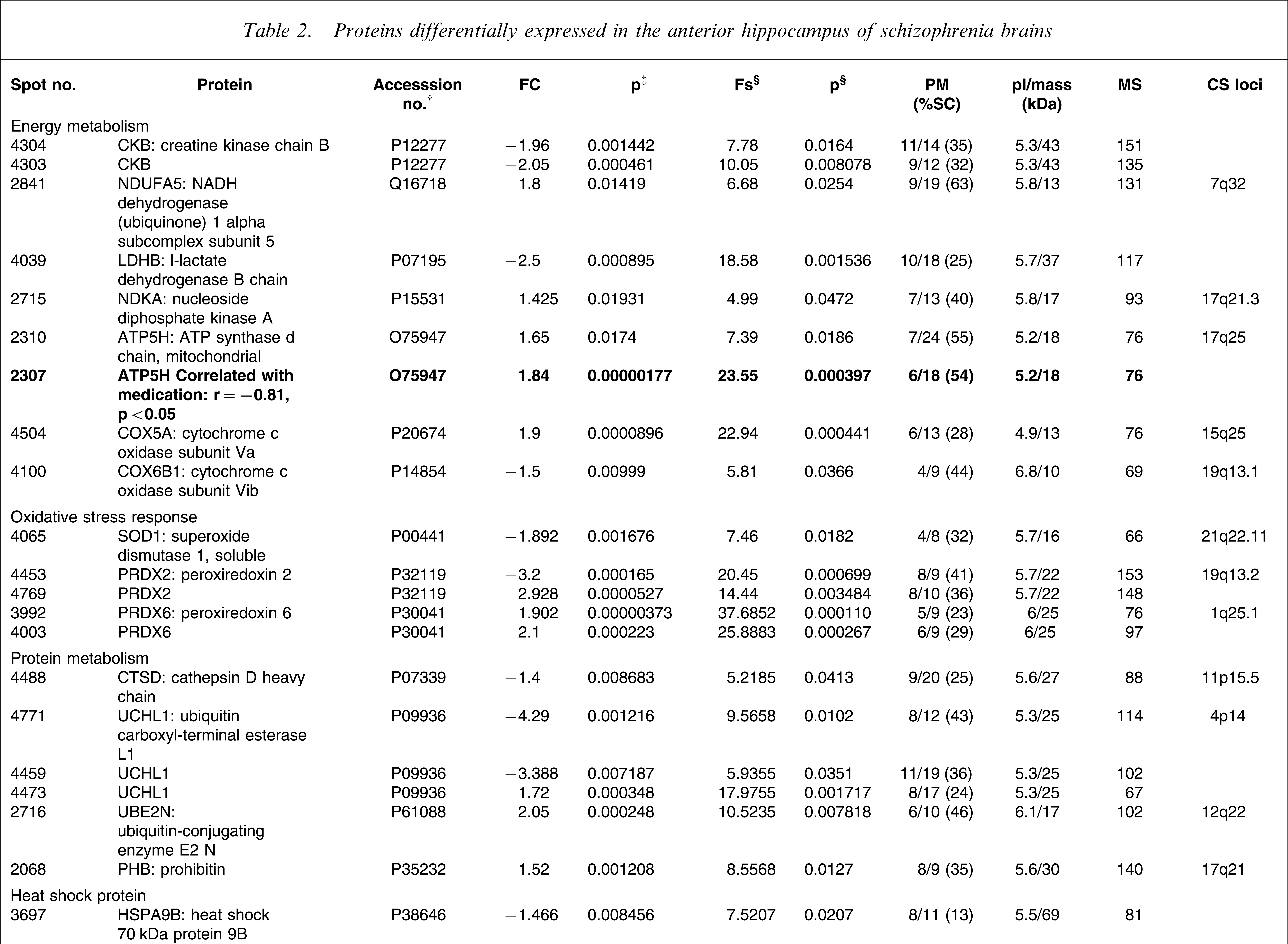
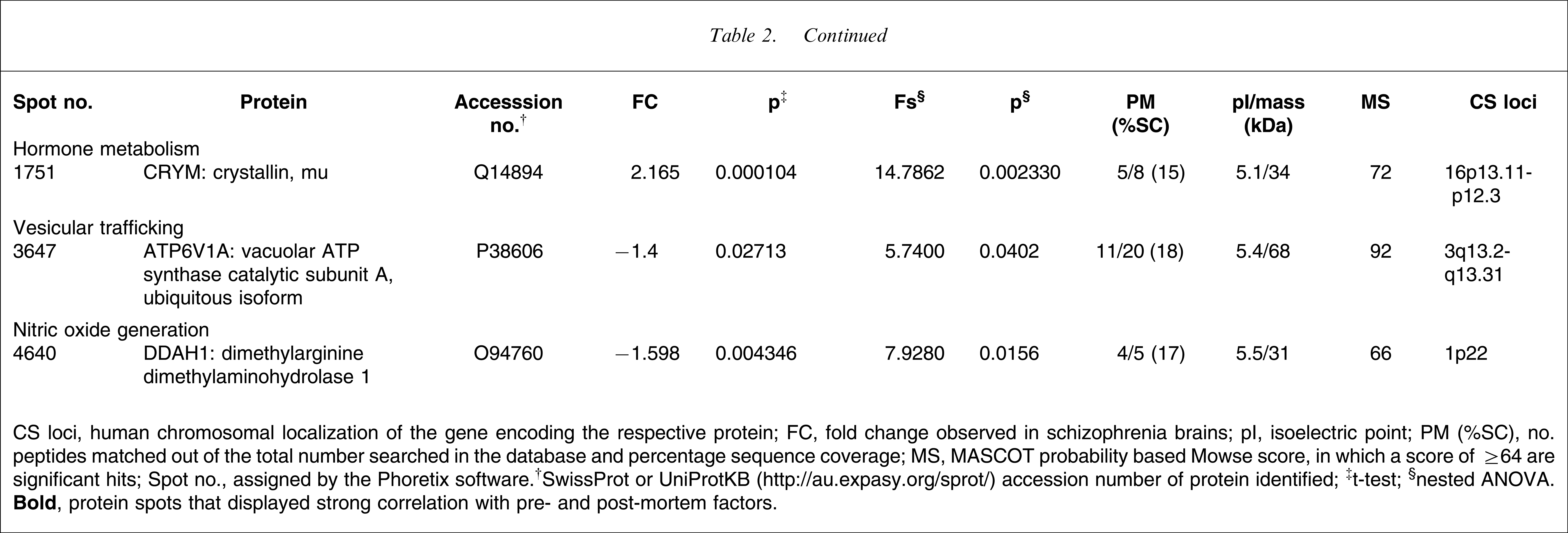
CS loci, human chromosomal localization of the gene encoding the respective protein; FC, fold change observed in schizophrenia brains; pI, soelectric point; PM (%SC), no. peptides matched out of the total number searched in the database and percentage sequence coverage; MS, MASCOT probability based Mowse score, in which a score of ≥64 are significant hits; Spot no., assigned by the Phoretix software. †SwissProt or UniProtKB (http://au.expasy.org/sprot/) accession number of protein identified; ‡t-test; §nested ANOVA.
Proteins differentially expressed in the posterior hippocampus of schizophrenia brains
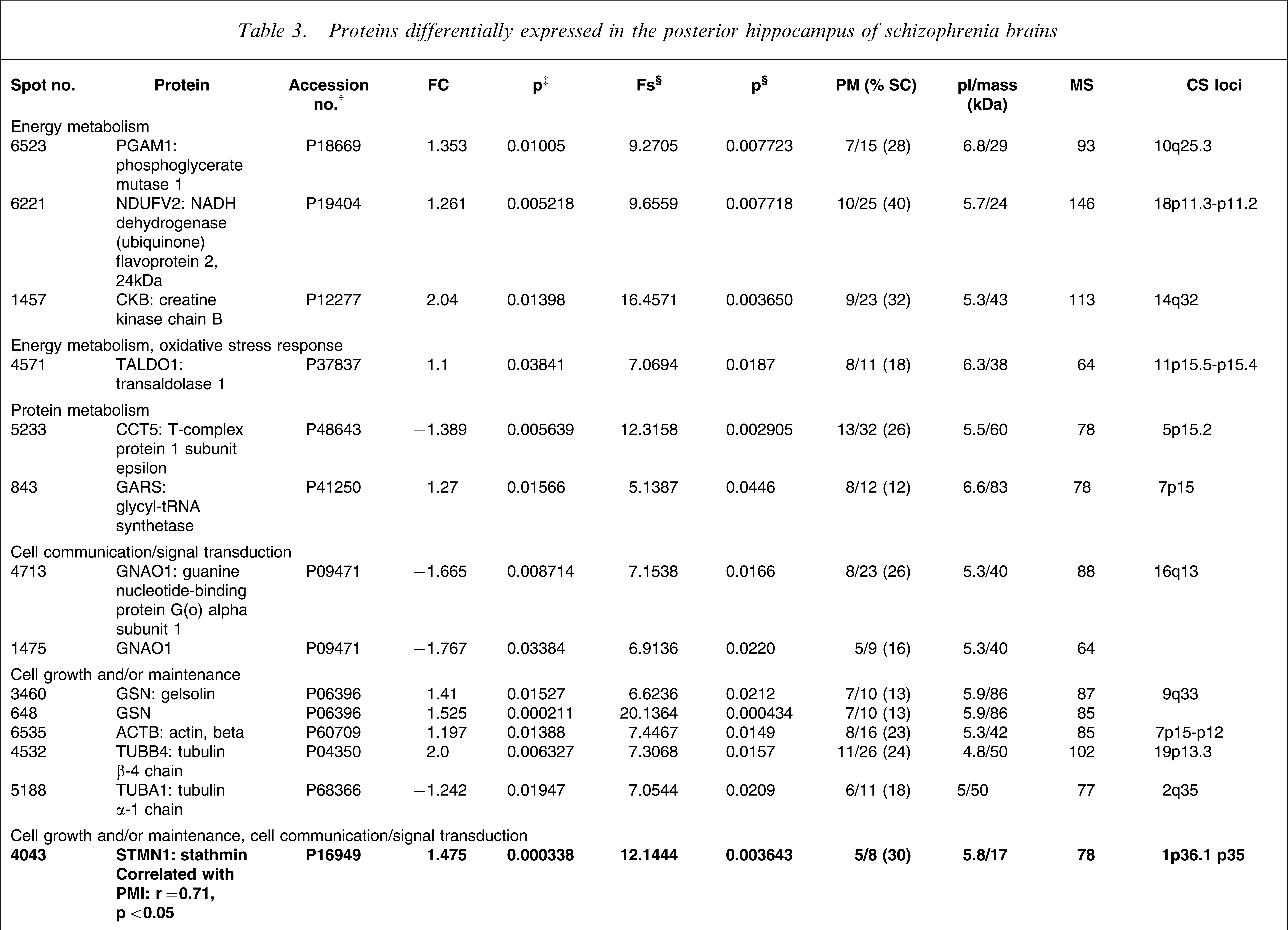
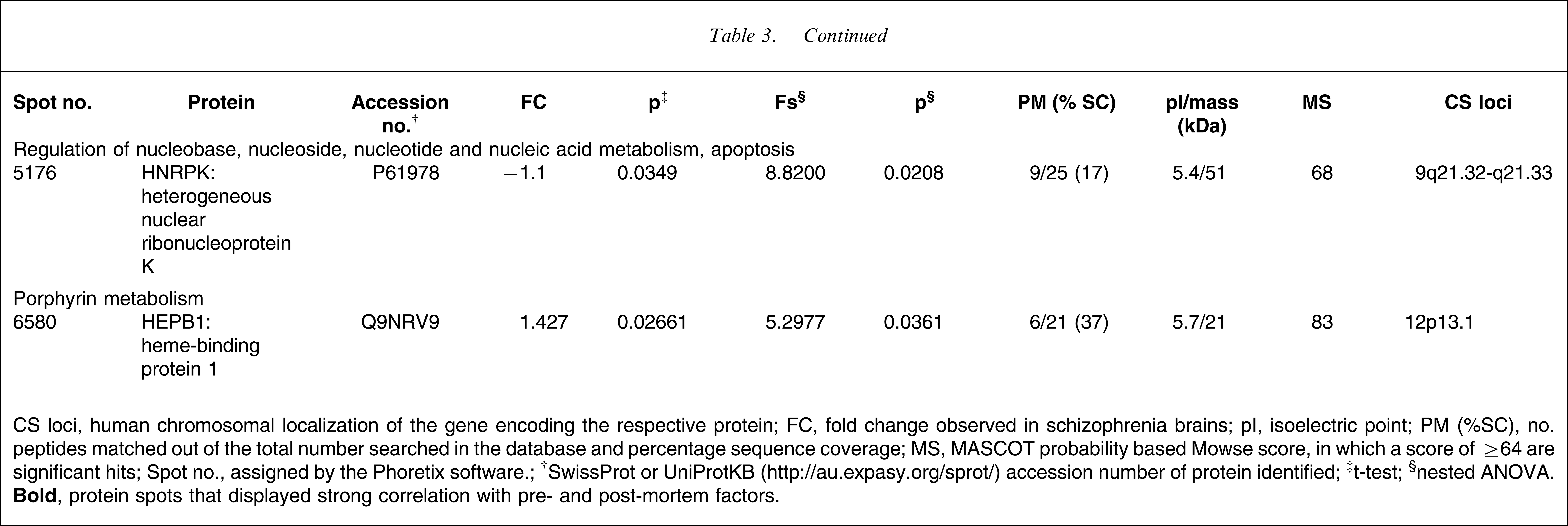
CS loci, human chromosomal localization of the gene encoding the respective protein; FC, fold change observed in schizophrenia brains; pI, isoelectric point; PM (%SC), no. peptides matched out of the total number searched in the database and percentage sequence coverage; MS, MASCOT probability based Mowse score, in which a score of ≥64 are significant hits; Spot no., assigned by the Phoretix software. †SwissProt or UniProtKB (http://au.expasy.org/sprot/) accession number of protein identified; ‡t-test; §nested ANOVA.
The 43 differentially expressed protein spots in the AH represent 34 unique proteins involved in a number of functions (Table 2), classified using the Human Protein Reference Database (http://www.hprd.org). Seven unique proteins were identified in more than one protein spot, reflecting either different isoforms or post-translation modification such as phosphorylation or glycosylation. Similarly the 14 unique proteins differentially expressed in the PH are involved in a number of functions (Table 3).
Variation in protein expression levels caused by post-mortem factors, brain pH and PMI and pre-mortem factors, age, DOI and medication were assessed using Pearson correlation test. The expression levels of two protein spots, 2307 and 4241 (Table 2), displayed strong correlation with medication (r = − 0.81, p < 0.05 and r = 0.76, p < 0.05, respectively). Protein spot 4043, identified as differentially expressed in the PH, showed strong correlation with PMI (Table 3, r = 0.71, p < 0.05). The difference in expression of spot 4043 between schizophrenia and controls remained significant (ANCOVA p = 0.032) after co-varying for PMI.
Discussion
2DGE is a proteomics tool that enables the investigation of high-abundance, hydrophilic components of a protein mixture 10–120 kDa in size [25]. In the present study we used 2DGE and MALDI-TOF-MS to profile the brain proteome of the AH and PH and identify differentially expressed proteins between schizophrenia and control brains. Proteomic analysis indicated that the AH and PH display different protein profiles (Figure 1), which may be explained by the different connectivity and functions of the two regions of the hippocampus [5]. Importantly, the larger number of molecular changes in the AH (n = 43) compared to the PH (n = 16) reflect greater perturbation in the AH in schizophrenia. Of these changes in protein expression levels, the measured intensity of protein spot adenosine triphosphate (ATP) synthase d chain (mitochondrial) was inversely correlated with antipsychotic medication, while vimentin (VIM) was positively correlated with medication. The difference in expression levels of these two proteins in the AH could be due to a change in response to medication.
Previous findings of altered protein and mRNA expression in the schizophrenia hippocampus of creatine kinase chain B (CKB), ubiquitin carboxyl-terminal esterase L1 and visinin-like protein 1 (VSNL1) were replicated in the present study in the AH [26–29]. Furthermore single nucleotide polymorphisms in the genes encoding superoxide dismutase 1, heat shock 70 kDa protein 1 and 14-3-3 zeta (YWHAZ) identified as differentially expressed in the present study, have been reported to show association with schizophrenia in previous studies [30–32]. Three abnormal pathways identified in both the AH and PH: altered glutamatergic and GABAergic neurotransmission, mitochondrial dysfunction and abnormal neuronal and glial cytoarchitecture, are discussed in the following section.
Altered glutamatergic excitatory and GABAergic inhibitory neurotransmission in the AH, consistent with previous findings in schizophrenia [33–35], were indicated by decreased expression of vacuolar ATP synthase catalytic subunit; increased expression of annexin V, proteins involved in uptake and release of neurotransmitters into the synaptic cleft [36, 37] and increased expression of YWHAZ, a protein that interacts directly with GABAB receptor 1 [38]. Abnormal neurotransmission and signal transduction was also indicated by decreased expression of guanine nucleotide-binding protein G(o) alpha subunit 1 in the PH, a protein that plays an important role in mediating GABAergic, cholinergic and dopaminergic neurotransmission [39], systems that have been implicated in schizophrenia pathogenesis [40]. Increased expression of VSNL1, thought to contribute to processes of synaptic plasticity, is suggestive of altered hippocampal circuitry [41–43].
Consistent with decreased energy metabolism in schizophrenia hippocampus [15, 44] altered expression of CKB, l-lactate dehydrogenase B chain, phosphoglycerate mutase 1 and nicotinamide adenine dinucleotide H (NADH) dehydrogenase (ubiquinone) flavoprotein 2, 24 kDa were observed in both the AH and PH. Among the abnormal activities of the various mitochondrial complexes of oxidative phosphorylation in schizophrenia, the activities of complex I and IV are reported to be most affected [45]. The present findings of increased expression of NADH dehydrogenase (ubiquinone) 1 alpha subcomplex subunit 5, a subunit of complex I [46] and altered expression of cytochrome c oxidase subunit Va and subunit VIb isoform 1, subunits of the cytochrome oxidase complex IV, a rate-limiting enzyme in oxidative phosphorylation, are consistent with that report.
Altered expression of proteins of neuron and glial cytoarchitecture in the AH and PH (beta actin, neurofilament medium polypeptide protein, VIM, nucleoside diphosphate kinase A, F-actin capping protein α-2 subunit, rho GDP-dissociation inhibitor 1, stathmin, 14-3-3 gamma, 14-3-3 epsilon, YWHAZ, gelsolin, tubulin β-4 chain, tubulin α-1 chain and T-complex protein 1 subunit epsilon), some of which if developmental in origin, may give rise to reduction in the extent of axonal or dendritic arborization, resulting in small neuron size and decreased neuropil volume in the hippocampus [4, 47–58]. The altered expression of these proteins is consistent with a previous study showing altered expression of microtubule-associated proteins in specific subfields of the hippocampal formation [59] and reduced neuron size in the PH in schizophrenia [60]. These changes are consistent with abnormal cytoarchitecture, diminished synaptic plasticity and connectivity in the hippocampus, and reports of reduced synaptic proteins and decreased density of mitochondria in axon terminal and dendrites [1, 5, 61, 62].
Altered expression of the proteins discussed here are consistent with the hypothesis that abnormal synaptic circuitry or ‘wiring’ within the AH and its extrinsic connections, especially with the prefrontal cortex (PFC), underlie cognitive dysfunction in schizophrenia [5, 6]. In support of this view a similar network of abnormal pathways and altered protein expression was also reported in the anterior cingulate cortex (ACC) grey matter [24], ACC white matter [63] and the genu of the corpus callosum [21]. Mitochondrial dysfunction and compromised oxidative stress response processes as identified in the present study were also reported in previous studies of the PFC and ACC [64–67]. The abnormal processes identified in the AH can lead to altered neuronal plasticity and brain connectivity, processes that are presently thought to be the core pathology of schizophrenia [68].
It should be noted that although some of these changes may be causative factors, others may be a consequence of the illness or compensatory such as response to medication, as we have shown. Genetic screening of the genes encoding these proteins for potential disease causing mutations, together with assessment of alterations in transcription and translation regulatory processes, may help to distinguish the causative factors. Future follow-up studies in addition to genetic screening, include assessment of the protein changes at the transcript level using quantitative polymerase chain reaction in a larger population and identification of the cellular and subcellular localization of the altered proteins using immunohistochemistry. Changes identified in the level of expressed transcripts will be followed up using quantitative in situ hybridization.
Footnotes
Acknowledgements
This work was supported by Schizophrenia Research Institute (SRI), utilizing infrastructure funding from NSW Health (SS) and the NSW Government BioFirst Award (IM). AH and PH tissues were provided by the NSW Tissue Resource Centre, which is supported by the University of Sydney, SRI, National Institute of Alcohol Abuse and Alcoholism (National Institutes of Health, USA) and NSW Department of Health and NHMRC. Mass spectrometry for the present study was carried out at the Biomedical Node of the Australian Proteome Analysis Facility and was supported in part by grants from the Major National Research Facilities Program and the University of Sydney. Note: Schizophrenia Research Institute was formally known as the Neuroscience Institute of Schizophrenia and Allied Disorders (NISAD)